Introduction
Acute lung injury (ALI) and its severe form, acute
respiratory distress syndrome (ARDS), is a clinical syndrome of
severe lung failure defined by acute onset, bilateral opacities on
the chest radiograph, respiratory failure not fully explained by
cardiac failure or fluid overload, and a ratio of arterial oxygen
to inspired oxygen of <200 mmHg with a positive end-expiratory
pressure of ≥5 cm H2O. ALI/ARDS is characterized by a
disruption of the endothelium and alveolar injury, resulting in an
uncontrolled inflammatory response, including increasing release of
reactive oxygen species (ROS), inflammatory cytokines, protein
content and neutrophil accumulation. Despite numerous studies that
have been performed in recent years, the underlying mechanisms of
ALI/ARDS remain unclear, there are no effective therapies for the
disease and the mortality rate of intensive care patients with
ALI/ARDS is as high as 40–60%, which is a problem in respiratory
medicine (1). Multiple factors may
be involved in the increased vascular permeability, including
endothelium injury, increased levels of pro-inflammatory cytokines
TNF-α (tumor necrosis factor alpha), interleukin 1 (IL-1), or IL-6
and IL-8, and endovascular occlusion associated with the
accumulation of erythrocytes with reduced deformability,
leukocytes, and platelets (2).
Nuclear factor (NF)-κB is a key protein in numerous
signal transduction pathways, the overactivation of which followed
by activation and the response of inflammatory cells serves an
important role in ALI/ARDS. The mammalian NF-κB family consists of
p65 (or RelA), RelB, c-Rel, p50 (or NF-κB1), and p52 (or NF-κB2),
which bind to the κB sites in the DNA of their target genes as
homo- or heterodimers through the conserved Rel homology domain
(RHD) (3). RNA interference is a
specific and effective gene silencing technology, which is able to
specifically inhibit target gene expression and reduce the
corresponding protein level. The present study was primarily aimed
at observing the NF-κB p65 silencing effect through small
interfering RNA (siRNA) targeted to the NF-κB p65 gene, which
prevents monocyte and phorbol myristate acetate (PMA)-induced THP-1
macrophages treated by lipopolysaccharide (LPS) from releasing
IL-1β and TNF-α, providing a basis for novel treatments for
ALI/ARDS.
Materials and methods
Cell culture and plasmids
The pSUPER. retro. neo (VEC-PRT-0003 linear) plasmid
DNA and the retrovirus packaging cell line 293A were kindly
provided by Dr Yang Yizeng (NIH Center for Molecular Studies in
Digestive and Liver Diseases, Perelman School of Medicine,
University of Pennsylvania, Philadelphia, PA, USA). The human
monocyte THP-1 cell line was kindly provided by the Department of
Pharmacology, Xuanwu Hospital of Capital Medical University
(Beijing, China), and the NIH3T3 cell line was provided by
Institute of Neurology, Basic Medical Sciences of Peking University
Health Science Center (Beijing, China). THP-1 and NIH3T3 cell lines
were cultured in RPMI 1640 medium supplemented with 10% fetal
bovine serum (FBS) and penicillin (100 U/ml) streptomycin (100
µg/ml). 293A cells were cultured in Dulbecco's modified Eagle's
medium supplemented with 10% FBS and penicillin (100 U/ml)
streptomycin (100 µg/ml). DMEM and RPMI-1640 medium were purchased
from Hyclone (GE Healthcare Life Sciences, Logan, UT, USA), and FBS
was purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham,
MA, USA).
Two looped specific NF-κB P65 siRNA sequences and
one scramble control sequence (list in Table I) were synthesized by Beijing
Sunbiotech Co., Ltd. (Beijing, China) and ligated into
pSUPER.retro.neo vector according to the protocol described in a
previous study (4).
 | Table I.siRNA targeting human nuclear
factor-κB p65 and the siControl sequence. |
Table I.
siRNA targeting human nuclear
factor-κB p65 and the siControl sequence.
| Group | Sequence |
|---|
| siRNA1 |
5′-GATCCCCAGCATCCCAGGCGAGAGGATTCAAGAGATCGTAGGGTCCGCTCTCCTTTTTTA-3′ |
|
|
3′-GGGTCGTAGGGTCCGCTCTCCTAAGTTCTCTAGCATCCCAGGCGAGAGGAAAAAATTCGA-5′ |
| siRNA1 |
5′-GATCCCCGGACATATGAGACCTTCAATTCAAGAGATTGAAGGTCTCATATGTCCTTTTTA-3′ |
|
|
3′-GGGCCTGTATACTCTGGAAGTTAAGTTCTCTACTTCCAGAGTATACAGGAAAAATTCGA-5′ |
| siControl |
5′-GATCCCCAACGAGTGTGCCTACATCCTTCAAGAGAGGATGTAGGCACACTCGTTTTTTTA-3′ |
|
|
3′-GGGTTGCTCACACGGATGTAGGAAGTTCTCTCCTACATCCGTGTGAGCAAAAAAATTCGA-5′ |
The NF-κB p65 siRNA vectors were transfected into
293A package cells using Lipofectamine2000™ (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's instructions, and the virus suspension was collected
following 24, 48 or 72 h. Virus titration was performed with NIH3T3
cells. The viral titer was 3.72×106 colony forming units
(CFU)/ml for p65 siRNA1, 3.56×106 CFU/ml for p65 siRNA2
and 3.66×106 CFU/ml for p65 si-Control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNAs were exacted from the cell lines at
80–90% confluence by using TRIzol (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 1 µg RNA was reverse transcribed
using a RevertAid first strand cDNA synthesis kit (Thermo Fisher
Scientific, Inc.). The qPCR was performed using QuantiTect SYBR
Green PCR kit (Qiagen, Inc., Valencia, CA, USA) according to the
manufacturer's protocol. All the primers (listed in Table II) were obtained from Sanbio B.V.
(Uden, The Netherlands). All the PCR reactions were initiated with
incubation at 94°C for 2 min, followed by 29 cycles of 94°C for 30
sec, 58°C for 30 sec and 72°C for 2 min. Reactions were finished
with a 72°C 10 min extension. Data were normalized using the
2−ΔΔCq method (5).
| Table II.Specific primers for NF-κB, IL-1β,
TNF-α and β-actin. |
Table II.
Specific primers for NF-κB, IL-1β,
TNF-α and β-actin.
| Gene | Direction | Primer sequence
(5′-3′) | Size (bp) |
|---|
| NF-κB p65 | Forward |
GTGTTCACAGACCTGGCATCC | 230 bp |
| NF-κB p65 | Reverse |
TCCGCAATGGAGGAGAAGTCT |
|
| IL-1β | Forward |
TGTACCTGTCCTGCGTGTTG | 316 bp |
| IL-1β | Reverse |
GCCCTAGGGATTGAGTCCAC |
|
| TNF-α | Forward |
CCCATCCCCAATAACAATCCA | 262 bp |
| TNF-α | Reverse |
GAGCTCTGCAGTTGGGACAGT |
|
| β-actin | Forward |
GGGAAATCGTGCGTGACAT | 385 bp |
| β-actin | Reverse |
TCAGGAGGAGCAATGATCTTG |
|
Western blotting
Total protein were isolated from cultured cells by
300 µl ice cold lysis buffer containing 1% NP-40, 50 mmol/l Tris
(pH 7.4), 150 mmol/l NaCl, 0.1% SDS, 0.5% deoxycholate, 200 µg/ml
phenylmethanesulfonyl fluoride and 50 µg/ml aprotinin. Insoluble
materials were removed by ultracentrifugation at 15,000 × g for 30
min for 4°C. The concentration of the extracted protein was
measured spectrophotometrically with Coomassie G-250. Clarified
protein lysates (50 µg) were electrophoretically resolved on
denaturing SDS-PAGE (8–12%). The proteins were transferred onto
polyvinylidene fluoride membranes using a wet transfer method
following PAGE, and subsequently blocked with 3% bovine serum
albumin (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 1 h
at room temperature and washed with Tris-buffered saline and
Tween-20 three times. Antibodies specific for NF-κB p65 (mouse
anti-human IgG; 1:500; cat. no. sc-8008) and GAPDH (mouse
anti-human IgG; 1:10,000; cat. no. sc-47724) were used to probe
membranes for 2 h at room temperature, followed by
peroxidase-conjugated secondary antibodies (goat anti-mouse IgG;
1:5,000; cat. no. sc-3697) for 1 h at room temperature and enhanced
chemiluminescence detection (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). All primary and secondary antibodies were obtained from
Santa Cruz Biotechnology, Inc. For quantification of band
intensity, appropriate films were scanned and band densities were
determined using Quantity One software (version 4.6.6; Bio-Rad
Laboratories, Inc., Hercules, CA, USA), normalized against GAPDH,
and presented as a ratio of control.
ELISA
Secretory levels of IL-1β and TNF-α in culture
supernatants were determined by the Quantikine ELISA kit (R&D
Systems, Inc. Minneapolis, MN, USA, cat. nos. DLB50 and DTA00C).
The color generated was determined by measuring the OD at 450 nm
using a spectrophotometric microplate reader. A standard curve was
run on each assay plate using serial dilutions of recombinant IL-1β
and TNF-α. The experiment was repeated three times and results were
presented as the mean value.
Statistical analysis
Data are presented as the mean ± standard deviation.
All data analyses were performed using SPSS software version 13.0
for Windows (SPSS, Inc., Chicago, IL, USA). One-way analysis of
variance (ANOVA) and independent-samples t-tests were conducted.
Tukey's test was used for post-hoc testing following the ANOVA. All
experiments were performed at least three times with similar
results. P<0.05 was considered to indicate a statistically
significant difference.
Results
Inhibition of mRNA expression by p65
siRNA retroviruses
In a previous study, the authors successfully
constructed the p65 siRNA retroviruses using gene recombination
technology. In the present study, the effect of p65 siRNA
retroviruses on the expression of NF-κB p65 mRNA in THP-1 cells was
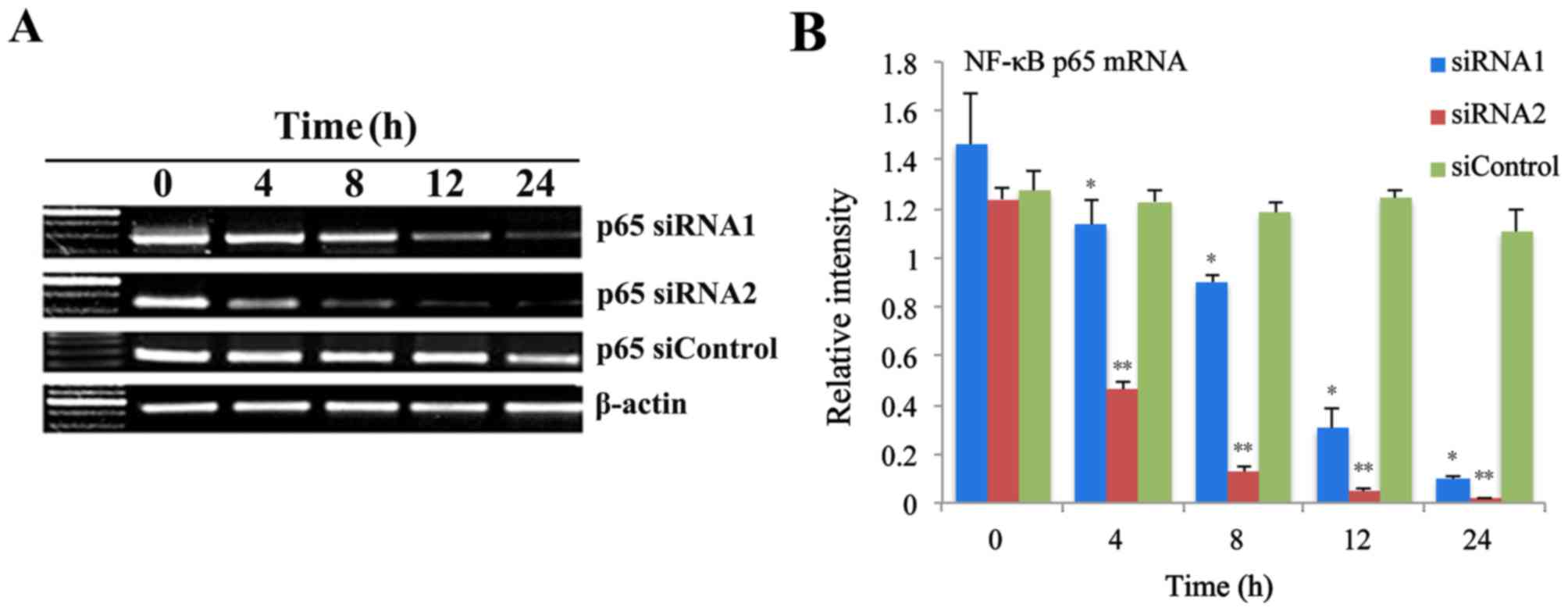
investigated. Fig. 1A revealed the
levels of p65 mRNA at 0, 4, 8, 12 and 24 h following infection with
p65 siRNA retroviruses, as measured by RT-qPCR. Statistical
analysis demonstrated that the p65 mRNA expression levels were
significant reduced at 4, 8, 12 and 24 h following infection with
p65 siRNA1 and p65 siRNA2 retroviruses, while no significant
difference in THP-1 cells infected with siControl retroviruses
(Fig. 1B).
Inhibition of protein expression by
p65 siRNA retroviruses
To further investigate the inhibitory effects of p65
siRNA retroviruses on the NF-κB signaling pathway, the expression
of NF-κB p65 protein was detected using western blot analysis. The
level of p65 protein in THP-1 cells infected with p65 siRNA
retroviruses at 0, 4, 8, 12, 24, 48 and 72 h was presented in
Fig. 2A. Statistical analysis
revealed that the protein expression levels of p65 in THP-1 cells
was significantly decreased from 12 h to 72 h following infection
with p65 siRNA1 retroviruses, while decreased from 8 h to 72 h
following infection with p65 siRNA2 retroviruses. There was no
significant difference of p65 protein expression at different time
in THP-1 cells following infection with siControl retroviruses
(Fig. 2B).
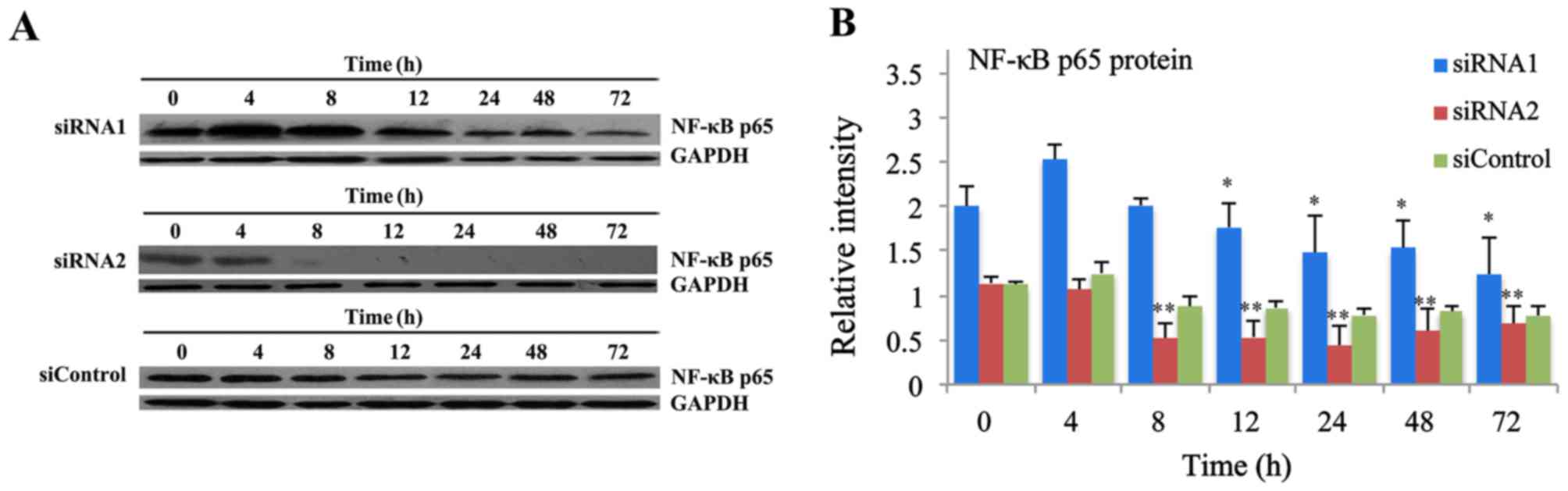 | Figure 2.Inhibitory effects of p65 siRNA
retroviruses on p65 protein expression levels. (A) The expression
levels of p65 protein at 0, 4, 8, 12, 24, 48 and 72 h in THP-1
cells transfected with p65 siRNA retroviruses were measured by
western blot analysis. (B) The protein expression levels of p65
were significantly reduced at 12, 24, 48 and 72 h in THP-1 cells
following transfection with p65 siRNA1 retroviruses, while
decreased at 8, 12, 24, 48 and 72 h following transfection with p65
siRNA2 retroviruses. There was no significant difference of protein
expression at different times in THP-1 cells infected with
siControl retroviruses. Data are expressed as the mean ± standard
deviation (n=5). *P<0.05, **P<0.01 vs. 0 h. siRNA, small
interfering RNA; NF-κB, nuclear factor-κB. |
The RT-qPCR and western blot analysis revealed that
the siRNA2 was the most effective in decreasing p65 expression, so
p65 siRNA2 was used for further research.
Inhibition of pre-inflammation
cytokine IL-1β release by p65 siRNA retroviruses
In order to investigate the effect of p65 siRNA
retroviruses on the pro-inflammatory cytokines release stimulated
by LPS, RT-qPCR was used to detect the expression of IL-1β in THP-1
cells. The level of IL-1β mRNA in THP-1/p65(−) and THP-1/siControl
cells stimulated by LPS (0.1 µg/ml) at 0, 4, 8, 12 and 24 h is
presented in Fig. 3. Statistical
analysis revealed that the mRNA expression levels of p65 was
significantly decreased at 12 h and 24 h in THP-1/p65(−) cells
following stimulation with LPS, while increased at 12 h and 24 h in
THP-1/siControl cells (Fig. 3B and
D).
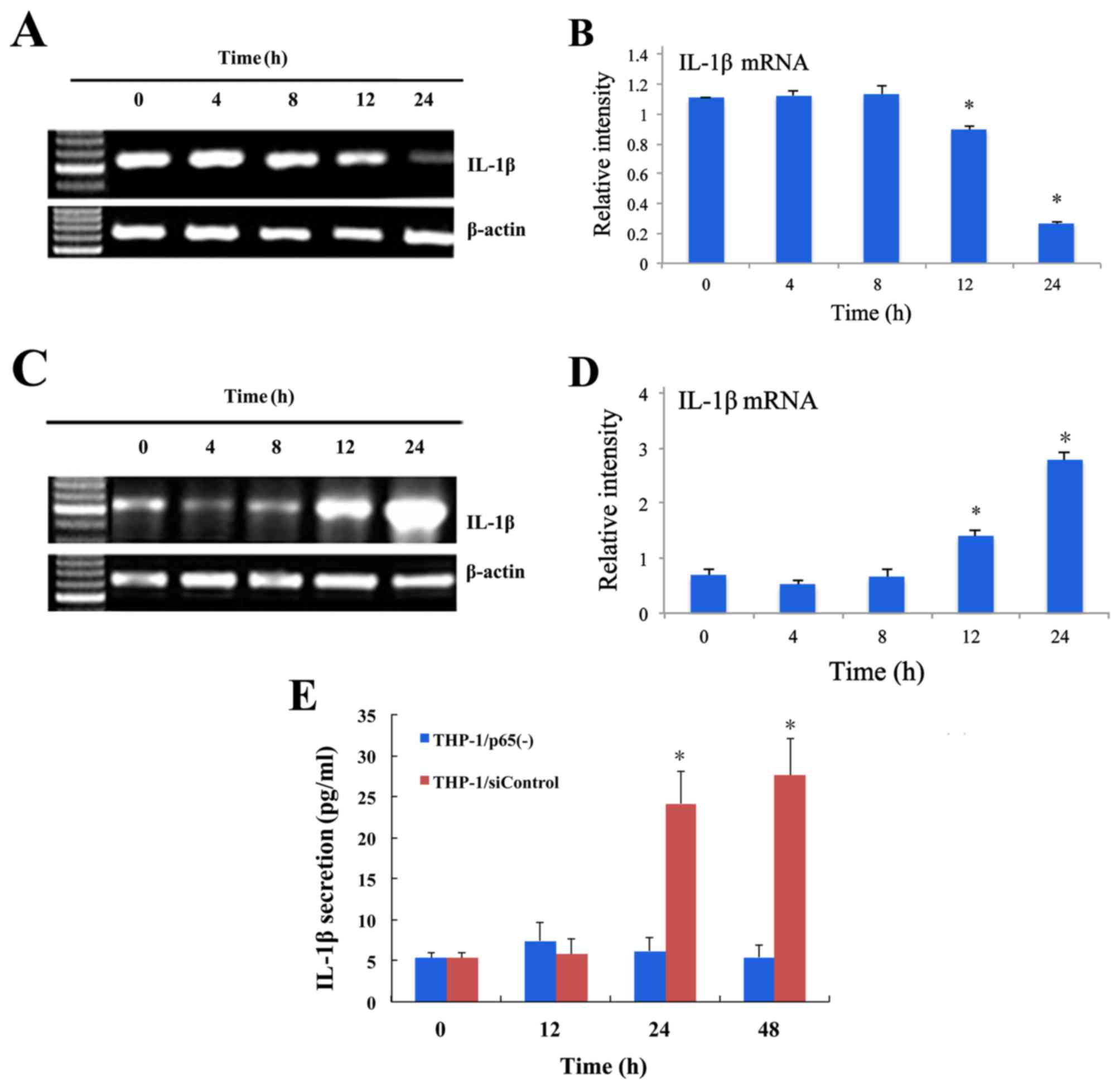 | Figure 3.Inhibitory effects of p65 siRNA
retroviruses on IL-1β expression, as assessed by RT-qPCR and ELISA
in THP-1/p65(−) and THP-1/siControl cells stimulated by LPS. (A)
The THP-1/p65(−) cells were treated with LPS (0.1 g/ml) and mRNA
levels of IL-1β at 0, 4, 8, 12 and 24 h were measured by RT-qPCR.
(B) The mRNA expression levels of IL-1β were significantly reduced
at 12 h and 24 h compared with at 0 h. Data are expressed as the
mean ± standard deviation (n=5). (C) The THP-1/siControl cells were
treated with LPS (0.1 g/ml) and mRNA levels of IL-1β at 0, 4, 8, 12
and 24 h were measured by RT-qPCR. (D) The mRNA expression levels
of IL-1β were significant increased at 12 h and 24 h compared with
at 0 h. Data are expressed as the mean ± standard deviation (n=5).
(E) Inhibitory effect of p65 siRNA retroviruses on IL-1β secretion
assessed by ELISA in THP-1 cells stimulated by LPS. The
THP-1/p65(−) and THP-1/siControl cells were treated with LPS (0.1
g/ml) and levels of IL-1β at 0, 12, 24 and 48 h was measured by
ELISA. Treatment with LPS resulted in a significant increase in the
secretion of IL-1β at 24 h and 48 h in THP-1/siControl cells.
However, the secretion of IL-1β sustained a lower level and there
was no significant difference at 0, 12, 24 and 48 h in THP-1/p65(−)
cells stimulated by LPS. Data are expressed as the mean ± standard
deviation (n=5). *P<0.05 vs. 0 h. THP-1/p65(−) cells, THP-1
cells infected with p65 siRNA2 retrovirus. THP-1/siControl cells,
THP-1 cells infected with siControl retroviruses. siRNA, small
interfering RNA, IL, interleukin; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; LPS,
lipopolysaccharide. |
To further evaluate whether p65 siRNA retroviruses
could suppress IL-1β production, ELISA was used to measure the
secretion of IL-1β in THP-1/p65(−) and THP-1/siControl cells
stimulated by LPS. As presented in Fig. 3E, treatment with LPS resulted in a
significant increase in the secretion of IL-1β at 24 h and 48 h in
THP-1/siControl cells. However, the secretion of IL-1β sustained a
lower level and there was no significant difference at 0 h, 12 h,
24 h and 48 h in THP-1/p65(−) cells following stimulation with LPS
(Fig. 3E).
Inhibition of proinflammatory cytokine
TNF-α release by p65 siRNA retroviruses
To explore the effect of p65 siRNA retroviruses on
the production of TNF-α, THP-1 differentiated macrophage-like
(THP-1/M) cells were used. When the THP-1 cells were pretreated
with PMA (100 nM/ml) for 48 h, the THP-1 cells were almost
completely induced to differentiate into macrophage-like cells
(Fig. 4).
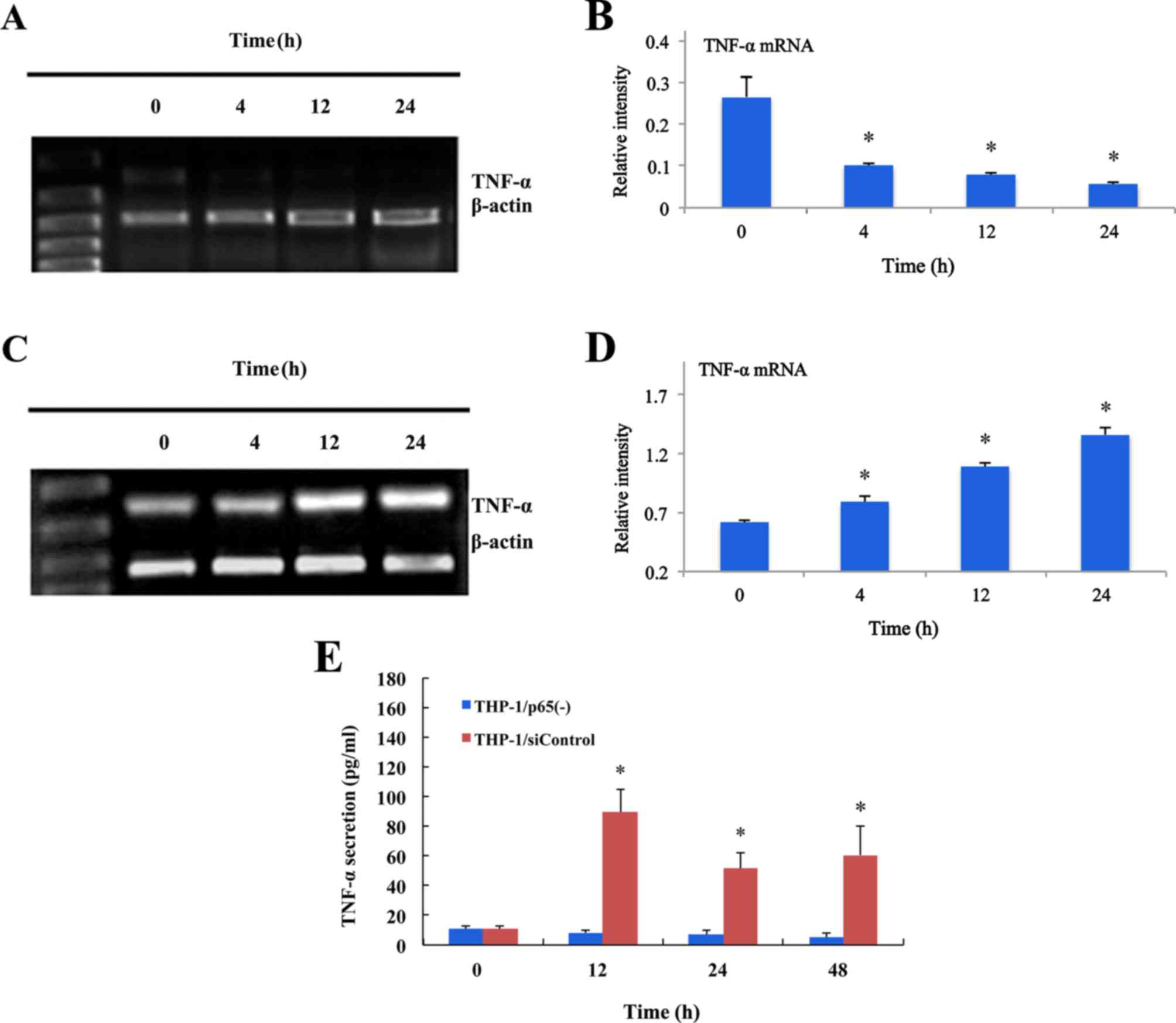 | Figure 4.Inhibitory effect of p65 siRNA
retroviruses on TNF-α expression assessed by RT-qPCR and ELISA in
THP-1 cells stimulated by LPS. (A) The THP-1/siControl cells were
treated with LPS (0.1 g/ml) and mRNA levels of TNF-α at 0, 4, 12
and 24 h were measured by RT-qPCR. (B) The mRNA expression levels
of TNF-α were significant reduced at 4, 12 and 24 h compared with
at 0 h (n=4). (C) The THP-1/siControl cells were treated with LPS
(0.1 g/ml) and mRNA levels of TNF-α at 0, 4, 12 and 24 h were
measured by RT-qPCR. (D) The mRNA expression levels of TNF-α were
significantly increased at 4, 12 and 24 h compared with at 0 h
(n=4). (E) Inhibiting effect of p65 siRNA retroviruses on TNF-α
secretion assessed by ELISA in THP-1 cells stimulated by LPS. The
THP-1/p65(−) and THP-1/siControl cells were treated with LPS (0.1
g/ml) and secretory levels of TNF-α at 0, 12, 24 and 48 h were
measured by ELISA. Treatment with LPS resulted in a significant
increase in the secretion of TNF-α at 12, 24 and 48 h in
THP-1/M/siControl cells. However, the secretion of TNF-α sustained
a lower level and there was no significant difference at 0, 12, 24
and 48 h in THP-1/M/p65(−) cells (n=5). Data are expressed as the
mean ± standard deviation. *P<0.05 vs. 0 h. THP-1/p65(−) cells,
THP-1 cells infected with p65 siRNA2 retroviruses; THP-1/siControl
cells, THP-1 cells infected with siControl retroviruses. siRNA,
small interfering RNA; TNF-α, tumor necrosis factor-α; LPS,
lipopolysaccharide. |
The level of TNF-α mRNA in THP-1/M/p65(−) and
THP-1/M/siControl cells stimulated by LPS (0.1 µg/ml) at 0, 4, 12
and 24 h was presented in Fig. 4A and
C, as measured by RT-qPCR. Statistical analysis revealed that
the mRNA expression levels of TNF-α was significantly decreased in
THP-1/M/p65(−) cells at 4, 12 and 24 h stimulated by LPS, while
increased in THP-1/M/siControl cells at 4, 12 and 24 h stimulated
by LPS (Fig. 4B and D).
In addition, to confirm whether the inhibition of
TNF-α mRNA correspond to a decrease in TNF-α protein, the secretion
of TNF-α in THP-1/M/p65(−) and THP-1/M/siControl cells stimulated
by LPS (0.1 µg/ml) was measured by ELISA. As demonstrated in
Fig. 4E, treatment with LPS
resulted in a significant increase in the secretion of TNF-α at 12,
24 and 48 h in THP-1/M/siControl cells. However, the secretion of
TNF-α sustained a lower level and there was no significant
difference at 0, 12, 24 and 48 h in THP-1/M/p65(−) cells stimulated
by LPS (Fig. 4E).
Discussion
ALI is a condition of acute respiratory failure,
characterized by diffuse pulmonary infiltrates and severe hypoxemia
(6). During ALI, the acute phase
of inflammation induces the recruitment of activated inflammatory
cells, including macrophages and lymphocytes, to the damaged
lesions (7). Macrophages and
lymphocytes are circulating immune cells that serve a crucial role
in secreting proinflammatory cytokines and inactivating
inflammatory mediators in the ischemic region (8).
NF-κB is a critical signal transcription factor for
regulating immune and inflammatory responses (9). Importantly, the activity of NF-κB is
regulated by its subcellular localization, and NF-κB is retained in
the cytosol when bound to its inhibitor, IκB. Activation of the IκB
proteins, which can be induced by a variety of stimuli, such as
pro-inflammatory cytokines, allows NF-κB to be released from IκB
and translocate to the nucleus where it can initiate transcription
by binding to numerous specific gene promoter elements. The
activation of the nuclear transcription factor NF-κB is closely
associated with the excessive release of pro-inflammatory and
inflammatory factors, such as TNF-α and IL-1β (10).
IL-1β is a member of inflammatory factors that is
implicated in the pathogenesis of acute respiratory distress
syndrome. It has been reported that IL-1β is one of the most
biologically active cytokines in early phase of ALI, which is
elevated in plasma, and that IL-1β is a potent inducer of lung
which causes release of a variety of pro-inflammatory cytokines,
such as monocyte chemotactic protein-1, macrophage inflammatory
protein-1, IL-6 and IL-8 with subsequent recruitment of
inflammatory cells into the airspaces as well as being able to
alter endothelial-epithelial barrier permeability and fluid
transport leading to edema (11).
TNF-α, a pro-inflammatory cytokine, is secreted in the pulmonary
tissue by activated immune cells, which control the inflammatory
process and accelerate secondary inflammatory processes by inducing
inflammatory molecules, such as intercellular adhesion molecules,
vascular cell adhesion molecules-1 and selectin (12,13).
TNF-α is also responsible for the accumulation of inflammatory
cells in the peripheral nucleus of pulmonary tissue, and it induces
a second inflammatory response.
Several studies have proven that the inhibition of
NF-κB activity leads to the reduction of the excessive release of
inflammatory factors and may be a potential method for the clinical
treatment of ALI (14,15). However, an effective drug target to
inhibit NF-κB has not been reported so far. RNA interference, which
was first identified in Caenorhabditis elegans in 1998, is a
process of sequence-specific, post-transcriptional gene silencing
in animals and plants, initiated by siRNA homologous in sequence to
the silenced gene. RNA interference technique is now extensively
employed in genetic engineering as a simple and effective gene
knockdown tool (16,17). The results demonstrated that p65
siRNA could significantly suppress the expression of NF-κB p65 mRNA
and protein level, which confirmed that p65 siRNA could
significantly inhibit the activity of NF-κB.
As the NF-κB signaling pathway is activated during
ALI, the authors also investigated the effect of siRNA retroviruses
on the activity of the NF-κB signaling pathway. The results showed
that the expression level of NF-κB p65 mRNA and protein significant
decreased in THP-1 cells infected by p65 siRNA retroviruses, which
indicated that p65 siRNA may inhibit the expression of NF-κB
p65.
Furthermore, the authors detected the effect of
siRNA retroviruses on the release of inflammatory cytokines in
THP-1 and THP-1/M cells stimulated by LPS. LPS, a component of the
outer membrane of Gram-negative bacteria, is considered to be the
most potent activator of monocytes and macrophages. Monocytes and
macrophages serve an essential role in inflammation and
mobilization of the host defense against bacterial infection
(18).
As a result, the expression levels of IL-1β and
TNF-α mRNA and secreted protein were significantly increased in
THP-1 and THP-1/M cells, which suggested that LPS may promoted the
release of inflammation cytokines, such as IL-1β and TNF-α. For the
siRNA group, the results indicated that IL-1β and TNF-α mRNA
expression levels was decreased, while the secreted protein
expression levels exhibited no significant difference at different
times in THP-1/p65(−) cells stimulated by LPS. The authors
speculated that the protein secretion levels of IL-1β and TNF-α
were very low without LPS stimulation, so the p65 siRNA may only
inhibit the release of IL-1β and TNF-α protein secretion stimulated
by LPS, rather than reduce the expression levels of secreted
protein in THP-1 cells.
The results proved that NF-κB serves an essential
role in regulating the release of inflammatory factors. The
inhibition of the excessive release of inflammatory factors through
NF-κB signal pathway may be an important method for the clinical
treatment of ALI.
In summary, the results demonstrated that
suppressing the expression of the NF-κB signaling pathway through
NF-κB p65 siRNA causes an inhibition to release of inflammatory
factors induced by LPS. The results suggested that disruption of
the NF-κB signaling pathway represents an opportunity for rational
drug design for ALI prevention or treatment. These insights may
help develop NF-κB activity based pharmacological strategies to
regulate inflammation but further study is required in
vivo.
References
|
1
|
Wheeler AP and Bernard GR: Acute lung
injury and the acute respiratory distress syndrome: A clinical
review. Lancet. 369:1553–1564. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kong MY, Gaggar A, Li Y, Winkler M,
Blalock JE and Clancy JP: Matrix metalloproteinase activity in
pediatric acute lung injury. Int J Med Sci. 6:9–17. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Harbi NO, Imam F, Al-Harbi MM, Ansari
MA, Zoheir KM, Korashy HM, Sayed-Ahmed MM, Attia SM, Shabanah OA
and Ahmad SF: Dexamethasone attenuates LPS-induced acute lung
injury through inhibition of NF-κB, COX-2, and Pro-inflammatory
mediators. Immunol Invest. 45:349–369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu CT, Zhu GF, Zhao JH, Zhang XF, Jin LY
and Yan SF: Effect of retroviral vector mediating RNA interference
on the release of tumor necrosis factor-alpha in
lipopolysaccharide-stimulating macrophage. Zhonghua Yi Xue Za Zhi.
90:1283–1287. 2010.(In Chinese). PubMed/NCBI
|
|
5
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang X, Sun CY, Zhang YB, Guo HZ, Feng
XX, Peng SZ, Yuan J, Zheng RB, Chen WP, Su ZR and Huang XD: Kegan
Liyan oral liquid ameliorates lipopolysaccharide-induced acute lung
injury through inhibition of TLR4-mediated NF-κB signaling pathway
and MMP-9 expression. J Ethnopharmacol. 186:91–102. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim YS, Hwang JW, Jang JH, Son S, Seo IB,
Jeong JH, Kim EH, Moon SH, Jeon BT and Park PJ: Trapa japonica
pericarp extract reduces LPS-induced inflammation in macrophages
and acute lung injury in mice. Molecules. 21:3922016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun CY, Xu LQ, Zhang ZB, Chen CH, Huang
YZ, Su ZQ, Guo HZ, Chen XY, Zhang X, Liu YH, et al: Protective
effects of pogostone against LPS-induced acute lung injury in mice
via regulation of Keap1-Nrf2/NF-κB signaling pathways. Int
Immunopharmacol. 32:55–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qiu J, Yu L, Zhang X, Wu Q, Wang D, Wang
X, Xia C and Feng H: Asiaticoside attenuates
lipopolysaccharide-induced acute lung injury via down-regulation of
NF-κB signaling pathway. Int Immunopharmacol. 26:181–187. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang J, Liu YT, Xiao L, Zhu L, Wang Q and
Yan T: Anti-inflammatory effects of apigenin in
lipopolysaccharide-induced inflammatory in acute lung injury by
suppressing COX-2 and NF-κB pathway. Inflammation. 37:2085–2090.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang L, Li D, Zhuo Y, Zhang S, Wang X and
Gao H: Protective role of liriodendrin in sepsis-induced acute lung
injury. Inflammation. 39:1805–1813. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lucas SM, Rothwell NJ and Gibson RM: The
role of inflammation in CNS injury and disease. Br J Pharmacol. 147
Suppl 1:S232–S240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perera M Nilupul, Ma HK, Arakawa S,
Howells DW, Markus R, Rowe CC and Donnan GA: Inflammation following
stroke. J Clin Neurosci. 13:1–8. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang D, Sun Y, Shen Y, Li F, Song X, Zhou
E, Zhao F, Liu Z, Fu Y, Guo M, et al: Shikonin exerts
anti-inflammatory effects in a murine model of
lipopolysaccharide-induced acute lung injury by inhibiting the
nuclear factor-kappaB signaling pathway. Int Immunopharmacol.
16:475–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu T, Wang DX, Zhang W, Liao XQ, Guan X,
Bo H, Sun JY, Huang NW, He J, Zhang YK, et al: Andrographolide
protects against LPS-induced acute lung injury by inactivation of
NF-κB. PLoS One. 8:e564072013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fire A, Xu S, Montgomery MK, Kostas SA,
Driver SE and Mello CC: Potent and specific genetic interference by
double-stranded RNA in Caenorhabditis elegans. Nature. 391:806–811.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Elbashir SM, Harborth J, Lendeckel W,
Yalcin A, Weber K and Tuschl T: Duplexes of 21-nucleotide RNAs
mediate RNA interference in cultured mammalian cells. Nature.
411:494–498. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Greenlee-Wacker MC: Clearance of apoptotic
neutrophils and resolution of inflammation. Immunol Rev.
273:357–370. 2016. View Article : Google Scholar : PubMed/NCBI
|