Introduction
Anti-glomerular basement membrane glomerulonephritis
(anti-GBM GN) is an aggressive form of autoimmune diseases of the
rapidly progressive glomerulonephritis (RPGN) category, which is
characterized by a rapid decline in renal function and the
formation of glomerular crescents (1). Previous studies have reported that
CD4+ T cell-mediated immunity serves an important role
in the pathogenesis of anti-GBM GN (2–5).
CD4+ T cells may differentiate into four major subsets,
including T helper (Th)1, Th2, Th17 and regulatory T (Tregs) cells,
as defined by their pattern of cytokine production and function
(6–8). Our previous study revealed that
anti-GBM GN was driven by Th1 cell-mediated immune response and
Th17 cells also contributed to the progress of anti-GBM GN
(4). However, the combined actions
of Th1 and Th17 cells lead to renal injury from a number of
activated mechanisms. The immunopathology of Th17 cell-mediated
immune response develops early and may be followed later by Th1
cell-mediated injury (9). Tregs
are a special subset of T cell that serve a protective role in
anti-GBM GN and significantly reduce renal injury (10). The activity of Th1 and Th17 cells
may be attenuated by the anti-inflammatory actions of Tregs, which
inhibits T cell proliferation and T effector function (11). Although Th17 cells and Tregs serve
different functions, they have the same precursor cells, which may
be differentiated to different cell types through pleiotropic
cytokines (12,13). Interleukin (IL)-6 and transforming
growth factor (TGF)-β1 work together to direct T cells to a Th17
phenotype, whereas TGF-β1 alone instructs T cells to function as
Tregs (14,15). In certain conditions, Tregs may be
converted to Th17 cells. Therefore, investigating the gene
regulatory mechanisms of Th17 and Treg cell differentiation may
contribute to the discovery of crucial factors that are involved in
activation of the immune responses in anti-GBM GN.
Inhibitor of DNA binding 3 (ID3) is a 13 kDa nuclear
protein that is upregulated in a number of cell types upon
stimulation by several growth and differentiation signals (16,17),
including T cell receptor (TCR)- and B cell receptor (BCR)-mediated
signals in T and B lymphocytes, respectively (18,19).
As a member of the basic-helix-loop-helix (bHLH) transcription
factor family, ID3 is unique in that it lacks the basic region
required for DNA binding (20);
however, ID3 retains the functional dimerization domain, and is an
inhibitor of the bHLH protein E2A by binding to its target genes
and forming inactive heterodimers (20,21).
Previous studies reported that ID3, as a transcription factor that
is involved in T cell development, regulates the TGF-β1-mediated
reciprocal differentiation of Tregs and Th17 cells in mice
(21,22). However, how ID3 is involved in the
progression of anti-GBM GN and the mechanisms by which ID3 may
alter the profiles of Tregs and Th17 cells in anti-GBM GN remain
unknown.
The present study demonstrated that the Th17- and
Treg cell-mediated immune response contributed to anti-GBM GN in
mice. With the development of anti-GBM GN, ID3 involvement in
CD4+ T cell differentiation led to a downregulation in
Th17 cells, which may affect the delicate balance between Th17
cells and Tregs by skewing CD4+ T cell differentiation
towards Tregs. This effect of ID3 may depend on binding with E2A.
The present results suggested that ID3 may protect mice against
anti-GBM GN.
Materials and methods
Mice
Male C57BL/6 mice (age, 6–8 weeks; weight, 20–22 g;
n=40) were obtained from Beijing HFK Bioscience Co., Ltd. (Beijing,
China) and raised in the Animal Care Unit of Tongji Medical
College, Huazhong University of Science and Technology (Wuhan,
China). All mice were provided with water and a standard laboratory
diet ad libitum, and were housed at room temperature
(23–26°C) with 50% humidity and a 12-h light/dark cycle, receiving
humane care in accordance with governmental and institutional
guidelines. All experimental procedures were approved by the Animal
Care and Use Committee of Tongji Medical College. Each experiment
was replicated three times.
Induction of anti-GBM GN in mice
Anti-GBM serum was harvested from rabbits immunized
by C57BL/6 mice GBMs, according to standard laboratory protocols
(4). All mice were randomly
divided into two groups (n=20/group), the Control group and the
anti-GBM GN group. All mice were preimmunized with a subcutaneous
injection of rabbit immunoglobulin (Ig)G (0.02 mg/g; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) and complete Freund's adjuvant
(Sigma-Aldrich; Merck KGaA) 10 days prior to the induction of
nephritis. A total of 10 days later, anti-GBM serum (0.02 ml/g) was
administered through the tail vein to induce nephritis in mice in
the anti-GBM GN group, whereas the Control group mice received
normal rabbit IgG (0.02 ml/g). Mice were sacrificed at day 7, 14,
21 and 28.
Renal function analysis
Following sacrifice, blood and urine samples were
collected at day 7, 14, 21 and 28, and tested with the Serum
Creatinine (SCR) Assay kit and the Urea Assay kit (BioAssay
Systems, Hayward, CA, USA). Urine albumin excretion was determined
with the Bicinchoninic Acid (BCA) Protein Assay kit (Beyotime
Institute of Biotechnology, Shanghai, China). The absorbance of the
final reactant was determined at 450 nm with an ELISA plate reader
(BioTek Instruments, Inc., Winooski, VT, USA). All assays were
performed according to the kit manufacturer's protocols.
Histology
Following sacrifice, the renal cortex was
immediately harvested from each mouse, and fixed with 4%
paraformaldehyde overnight at room temperature, dehydrated in a
series of ethanol (80, 95 and 100%, respectively; 5 min each), and
embedded in paraffin. Renal tissues were sectioned (3 µm) and
stained with periodic acid-Schiff (PAS) reagent at room temperature
for 10 min for histological analysis. Glomerular crescent formation
and glomerular sclerosis (deposition of PAS-positive material) were
assessed in 30 glomeruli per slide in a blinded manner in
PAS-stained paraffin sections. Specimens were examined with an
Olympus electron microscope (Olympus Corporation, Tokyo,
Japan).
To analyze the deposition of rabbit IgG and mouse
IgG in kidneys, snap-frozen renal cortex sections (5 µm; frozen in
liquid nitrogen and stored at −80°C) were fixed in ice cold acetone
for 5 min at −20°C, air dried for 30 min, and then were stained
directly with fluorescein isothiocyanate (FITC)-conjugated sheep
anti-rabbit IgG (cat. no. F6005; 1:100; Sigma-Aldrich; Merck KGaA)
and FITC-conjugated sheep anti-mouse IgG (cat. no. F3008; 1:100;
Sigma-Aldrich; Merck KGaA) antibodies at room temperature for 25
min. At least 10 cortical interstitial fields (excluding
perivascular areas; magnification, ×400) were examined per sample.
Specimens were examined with an Olympus electron microscope
(Olympus Corporation).
Leukomonocyte isolation from
spleen
Leukomonocytes were isolated from mouse spleens
according to a previously published protocol (4). Briefly, tissues from 3 mice per in
vitro experiment, were homogenized and sequentially passed
through 70 and 40 µm nylon mesh. In case of spleen single-cell
suspension, erythrocytes were lysed using Red Blood Cell Lysis
Buffer (Biolegend, Inc., San Diego, CA, USA). Subsequently, cells
were washed several times with PBS and resuspended in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.).
The viability of the cells was assessed by trypan blue staining
prior to fluorescence-activated cell sorting (FACS; as described in
the following Flow cytometry subsection).
CD4+ T cell isolation and
Treg and Th17 cell differentiation in vitro
CD4+ T cells were isolated from spleens
of C57BL/6 mice by negative magnetic cell sorting (MACS) using the
EasySep Mouse CD4+ T Cell Enrichment kit (Stemcell
Technologies, Inc., Vancouver, BC, Canada), according to
manufacturer's protocol. The purity of CD4+ T cells was
>90%, as confirmed by FITC-conjugated anti-CD4 staining and
subsequent FACS analysis (as described in the following Flow
cytometry subsection). Purified CD4+ T cells were
incubated with 10 µM carboxy-fluorescein diacetate, succinimidyl
ester (CFDA SE) from the Vybrant CFDA SE Cell Tracer kit
(Invitrogen; Thermo Fisher Scientific, Inc.), according to
manufacturer's protocol, and subsequently cultured in 96-well
plates with RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (Thermo Fisher Scientific, Inc.),
anti-CD3 (2.5 µg/ml) and anti-CD28 (5 µg/ml) at 37°C in a
humidified atmosphere (5% CO2). CD4+ T cell
proliferation was examined. Purified CD4+ T cells
(2×106 cells/ml; in RPMI-1640 medium with 10% FBS) were
differentiated into Th17 cells or Tregs in the presence of
different cytokines and antibodies for 6 days at 37°C. For Th17
cells differentiation, the culture medium was supplemented with
IL-6 (30 ng/ml), TGF-β1 (3 ng/ml), IL-1β (20 ng/ml), tumor necrosis
factor-α (20 ng/ml), IL-23 (20 ng/ml), anti-interferon (IFN)-γ (10
µg/ml) and anti-IL-4 (10 µg/ml) neutralizing antibodies. For Treg
cell differentiation, the cultures were supplemented with TGF-β1
(10 ng/ml), anti-IFN-γ (10 µg/ml) and anti-IL-4 (10 µg/ml).
CD4+ T cell
transfection
Isolated CD4+ T cells (1×106
cells/well) were seeded in 12-well plates and were transfected with
100 nM of either ID3-targeted small interfering (si)RNA (cat. no.
sc-38003; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) or
Scrambled siRNA (cat. no. sc-37007; Santa Cruz Biotechnology, Inc.;
sequences commercially unavailable) using the Amaxa Mouse T Cell
Nucleofector kit (cat. no. VPA-1006; Lonza Group, Ltd., Basel,
Switzerland), according to the manufacturer's protocol. Cells were
gently transferred into 12-well plates (final volume, 2 ml
media/well) and incubated for 24 h at 37°C; transfected
CD4+ T cells were harvested by centrifugation at 71.55 ×
g for 3 min at room temperature and the supernatants were
discarded. The cells were then washed with PBS and collected for
subsequent experiments.
Flow cytometry
For FACS flow cytometric analysis, the following
antibodies were used: FITC-conjugated anti-CD4 (cat. no. 11-0041;
1:200); phycoerythrin (PE)-conjugated anti-CD25 (cat. no. 12-0251;
1:100); PE-conjugated anti-IL-17A (cat. no. 12-7177; 1:100) and
allophycocyanin-conjugated anti-forkhead box P3 (FoxP3; cat. no.
71-5775; all eBioscience; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. All procedures were performed
at 4°C for 25 min, away from light. For staining of intracellular
IL-17A, splenocytes (1×105) were incubated at 37°C in 5%
CO2 for 5 h with phorbol 12-myristate 13-acetate (50
ng/ml; Sigma-Aldrich; Merck KGaA) and ionomycin (1 µg/ml;
Calbiochem; Merck KGaA) in RPMI-1640 with 10% FBS. Brefeldin A (10
µg/ml; Sigma-Aldrich; Merck KGaA) was then added for 30 min at 4°C.
Cells were washed several times over 30 min with PBS and stained
with the cell surface marker FITC-anti-CD4 for 25 min at 4°C. Cells
were incubated for 20 min in the dark at room temperature in
Fixation Buffer (Biolegend, Inc., San Diego, CA, USA) to fix cell
surface marker, and Permeabilization Wash Buffer (Biolegend, Inc.)
was used to permeabilize cell membranes. Subsequently,
intracellular IL-17A was stained using an PE-anti-mouse IL-17A
antibody for 25 min at 4°C; endonuclear FoxP3 staining was
performed using the anti-mouse FoxP3 Staining kit (eBioscience;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Flow cytometry was performed on a BD FACSCalibur Flow
Cytometry System (BD Biosciences, Franklin Lakes, NJ, USA) with FCS
Expression v3 software (De Novo Software, Glendale, CA, USA).
ELISA
The levels of IL-17A, IL-10 and TGF-β1 in serum and
cell supernatants were quantified using ELISA Assay kits (cat. nos.
DKW12-2170, DKW12-2100 and DKW12-2710; Dakewe Bioengineering Co.,
Ltd. Beijing, China), according to the manufacturer's protocol. The
absorbance of the final reactant was detected at 450 nm with an
ELISA Microplate Reader (Thermo Fisher Scientific, Inc.).
Western blotting
Nuclear and total protein extracts were obtained
from cultured CD4+ T cells (in vitro), kidneys
and spleens (in vivo) using Nuclear and Total Protein
Extraction kits (Beyotime Institute of Biotechnology), according to
the manufacturer's protocol. Protein was determined using a BCA
Protein Assay kit (Beyotime Institute of Biotechnology). Equal
amounts of cell culture or tissue protein extracts (30 and 60 µg,
respectively) were separated by 10% SDS-PAGE and blotted onto
polyvinylidene fluoride membranes (Thermo Fisher Scientific, Inc.).
Following blocking for 1 h at room temperature with 5% fat-free dry
milk in TBS containing 0.1% Tween-20, membranes were incubated at
4°C overnight with the following primary antibodies: Mouse anti-ID3
(1:250; cat. no. 556524; BD Biosciences); rabbit anti-E2A (1:250;
cat. no. AP13991b; Abgent, Inc., San Diego, CA, USA); mouse
anti-FoxP3 (1:500; cat. no. AO1042a; Abgent, Inc.); mouse
anti-RAR-related orphan receptor (ROR)-γt (1:250; cat. no. 562663;
BD Biosciences); rabbit anti-Lamin A/C (1:2,000; cat. no. ab108922;
Epitomics; Abcam, Cambridge, MA, USA) and mouse anti-GAPDH
(1:5,000; cat. no. A01020; Wuhan, China). Membranes were then
incubated with the following secondary antibodies for 1 h at 37°C:
Alkaline phosphatase-conjugated anti-rabbit IgG (cat. no.
A120-201AP), anti-rat IgG (cat. no. A110-106AP) or anti-mouse IgG
(cat. no. A90-105AP; all Dakewe Bioengineering Co., Ltd.). Specific
bands were visualized using a
5-bromo-4-chloro-3′indolyphosphate/nitro-blue tetrazolium Alkaline
Phosphatase Color Development kit (Dakewe Bioengineering Co.,
Ltd.), according to the manufacturer's instructions, under
protection from direct light. The optical density of the bands was
quantified using Image J v2.1.4.7 (National Institutes of Health,
Bethesda, MD, USA). Protein expression was normalized to that of
GAPDH.
Co-immunoprecipitation (IP)
Co-IP was performed as previously described
(23). Total proteins (800 µg) of
renal tissues and spleens were homogenized in IP buffer (Tris, pH
7.4, 10% glycerol, 1% Nonidet P-40, protease inhibitors and 500
µmol/l sodium vanadate) containing complete protease inhibitor
cocktail and phosphate inhibitor cocktail A (cat. no. sc-45044;
Santa Cruz Biotechnology, Inc.), and incubated on ice for 30 min.
Supernatants were collected followed by centrifugation (12,000 × g
for 5 min at 4°C). Total proteins (800 µg) was incubated with 4 µg
of either anti-E2A antibody (cat. no. sc-349; Santa Cruz
Biotechnology, Inc.) or rabbit IgG (Abmart, Inc., Shanghai, China)
on ice for 1 h. Protein A/G PLUS-Agarose Beads (100 µl; Abmart,
Inc.) were added and the beads bound to the antigen-antibody
complex were precipitated overnight in a clinical rotor at 4°C. The
beads were washed with IP buffer extensively, and 0.25 ml of SDS
sample loading buffer containing 10% β-mercaptoethanol was added to
the agarose beads and heated at 100°C for 10 min. The denatured
proteins were analyzed by western blotting with mouse anti-ID3
(1:250; cat. no. 556524; BD Biosciences).
Revere transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the splenic and renal
tissues of each mouse (n=6/group) with TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.). A total of 1 µg RNA was used to
synthesize cDNA with the GoScript Reverse Transcription System
(Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocol. The temperature protocol for RT-PCR was as
follows: 94°C for 2 min, followed by 35 cycles of 94°C for 30 sec,
60°C for 30 sec and 72°C for 1 min, and then 72°C for 5 min. qPCR
was performed with 1 µl cDNA in the presence of 1.5 µl (0.3 µM)
specific murine primers and 10 µl Maxima SYBR-Green qPCR Master Mix
(Thermo Fisher Scientific, Inc.). The levels of GAPDH mRNA were
measured as an internal standard for calibration. The thermocycling
conditions for qPCR were as follows: 95°C for 10 min, followed by
40 cycles of 15 sec at 95°C, 15 sec at 58°C and 45 sec at 72°C.
Melting curve analysis was also included (1 cycle of 95°C for 1
min, 55°C for 30 sec and 95°C for 30 sec), in order to verify the
specificity of the amplified PCR products. Primers used were as
follows: ID3 forward, 5′-TTAACCCAGCCCTCTTCACTTAC-3′ and reverse,
5′-CCATTCTCGGAAAAGCCAGT-3′; E2A forward,
5′-GGATCTGAGGTTAATGGCTCGCTC-3′ and reverse
5′-CCTGCATCGTAGTTGGGGGATAAG-3′; FoxP3 forward,
5′-GCAACTCAAGATGCTGTCCA-3′ and reverse, 5′-GGCTGGAAGAGACAGACAGG-3′;
GAPDH forward, 5′-CATCTCCGCCCCTTCTGC-3′ and reverse,
5′-CATCACGCCACAGCTTTCC-3′. The primers for RORγt were purchased
from GeneCopoeia, Inc. (cat. no. MQP030076; Rockville, MD, USA).
Quantification was performed using the 2−ΔΔCq method
(24) and the results were
normalized to those of GAPDH.
Statistical analysis
Results are expressed as the mean ± standard error
of the mean. All data were analyzed by Student's t-test ore one-way
analysis of variance followed multiple comparisons with Tukey's
post hoc test. A value of P<0.05 was considered statistically
significant, and all experiments were repeated at least 3
times.
Results
Th17 and Tregs cell-mediated immune
response contributes to anti-GBM GN in mice
To investigate the kinetic profiles of Th17 and Treg
cells, anti-GBM GN and Control mice were sacrificed at days 7, 14,
21, 28 following the first immunization, and splenocytes were and
analyzed by FACS. Th17 cells (CD4+IL-17A+)
and Tregs (CD4+CD25+FoxP3+) were
increased in anti-GBM GN mice compared with Control mice (Fig. 1A). Notably, the Th17 cell
population significantly increased from day 7 and reached a peak at
day 14, whereas Tregs increased significantly from day 14 up to day
28. The infiltration of leukocytes has been associated with the
increased expression of certain cytokines in peripheral blood
(4). Therefore, quantification of
cytokine expression levels in blood samples were evaluated, which
revealed high levels of IL-17A, IL-10 and TGF-β1 in anti-GBM GN
compared with Control (Fig. 1B).
In addition, expression levels of the transcription factors FoxP3
and RORγt were increased both in renal tissues and spleens (data
not shown).
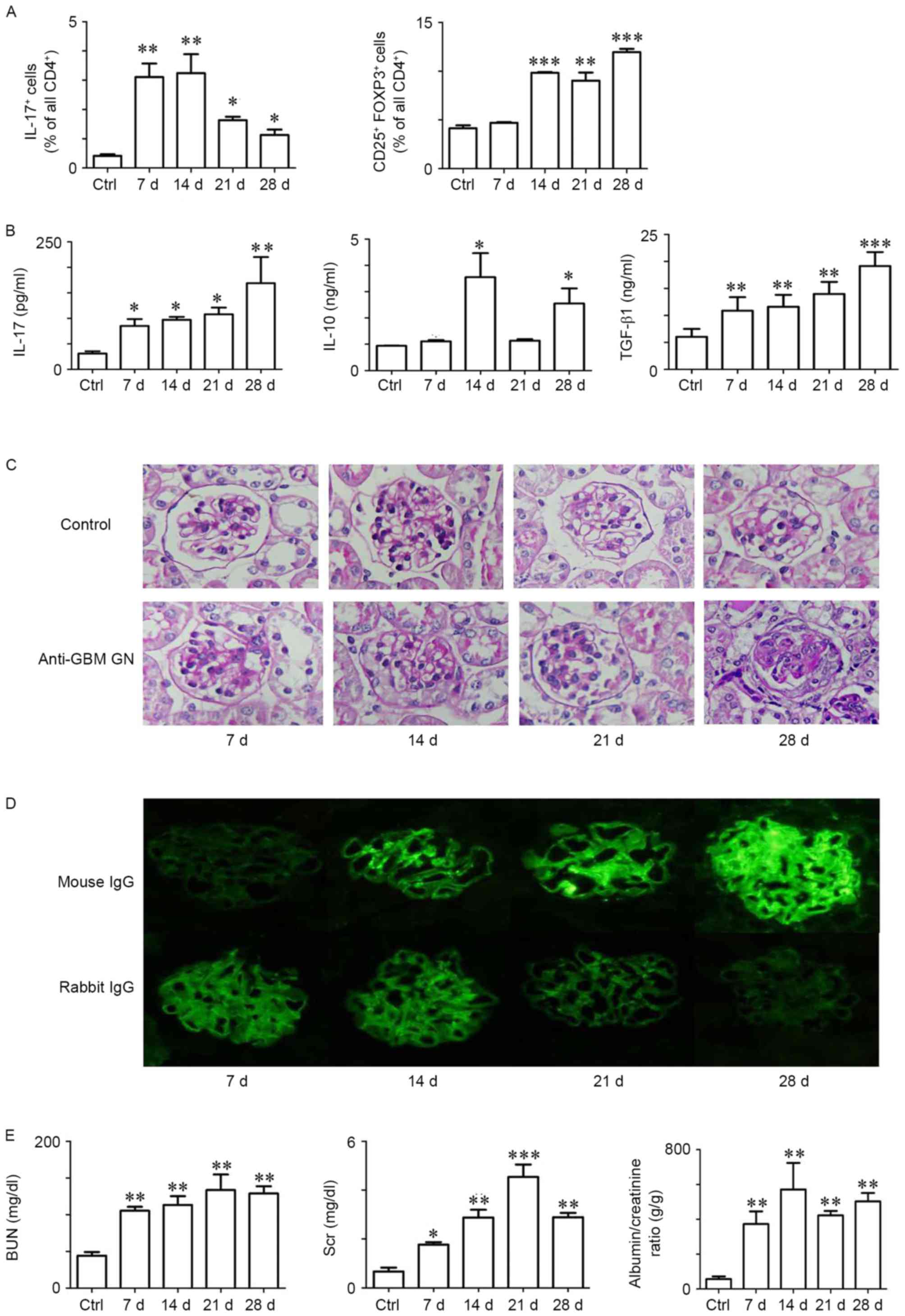 | Figure 1.Th17 and Treg cell-mediated immune
response contributes to anti-GBM GN in mice. (A) Quantification of
the percentage of IL-17A+ and CD25+
FoxP3+ cells in the CD4+ T cell subset during
the time course of the GN. Flow cytometric analysis of splenocytes
isolated from Control and anti-GBM GN mice at day 7, 14, 21 and 28
following GN induction. (B) Protein expression levels of IL-17A,
IL-10 and TGF-β1 in peripheral blood were detected by ELISA in
Control and nephritic mice at days 7, 14, 21 and 28. (C)
Representative photographs of periodic acid-Schiff-stained kidney
sections in Control and nephritic mice at day 7, 14, 21 and 28;
magnification, ×400. (D) Representative photographs of the
deposition of mouse IgG and rabbit IgG in the kidneys. (E) BUN and
Scr concentrations and albumin/creatinine ratio in urine from
Control and nephritic mice were analyzed in Control and nephritic
mice at day 7, 14, 21 and 28. Data are presented as the mean ±
standard error of the mean; n=3-4/group; *P<0.05, **P<0.01
and ***P<0.005 vs. Control. BUN, blood urea nitrogen; Ctrl,
control; d, day; FoxP3, forkhead box P3; GBM, glomerular basement
membrane; GN, glomerulonephritis; IgG, immunoglobulin G; IL,
interleukin; Scr, serum creatinine; TGF, transforming growth
factor; Th17, T helper 17; Treg, regulatory T. |
Following the infiltration of Th17 cells and Tregs,
the pathology of anti-GBM GN progressed. Renal structural damage
analyses in nephritic mice revealed severe glomerular crescent
formation from day 21. In addition, thickening and breakage of the
GBM, glomerular mesangial cell and matrix proliferation, protein
casts and inflammatory cell infiltration were observed in nephritic
mice from day 14 (Fig. 1C). Renal
tissue damage worsened with disease progression owing to Th17 and
Treg cell-mediated immune responses. Anti-GBM GN treatment induced
the deterioration of renal function and led to the deposition of
the mouse IgG in glomeruli at day 7 post-treatment (Fig. 1D and E).
ID3 involves in the differentiation of
Th17 cells and Tregs
Purified CD4+ T cells were isolated from
the spleens of C57BL/6 mice and stimulated by anti-CD3 and
anti-CD28 antibodies with the presence of TGF-β1 (differentiation
towards Tregs) or TGF-β1 plus IL-6 (differentiation towards Th17
cells; Fig. 2A and B). Following 6
days of incubation, the supernatants were collected for ELISA. The
expression of IL-10 increased in the presence of TGF-β1, and the
expression of IL-17A increased with TGF-β1 plus IL-6 (Fig. 2C and D). Cells from the different
groups were collected for western blotting and RT-qPCR. The levels
of ID3 increased when CD4+ T cells differentiated to
Tregs, and were reduced in Th17 cells (Fig. 2E-H). Above all, ID3 may be involved
in the differentiation of Th17 cells and Tregs in vitro.
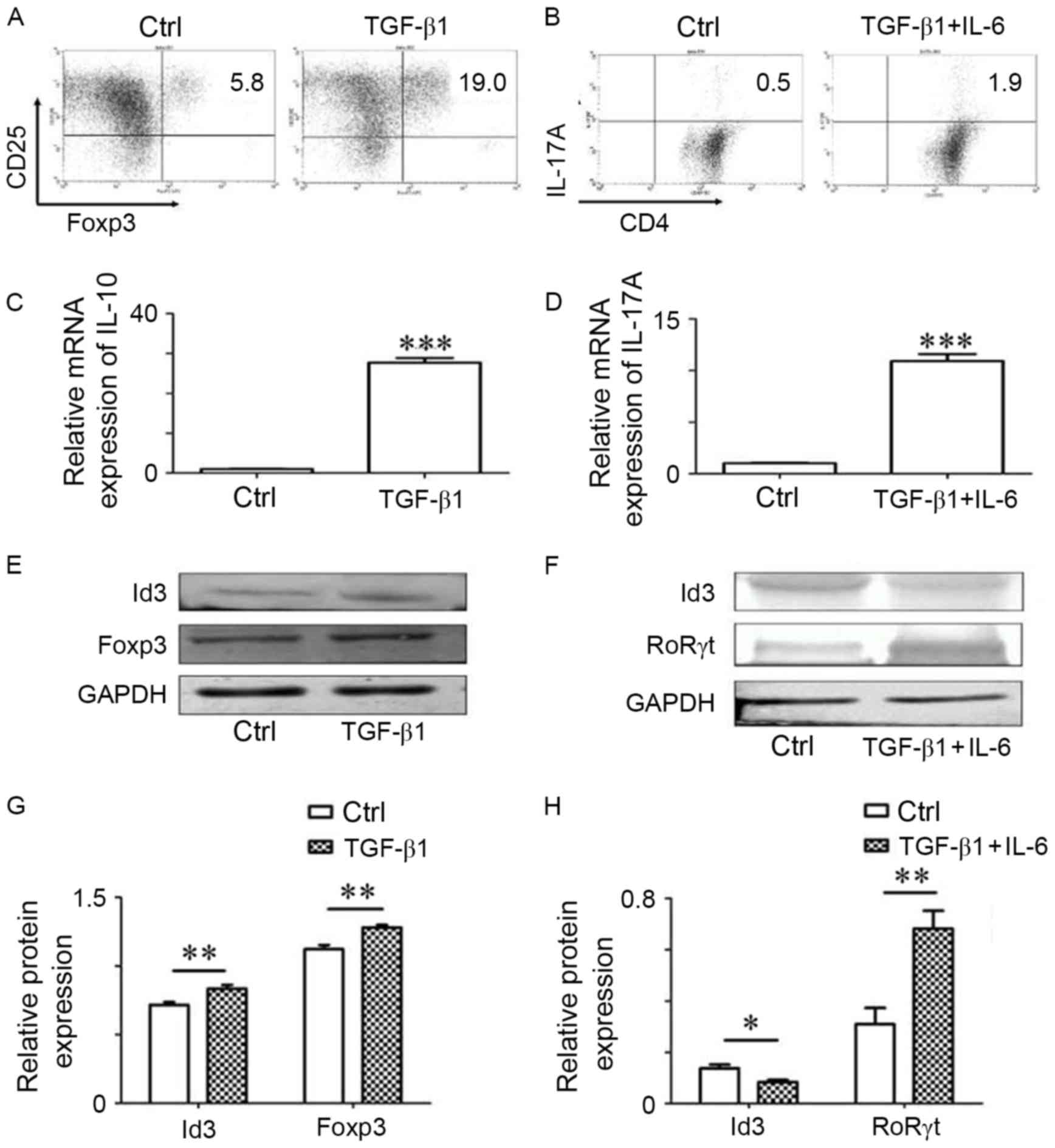 | Figure 2.ID3 serves a role the differentiation
of Th17 and Treg cells. Purified CD4+ T cells were
isolated from the spleens of C57BL/6 mice and subsequently
stimulated with anti-CD3 and anti-CD28 antibodies with or without
the presence of TGF-β1 or TGF-β1+IL-6. (A and B) Representative
gated data plots for CD4+ T cells. (C and D) The
expression levels of IL-10 and IL-17 were detected in the splenic
cell culture supernatants by ELISA. (E and F) Western blotting was
used to measure the protein expressions of ID3, FoxP3 and RORγt in
Th17 and Treg cells. (G and H) Bar graphs indicating the
quantification of ID3, FoxP3 and RORγt protein expression from (G
and H) Data are present as the mean ± standard error of the mean of
three independent experiments; *P<0.05, **P<0.01,
***P<0.005 vs. Ctrl group. Ctrl, control; FoxP3, forkhead box
P3; ID3, inhibitor of DNA binding 3; IL, interleukin; TGF,
transforming growth factor; RORγt, RAR-related orphan receptor γt;
Th17, T helper 17; Treg, regulatory T. |
ID3 negatively regulates Th17 cell
differentiation
As demonstrated, ID3 may be involved in the
differentiation of Th17 and Treg cells; however, it is still
unknown how ID3 alters the profiles of these cells. Therefore, ID3
was knocked down using siRNA to investigate the effects of reduced
ID3 on CD4+ T cell differentiation (Fig. 3A and B). Following transfection
with ID3-siRNA, the expression level of RoRγt mRNA was
significantly increased and the mRNA expression of FoxP3 was
decreased compared with expressions in Scram-siRNA transfected
cells (Fig. 3C). In addition, a
significant increase in the protein expression level of IL-17A was
observed when ID3 was knocked down (Fig. 3D), whereas there no significant
difference was identified in the protein expression levels of
TGF-β1 and IL-10 in ID3-siRNA group compared with the
Scramble-siRNA control group (Fig.
3D). These data indicated that ID3 may affect the delicate
balance between Th17 and Treg cells by skewing the differentiation
of CD4+ T cells towards Tregs.
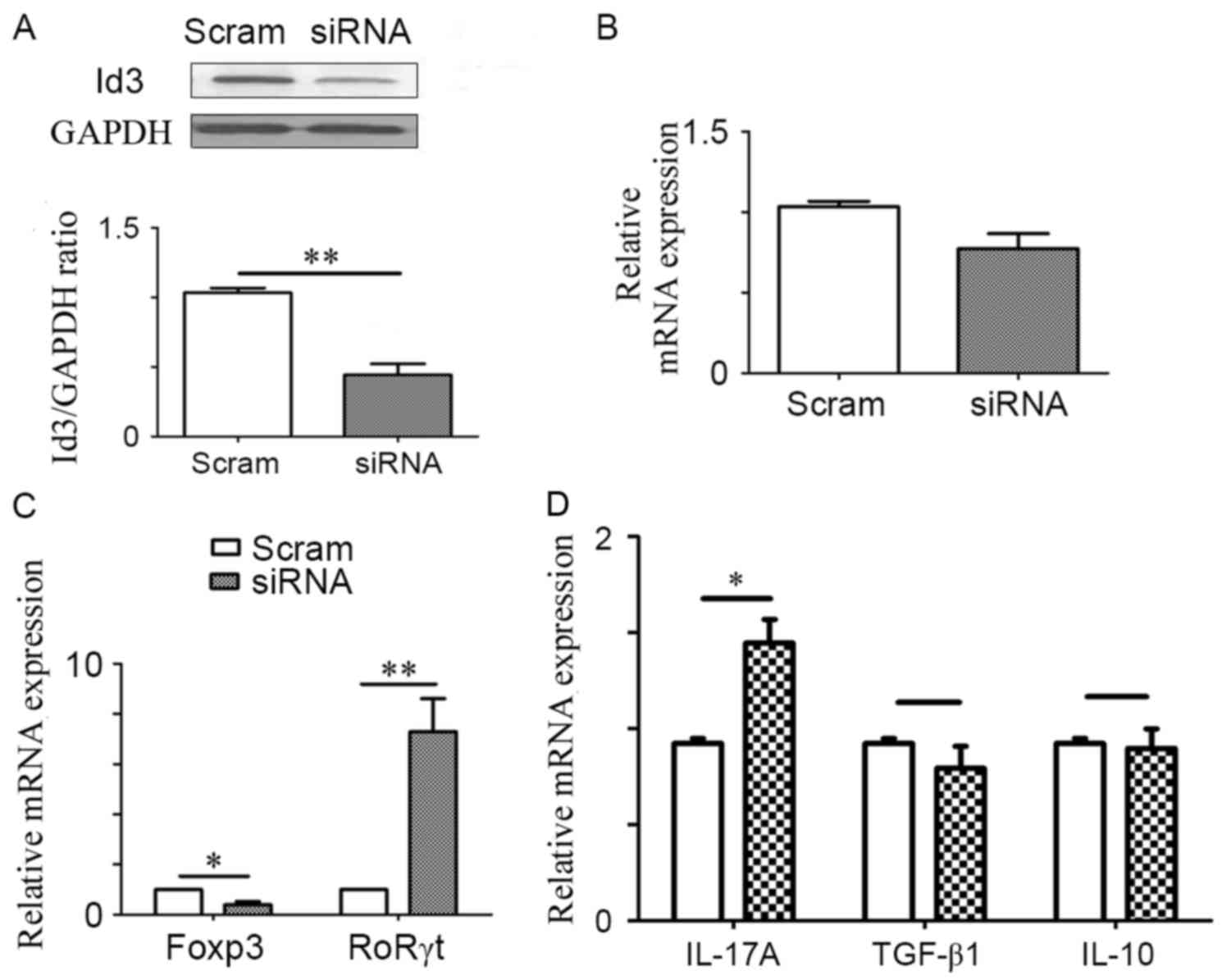 | Figure 3.ID3 negatively regulates Th17 cell
differentiation. Purified CD4+ T cells were isolated
from the spleens of C57BL/6 mice and treated with ID3-specific
siRNA or Scram-siRNA; subsequently, the expression of ID3 (A)
protein and (B) mRNA was measured. (C) Following ID3-siRNA or
Scram-siRNA treatment, CD4+ T cells were activated with
anti-CD3 and anti-CD28 for 24 h. Cells were harvested to assess the
mRNA expression levels of FoxP3 and RORγt mRNA. (D) Cell culture
supernatants were used to measure the protein expression levels of
IL-17A, TGF-β1 and IL-10 by ELISA. Data are presented as the mean ±
standard error of the mean of three independent experiments;
*P<0.05 and **P<0.01. FoxP3, forkhead box P3; ID3, inhibitor
of DNA binding 3; IL, interleukin; RORγt, RAR-related orphan
receptor γt; scram, scrambled; siRNA, small interfering RNA; TGF,
transforming growth factor; Th17, T helper 17. |
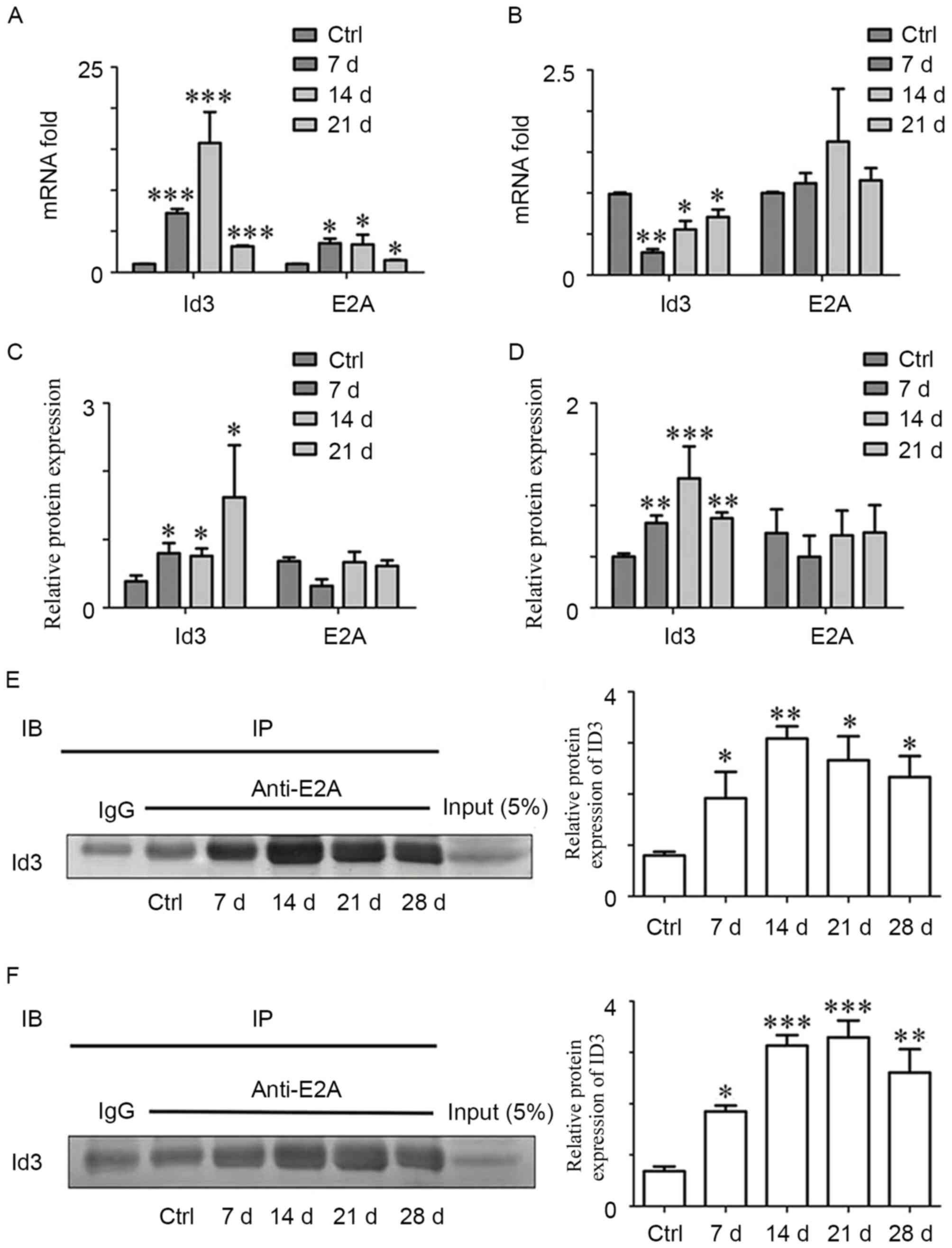
ID3 contributes to the regulation of
differentiation, which depends on the binding with E2A
To investigate how ID3 regulates the differentiation
of CD4+ T cells, the expression levels of ID3 and E2A in
both kidneys and spleens were detected by RT-qPCR and western
blotting. Renal ID3 mRNA expression increased between 3- and
20-fold from day 7 post-treatment, with a peak at day 14, compared
with the Control group (Fig. 4A).
Notably, splenic ID3 mRNA expression decreased significantly in the
early phase (day 7) and gradually increased in the later phase (day
14 and 21; Fig. 4B). ID3 protein
expression was also upregulated in the kidney on day 7 (Fig. 4C), and the protein expression of
ID3 was increased in spleen of nephritic mice, (Fig. 4D).
In renal tissues of anti-GBM GN mice, E2A mRNA
expression increased in the early phase and then decreased at day
21 (Fig. 4A). However, E2A mRNA
expression did not exhibit significant differences in the spleen
(Fig. 4B), nor was a significant
difference identified in E2A protein expression in kidney or spleen
(Fig. 4C and D, respectively). A
previous study reported that ID3 regulated the maturation of T
cells in the thymus by binding to the transcription factor E2A,
forming a non-DNA-binding dimer (25). Therefore, the present study
examined the relationship between ID3 and E2A at different phases
in anti-GBM GN progression by co-IP. The results indicated that ID3
was able to interact with E2A (Fig. 4E
and F). In addition, a significant increase in binding between
ID3 and E2A in anti-GBM GN was observed from day 7, which declined
at the later time points in both renal tissues and spleens.
Discussion
Anti-GBM glomerulonephritis is a type of autoimmune
disease that is frequently associated with systemic and
organ-specific autoimmunity (3).
There are two phases in anti-GBM GN pathogenesis, the heterologous
phase and the autologous phase (26). The heterologous phase corresponds
to the effects in the early phase of disease pathology, and the
autologous phase gradually dominates with the progression of
anti-GBM GN, which was characterized by the immune response of the
host against the heterologous antibodies due to the activated
CD4+ T cells and B cells (27). As expected, two distinct immune
phases were observed in anti-GBM GN, and the autologous phase
seemed to be more important. CD4+ T cells serve a
crucial role in the initiating the immune response, which leads to
the crescentic injury in anti-GBM GN (28,29).
Our previous study revealed that anti-GBM GN may be driven by Th1
and Th17 cell-mediated immune response (4). The time of Th17 cell-mediated immune
response is early in disease pathogenesis and is followed later by
Th1 cell-mediated injury (9).
Tregs serve a protective role in anti-GBM GN and may significantly
reduce renal injury (10). The
activity of Th1 and Th17 cells may be attenuated by the
anti-inflammatory action of Tregs, which inhibit T cell
proliferation and T effector function (11). In the present study, the number of
Th17 cells appeared to increase from day 7 and reached a peak at
day 14, whereas the number of Tregs increased significantly from
day 14 to day 28. However, although the infiltration of Th17 cells
was reduced, the renal tissues continued to deteriorate. These
results indicated that activated Th17 cells may be able to initiate
and amplify glomerular inflammation, which is in agreement with a
previous study (30). The
protection of Tregs was not enough to lead to a recovery from
autoimmune tissue damage in anti-GBM GN. Thus, it is necessary to
identify an effective method that is able to suppress Th17 cells
and increase Tregs, in order to exert a regulatory function to
protect the kidneys.
Although Th17 and Treg cells have different
functions, they have the same precursor cell, which may be
differentiated to different cell types by pleiotropic cytokines
(12,13). Under certain conditions, Tregs may
be converted to Th17 cells; therefore, investigating the gene
regulatory mechanisms of Th17 and Treg cell differentiation may
contribute to the identification of crucial factors that are
involved in activation of the immune responses in anti-GBM GN. A
previous study demonstrated that ID3, as a transcription factor
that is involved in T cell development, regulated TGF-β1-mediated
reciprocal differentiation of Treg and Th17 cells in mice (21). ID3 is a nuclear protein and
contains a basic-helix-loop-helix (bHLH) domain, and is upregulated
in a number of cell types upon stimulation by several growth and
differentiation signals (16,17),
including TCR- and BCR-mediated signals in T and B lymphocytes,
respectively (18,19). Therefore, the present study
hypothesized that ID3 may offer protection in mice against anti-GBM
GN by regulating the differentiation of Th17 and Treg cells. The
present data indicated that ID3 expression increased when
CD4+ T cells differentiated into Tregs, but decreased in
Th17 cells in vitro. Th17 and Treg cells were present
throughout the entire period of anti-GBM GN, which may differ at
different time points accompanied with the expression of ID3. These
resulted suggested that ID3 may be involved in the differentiation
of Th17 and Treg cells in anti-GBM GN. In addition, the expression
levels of RoRγt and IL-17A were increased when ID3 expression was
knocked down. No significant difference in the expression of TGF-β1
and IL-10 between the ID3-siRNA group and Scramble-siRNA control
group. In other words, ID3 may tilt the delicate balance between
Th17 and Treg cells by skewing CD4+ T cell
differentiation towards Tregs, which is of great importance for
maintaining tolerance of and the suppressors of anti-GBM GN
(10). However, glomerular
crescents and protein casts were still observed at day 21 and 28
post-treatment, which led to the deterioration of renal function.
This suggested that endogenous ID3 was not enough to reduce renal
injury in anti-GBM GN and in vivo administration of
exogenous ID3 in mice may be required to increase the number of
Tregs in order to protect renal tissues.
To further explore how ID3 affected the
differentiation of Th17 and Treg cells, the expression of E2A in
kidney and spleen was detected by RT-qPCR and western blotting. A
previous study reported that ID3 interacted with the transcription
factor E2A to form a non-DNA-binding dimer, which regulates the
maturation of T cells in the thymus (27). Consistent with those observations,
the present study demonstrated that ID3 binds with E2A in anti-GBM
GN. Furthermore, it was observed that the binding between ID3 and
E2A increased from day 7 and declined at later period in both renal
tissues and spleens. In peripheral lymphoid organs, E2A binds with
the promoters to induce the expression of FoxP3 and RoRγt, which
are the key transcription factors involved in Treg and Th17 cell
differentiation (21,31,32).
ID3 may prevent E2A binding to the promoters by forming a
non-DNA-binding dimer with E2A (21). Therefore, the present study
hypothesized that ID3 may affect the expression of FoxP3 and RoRγt
by binding with E2A, and subsequently regulating the
differentiation of Th17 cells and Tregs. However, a limitation to
the present study was that ID3 expression was not knocked down in
in vivo experiments, which may provide direct evidence to
support this hypothesis in our future studies.
In conclusion, the present study was the first, to
the best of our knowledge, to demonstrate the importance of ID3 in
anti-GBM GN, and that the Th17 and Treg cell-mediated immune
response contributed to anti-GBM GN in mice. As the development of
anti-GBM GN progressed, ID3 affected the differentiation of Th17
and Treg cells by downregulating the differentiation of Th17 cells,
which may subsequently affect the delicate balance between these
cells by skewing CD4+ T cell differentiation towards
Tregs; and effect that is probably associated with ID3 binding with
E2A.
Acknowledgements
The present study was supported by The National
Science Foundation of China (grant nos. 81241025, 81370817 and
81500546).
Glossary
Abbreviations
Abbreviations:
|
anti-GBM GN
|
anti-glomerular basement membrane
glomerulonephritis
|
|
bHLH
|
basic helix-loop-helix
|
|
GBM
|
glomerular basement membrane
|
|
ID3
|
inhibitor of DNA binding 3
|
|
IP
|
immunoprecipitation
|
|
MACS
|
magnetic-activated cell sorting
|
|
PAS
|
periodic acid-Schiff
|
|
RPGN
|
rapidly progressive
glomerulonephritis
|
|
SCR
|
serum creatinine
|
|
Tregs
|
regulatory T cells
|
References
|
1
|
Pedchenko V, Bondar O, Fogo AB, Vanacore
R, Voziyan P, Kitching AR, Wieslander J, Kashtan C, Borza DB,
Neilson EG, et al: Molecular architecture of the Goodpasture
autoantigen in anti-GBM nephritis. N Engl J Med. 363:343–354. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Phoon RK, Kitching AR, Odobasic D, Jones
LK, Semple TJ and Holdsworth SR: T-bet deficiency attenuates renal
injury in experimental crescentic glomerulonephritis. J Am Soc
Nephrol. 19:477–485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tipping PG and Holdsworth SR: T cells in
crescentic glomerulonephritis. J Am Soc Nephrol. 17:1253–1263.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Q, Luan H, Wang L, He F, Zhou H, Xu
X, Li X, Xu Q, Niki T, Hirashima M, et al: Galectin-9 ameliorates
anti-GBM glomerulonephritis by inhibiting Th1 and Th17 immune
responses in mice. Am J Physiol Renal Physiol. 306:F822–F832. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hünemörder S, Treder J, Ahrens S,
Schumacher V, Paust HJ, Menter T, Matthys P, Kamradt T,
Meyer-Schwesinger C, Panzer U, et al: TH1 and TH17 cells promote
crescent formation in experimental autoimmune glomerulonephritis. J
Pathol. 237:62–71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mosmann TR, Cherwinski H, Bond MW, Giedlin
MA and Coffman RL: Two types of murine helper T cell clone. I.
Definition according to profiles of lymphokine activities and
secreted proteins. J Immunol. 136:2348–2357. 1986.PubMed/NCBI
|
|
7
|
Harrington LE, Hatton RD, Mangan PR,
Turner H, Murphy TL, Murphy KM and Weaver CT: Interleukin
17-producing CD4+ effector T cells develop via a lineage distinct
from the T helper type 1 and 2 lineages. Nat Immunol. 6:1123–1132.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sakaguchi S, Sakaguchi N, Asano M, Itoh M
and Toda M: Immunologic self-tolerance maintained by activated T
cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a
single mechanism of self-tolerance causes various autoimmune
diseases. J Immunol. 155:1151–1164. 1995.PubMed/NCBI
|
|
9
|
Kitching AR and Holdsworth SR: The
emergence of TH17 cells as effectors of renal injury. J Am Soc
Nephrol. 22:235–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fehérvari Z and Sakaguchi S: CD4+ Tregs
and immune control. J Clin Invest. 114:1209–1217. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barbi J, Pardoll D and Pan F: Metabolic
control of the Treg/Th17 axis. Immunol Rev. 252:52–77. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Campbell DJ and Koch MA: Phenotypical and
functional specialization of FOXP3+ regulatory T cells. Nat Rev
Immunol. 11:119–130. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang XO, Nurieva R, Martinez GJ, Kang HS,
Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, et
al: Molecular antagonism and plasticity of regulatory and
inflammatory T cell programs. Immunity. 29:44–56. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu J, Yamane H and Paul WE:
Differentiation of effector CD4 T cell populations (*). Annu Rev
Immunol. 28:445–489. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee YK, Mukasa R, Hatton RD and Weaver CT:
Developmental plasticity of Th17 and treg cells. Curr Opin Immunol.
21:274–280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
17
|
Nakatsukasa H, Zhang D, Maruyama T, Chen
H, Cui K, Ishikawa M, Deng L, Zanvit P, Tu E, Jin W, et al: The
DNA-binding inhibitor Id3 regulates IL-9 production in CD4(+) T
cells. Nat Immunol. 16:1077–1084. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bain G, Cravatt CB, Loomans C,
Alberola-lla J, Hedrick SM and Murre C: Regulation of the
helix-loop-helix proteins, E2A and ID3, by the Ras-ERK MAPK
cascade. Nat Immunol. 2:165–171. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pan L, Sato S, Frederick JP, Sun XH and
Zhuang Y: Impaired immune responses and B-cell proliferation in
mice lacking the ID3 gene. Mol Cell Biol. 19:5969–5980. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murre C: Helix-loop-helix proteins and
lymphocyte development. Nat Immunol. 6:1079–1086. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maruyama T, Li J, Vaque JP, Konkel JE,
Wang W, Zhang B, Zhang P, Zamarron BF, Yu D, Wu Y, et al: Control
of the differentiation of regulatory T cells and T(H)17 cells by
the DNA-binding inhibitor ID3. Nat Immunol. 12:86–95. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miyazaki M, Miyazaki K, Chen S, Itoi M,
Miller M, Lu LF, Varki N, Chang AN, Broide DH and Murre C: Id2 and
ID3 maintain the regulatory T cell pool to suppress inflammatory
disease. Nat Immunol. 15:767–776. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kee BL, Quong MW and Murre C: E2A
proteins: Essential regulators at multiple stages of B-cell
development. Immunol Rev. 175:138–149. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jones ME and Zhuang Y: Acquisition of a
functional T cell receptor during T lymphocyte development is
enforced by HEB and E2A transcription factors. Immunity.
27:860–870. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Le Hir M: Histopathology of humorally
mediated anti-glomerular basement membrane (GBM) glomerulonephritis
in mice. Nephrol Dial Transplant. 19:1875–1880. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abbas AK, Murphy KM and Sher A: Functional
diversity of helper T lymphocytes. Nature. 383:787–793. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tipping PG, Huang XR, Qi M, Van GY and
Tang WW: Crescentic glomerulonephritis in CD4- and CD8-deficient
mice. Requirement for CD4 but not CD8 cells. Am J Pathol.
152:1541–1548. 1998.PubMed/NCBI
|
|
29
|
Huang XR, Tipping PG, Shuo L and
Holdsworth SR: Th1 responsiveness to nephritogenic antigens
determines susceptibility to crescentic glomerulonephritis in mice.
Kidney Int. 51:94–103. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robertson J, Wu J, Arends J, Zhou C,
Adrogue HE, Chan JT and Lou Y: Spontaneous recovery from early
glomerular inflammation is associated with resistance to anti-GBM
glomerulonephritis: Tolerance and autoimmune tissue injury. J
Autoimmun. 30:246–256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rivera RR, Johns CP, Quan J, Johnson RS
and Murre C: Thymocyte selection is regulated by the
helix-loop-helix inhibitor protein, ID3. Immunity. 12:17–26. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang F, Fuss IJ, Yang Z and Strober W:
Transcription of RORγt in developing Th17 cells is regulated by
E-proteins. Mucosal Immunol. 7:521–532. 2014. View Article : Google Scholar : PubMed/NCBI
|