Introduction
As an important sensory organ, the retina converts
photon energy into electrical impulses and transmits them to the
brain. As a result, the retina has a high metabolic function and
requires a continuous blood supply, which is provided by the
choriocapillaris and the central retinal artery (1). However, the high rate of blood flow
makes the retina susceptible to ischemia/reperfusion (I/R) injury.
Retinal I/R injury can result from a number of ocular diseases,
including retinal vascular occlusion, anterior optic neuropathy,
diabetic retinopathy and glaucoma (2–5). In
addition, ischemic events in the central nervous system can also
cause irreversible loss of neurons present in surrounding areas,
including the retina. Retinal I/R injury usually results in retinal
ganglion cell death due to their vulnerability to ischemia
(6,7). The time interval for retinal ischemia
to cause irreversible damage is ~1 h (8). It is widely accepted that I/R injury
is caused by the increased generation of reactive oxygen species
(ROS) during the process of I/R, including superoxide
(O2−), hydrogen peroxide
(H2O2) and hydroxyl radicals (OH) (9,10).
Excessive ROS can react with DNA, lipids and proteins, leading to
DNA breakage, lipid peroxidation and protein inactivation.
Edaravone has been shown to be neuroprotective in
cerebral ischemia and has been approved for the treatment of
cerebral infarction (11).
Previous investigations have found that edaravone can eliminate·OH
and other ROS, including O2− and nitric oxide
radicals, and inhibit H2O2-induced lipid
peroxidation (12,13). Edaravone can also activate
anti-oxidative enzymes, including superoxide dismutase (SOD),
catalase and guaiacol peroxidase (14). These findings suggest that
edaravone may be effective for the treatment of retinal I/R injury.
However, few investigations have examined the potential of
edaravone in the prevention or treatment of retinal I/R injury,
with the exception of a previous study by Song et al, which
indicated that edaravone protected the retina from I/R injury in
rats through reducing oxidative stress and inhibiting apoptosis of
retinal neurons (15). However,
the mechanism remains to be elucidated, and a detailed
understanding of the molecular events following I/R induced retinal
damage can facilitate the development of relevant treatments.
In the present study, retinal I/R injury was induced
in rats and the effects of edaravone on oxidative parameters,
including malondialdehyde (MDA), DNA fragmentation, total
antioxidant status (TAS), SOD and glutathione (GSH) in the retina
were investigated. Secondly, the retinal thickness and apoptotic
index (AI) in the ganglion cell layer (GCL) and inner nuclear layer
(INL) were measured to examine the protective effect of edaravone
against I/R injury. To investigate the underlying mechanism,
photoreceptor-derived 661W cells were treated with 1 mmol/l
H2O2 to induce oxidative injury and with
different concentrations of edaravone. The cell viability and
levels of cellular lactate dehydrogenase (LDH) were examined, and
involvement of the phosphatidylinositol 3-kinase (PI3K)/Akt
kinase/nuclear factor erythroid-2-related factor 2 (Nrf2) pathway
was investigated.
Materials and methods
Animals and drugs
Male Sprague-Dawley rats (8–12 weeks old, each
weighing 220±50 g) were used in the present study. The animals were
housed in the Hospital Animal Center of the Shanghai Tenth People's
Hospital in polycarbonate cages at 25°C on a 12-h light/dark cycle,
and were allowed free access to food and tap water. Ethical
approval was obtained from the ethics committee of Tongji
University (Shanghai, China) and performed in accordance with the
National Institute of Health Guide for the Care and Use of
Laboratory Animals (16). The rats
were randomly divided into five groups: Control (rats received no
treatment); Sham (rats received a sham retinal I/R surgery); I/R
(rats received retinal I/R surgery and normal saline injection);
I/R+edaravone (rats received a retinal I/R surgery and edaravone
injection); Edaravone (rats received edaravone injection only). The
edaravone injection was purchased from Nanjing Xiansheng
Pharmaceutical Co., Ltd. (Nanjing, China). The edaravone and the
normal saline were administered intraperitoneally at a dose of 6
mg/kg body weight.
Retinal I/R injury procedure
The procedure used to induce retinal I/R injury was
as previously described (17).
Initially, the rats were anesthetized with an intraperitoneal
injection of 1% pentobarbital sodium (10 mg/kg). Following corneal
analgesia with 0.4% oxybuprocainehydrochloride and dilation of the
pupil with 0.5% tropicamide and 0.5% phenylephrine, a 30-gauge
needle was cannulated with the anterior chamber of the right eye.
The other end of the needle was connected to a saline reservoir.
Secondly, the pressure of the reservoir was increased to 150 cm
above the eye, with the intraocular pressure maintained at 110 mmHg
to produce retinal ischemia, which was confirmed by corneal edema.
After 1 h, the pressure of the reservoir was decreased to the rat
eye level and the infusion needle was removed from the anterior
chamber to resume retinal blood supply. To prevent post-surgery
infection, erythromycin eye ointment was applied following
surgery.
Oxidative parameter measurement
Following completion of retinal reperfusion, the
retinas of the enucleated eyes were removed for the measurement of
oxidative parameters, MDA, DNA fragmentation, TAS, SOD and GSH.
Following tissue homogenization and centrifugation (5,000 × g, 5
min, 4°C), the supernatant was collected and detected using an MDA
detection kit (Beyotime Institute of Biotechnology, Haimen, China)
using a method similar to that described by Ohkawa et al
(18). The MDA level was
determined using athiobarbituric acid fluorometric method at 553 nm
with excitation at 515 nm, using 1,1,3,3-tetramethoxypropane as the
standard. DNA fragmentation was assessed by quantification of
cytosolic oligonucleosome-bound DNA using a Cell Death Detection
ELISA Plus kit (Roche Diagnostics GmbH, Mannheim, Germany). The
level of TAS in the supernatant was determined using an automated
measurement method with a commercially available kit developed by
Rel Assay Diagnostics (Gaziantep, Turkey). The results are
expressed as mmol Trolox equivalent per mg tissue protein. SOD
activity was measured using an SOD colorimetry assay kit from
Beyotime Institute of Biotechnology using a nitrobluetetrazolium
reduction assay method. A single unit of SOD was defined as the
quantity exhbiting 50% inhibition. GSH was measured using
5,5′-bis-dithionitrobenzoic acid reagent (19) and expressed as mg/mg tissue
protein.
Measurements of retinal thickness and
AI in ganglion cell layer (GCL) and inner nuclear layer (INL)
For the measurements of retinal thickness, the
retinas were fixed in formalin and embedded in paraffin. Thick
sections (5-µm) of the retinas were cut to include the full length
from superior to inferior along the vertical meridian and mounted
on microscope slides, followed by staining with hematoxylin and
eosin. Retinal thickness was measured in each section within 0.5–1
mm superior and inferior to the optic disc. Three measurements from
each section were obtained to determine the average value.
For the measurements of AI in the GCL and INL, the
sections were first incubated with proteinase K and hydrogen
peroxide at 37°C for 5 min. Subsequently, the sections were stained
using an apoptosis detection kit (EMD Millipore, Billerica, MA,
USA) to detect double-strand breaks in genomic DNA with
diaminobenzidine. The sections were analyzed in a blinded-manner in
10 microscopic fields from images captured using an Olympus digital
microscope (Olympus, Tokyo, Japan). The average numbers of
TUNEL-positive cells were counted in each image.
Cell culture and treatment
The 661W mouse photoreceptor cells (Shanghai Aulu
Biological Technology Co., Ltd., Shanghai, China) were cultured in
DMEM with 10% fetal bovine serum (FBS; Beyotime Institute of
Biotechnology Co., Ltd.) in a sterile humidified environment at
37°C in 95% O2 and 5% CO2 according to the
manufacturer's protocol. The cells were seeded into 6-well plates
12 h prior to treatment, and then divided into six groups: Control,
H2O2 group (treated with 1 mmol
H2O2 for 2 h), H2O2+25
µM Eda group (treated with 1 mmol H2O2 and 25
µM edaravone for 2 h), H2O2+50 µM Eda group
(treated with 1 mmol H2O2 and 50 µM edaravone
for 2 h), H2O2+100 µM Eda group (treated with
1 mmol H2O2 and 100 µM edaravone for 2 h) and
100 µM Eda group (treated with 100 µM edaravone for 2 h). Edaravone
was purchased from Yuanye Biotech (Shanghai, China). Different
doses of edaravone (25, 50 and 100 µM) were added to the culture 30
min prior to H2O2 treatment. In the second
phase of the cell experiment, the 661W cells were divided into five
groups: H2O2 group (treated with 1 mmol
H2O2 for 2 h),
H2O2+Eda+LY294002 group (treated with 1 mmol
H2O2, 50 µM edaravone and 20 µM LY294002 for
2 h), H2O2+Eda+Nrf2 small interfering (si)RNA
group (treated with 1 mmol H2O2, 50 µM
edaravone and Nrf2 siRNA for 2 h),
H2O2+Eda+triciribine group (treated with 1
mmol H2O2, 100 µM edaravone and 5 µM
triciribine for 2 h). In the third phase of the cell experiments,
the 661W cells were divided into six groups (control group and the
same groups as in the second phase of the cell experiments).
LY249002, purchased from Sigma-Aldrich; Merck Millipore (Darmstadt,
Germany) was added to the medium to reach a final concentration of
20 µM. Nrf2-siRNA was purchased from Qiagen, Inc. (Valencia, CA,
USA), including HP-validated siRNA and all stars Neg. siRNA AF488.
The following sense and antisense sequences were used for
Nrf2-siRNA forwards, 5′-GUAAGAAGCCAGAUGUUAAdUdU-3′ and reverse,
3′-dUdUCAUUCUUCGGUCUACAATT-5′. The 661W cells were transfected with
Nrf2-siRNA for 72 h using HiPerFect transfection reagent according
to the manufacturer's protocol (Qiagen, Inc.). Triciribine,
purchased from Sigma-Aldrich; Merck Millipore, was added to the
medium to reach a final concentration of 5 µM.
Cell viability and LDH leakage
assay
Cell viability was determined using an MTT assay kit
(Beyotime Institute of Biotechnology) similar to the method
described by Bai et al (20). The cells were cultured in a 96-well
plate (0.2×106 cells/ml) for treatment. Following
treatment of the cells, 100 µl MTT solution (1 mg/ml in medium
without serum and phenol red) was added to each well and the plates
were incubated at 37°C for 3 h. Following incubation, the medium on
top was removed and isopropanol was added to each well to dissolve
the formazan crystals. Finally, the absorbance values were measured
at 570 nm with an EIX-800 Micro elisa reader (BioTek Instruments,
Inc., Winooski, VT, USA). Cell survival rates were determined as
percentages of that of normal cells. The LDH leakage assay kit
(CytoTox 96® non-radioactive cytotoxicity assay) was a
product of Promega Corporation (Madison, WI, USA). The 661W cells
were seeded into 96-well plates 12 h prior to treatment. Following
treatment of the cells, 20 µl of the medium was transferred to a
new 96-well plate to measure LDH activity, according to the
manufacturer's protocol, as described by Chang et al
(21).
Western blot analysis
Following treatment of the 661W cells, the cells
were harvested and then re-suspended in lysis buffer (Cell lysis
buffer for Western and immunoprecipitation; Beyotime Institute of
Biotechnology). The samples were then centrifuged at 12,000 × g for
10 min at 4°C and the supernatants were collected. The total
protein levels were measured using a bicinchoninic assay (Roche
Diagnostics GmbH). The protein (25 µg) was separated by 10%
SDS-PAGE and transferred onto polyvinylidenedifluoride membranes
(EMD Millipore). The membranes were blocked in 5% nonfat milk and
then incubated with primary rabbit polyclonal antibodies against
Akt (cat. no. sc-5298; 1:1,500), phosphorylated-Akt (p-Akt; cat.
no. sc-293125; 1:2,000) and Nrf2 (cat. no. sc-365949; 1:2,000),
which were bought from Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA), in 5 ml of 5% bovine serum albumin wash buffer (Beyotime
Institute of Biotechnology) at 4°C overnight. The membranes were
then washed and incubated with secondary anti-rabbit immunoglobulin
G (1:2,000; cat. no. sc-2030; Santa Cruz Biotechnology, Inc.) in 5%
milk wash solution for 1 h at 25°C. Digitized images of protein
bands were quantitated using AlphaEaseFC™ software
(version 4.0.0; Witec, Littau, Switzerland). β-actin was used as an
internal control.
Statistical analysis
The data are expressed as the mean ± standard
deviation of triplicate experiments, with at least eight separate
experiments performed for each condition (n≥8). Differences among
means were assessed using a one-way analysis of variance followed
by the Student-Newman-Keuls post hoc test. P<0.05 was considered
to indicate a statistically significant difference. All statistical
analyses were performed using SPSS 17.0 (SPSS, Inc., Chicago, IL,
USA).
Results
Oxidative parameters are increased by
I/R and inhibited by edaravone
The changes in oxidative parameters (MDA, DNA
fragmentation, TAS, SOD and GSH) are exhibited in Table I. No significant alterations in
these parameters were found in the Sham rats. The rats exposed to
retinal I/R exhibited significant increases in MDA and DNA
fragmentation, and significant decreases in TAS, SOD and GSH,
compared with the control and sham group (P<0.05). However,
these changes in the parameters in the I/R group were all
significantly inhibited in the I/R+edaravone group: MDA and DNA
fragmentation were decreased; and levels of TAS, SOD and GSH were
increased (P<0.05). Compared with the control and sham groups,
no significant changes in these parameters were observed in the
edaravone group, with the exception of increased TAS
(P<0.05).
 | Table I.Changes in oxidative parameters
following treatment with Eda. |
Table I.
Changes in oxidative parameters
following treatment with Eda.
| Parameter | Control | Sham | I/R | I/R + Eda | Eda |
|---|
| MDA (µmol/mg
protein) |
9.02±1.12 |
8.59±1.24 |
16.69±2.33a |
11.12±1.05b |
9.12±1.26 |
| DNA fragmentation
(U/mg protein) |
2.14±0.36 |
2.07±0.25 |
3.69±0.27a |
2.26±0.29b |
2.05±0.21 |
| TAS (mmol trolox
equiv./mg protein) |
0.52±0.04 |
0.41±0.05 |
0.36±0.07a |
0.67±0.05b |
0.77±0.06 |
| SOD (%
inhibition/mg protein) |
33.29±2.54 |
31.14±2.63 |
21.18±2.29a |
28.64±2.04b |
35.26±3.22 |
| GSH (µg/mg
protein) |
3.15±0.26 |
3.08±0.22 |
2.36±0.17a |
2.89±0.18b |
3.19±0.31 |
Edaravone inhibits the changes in
retinal thickness, and AI in the GCL and INL induced by I/R
The changes of retinal thickness and AI in the GCL
and INL following treatment of edaravone are shown in Table II. No significant changes in these
parameters were observed in the Sham rats (P>0.05). The rats
exposed to retinal I/R exhibited a significant increase in retinal
thickness, and increased AI in the GCL and INL, compared with those
in the Control and Sham groups (P<0.05). These parameters in the
I/R+edaravone group were significantly lower, compared with those
in the I/R group (P<0.05). Compared with the Control and Sham
group, no significant changes were observed in these parameters in
the edaravone group (P>0.05).
| Table II.Changes in retinal thickness and AI
in the GCL and INL following treatment with EDA. |
Table II.
Changes in retinal thickness and AI
in the GCL and INL following treatment with EDA.
| Parameter | Control | Sham | I/R | I/R + Eda | Eda |
|---|
| Retinal thickness
(µm) |
159.5±14.2 |
167.6±16.3 |
266.1±18.4a |
184.6±20.7b |
162.5±13.2 |
| AI in GCL (%) |
15.23±1.64 |
14.46±1.25 |
27.42±1.58a |
14.56±2.02b |
17.63±2.11 |
| AI in INL (%) |
6.37±0.59 |
6.32±0.63 |
18.71±0.91a |
7.45±1.05b |
6.58±0.77 |
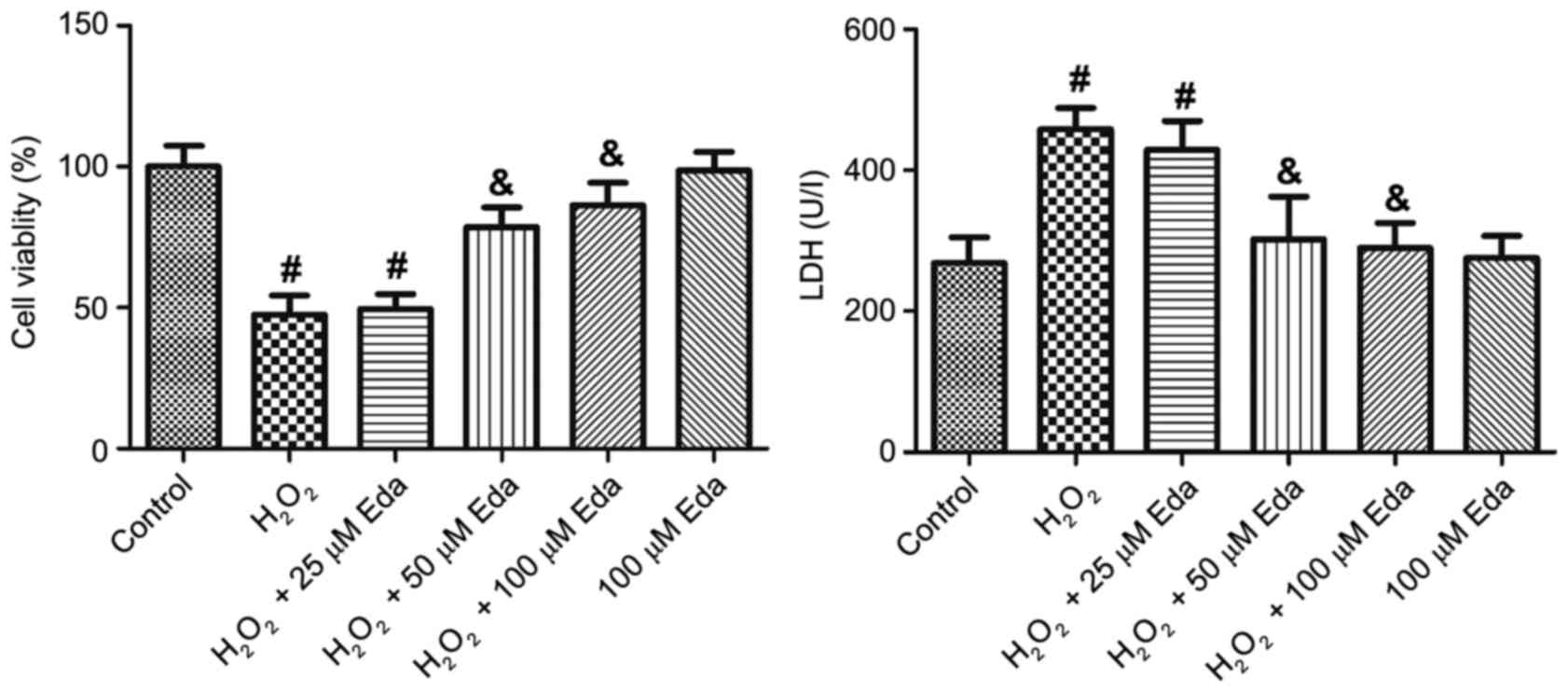
Edaravone protects cell viability and
membrane integrity of H2O2-treated 661W
cells
The results of the cell viability analysis showed
that: i) H2O2 treatment significantly
decreased the viability of the 661W cells (P<0.05); ii) 25 µM
edaravone had no significant effect on the cell viability of
H2O2-treated 661W cells, however, 50 and 100
µM edaravone increased cell viability (P<0.05); iii) edaravone
alone did not alter cell viability (Fig. 1A). The analysis of LDH leakage
demonstrated similar results: i) H2O2
treatment significantly increased LDH leakage (P<0.05); ii) 25
µM edaravone did not significantly affect LDH leakage, however, 50
and 100 µM edaravone significantly decreased leakage (P<0.05);
ii) edaravone alone did not affect LDH leakage (Fig. 1B).
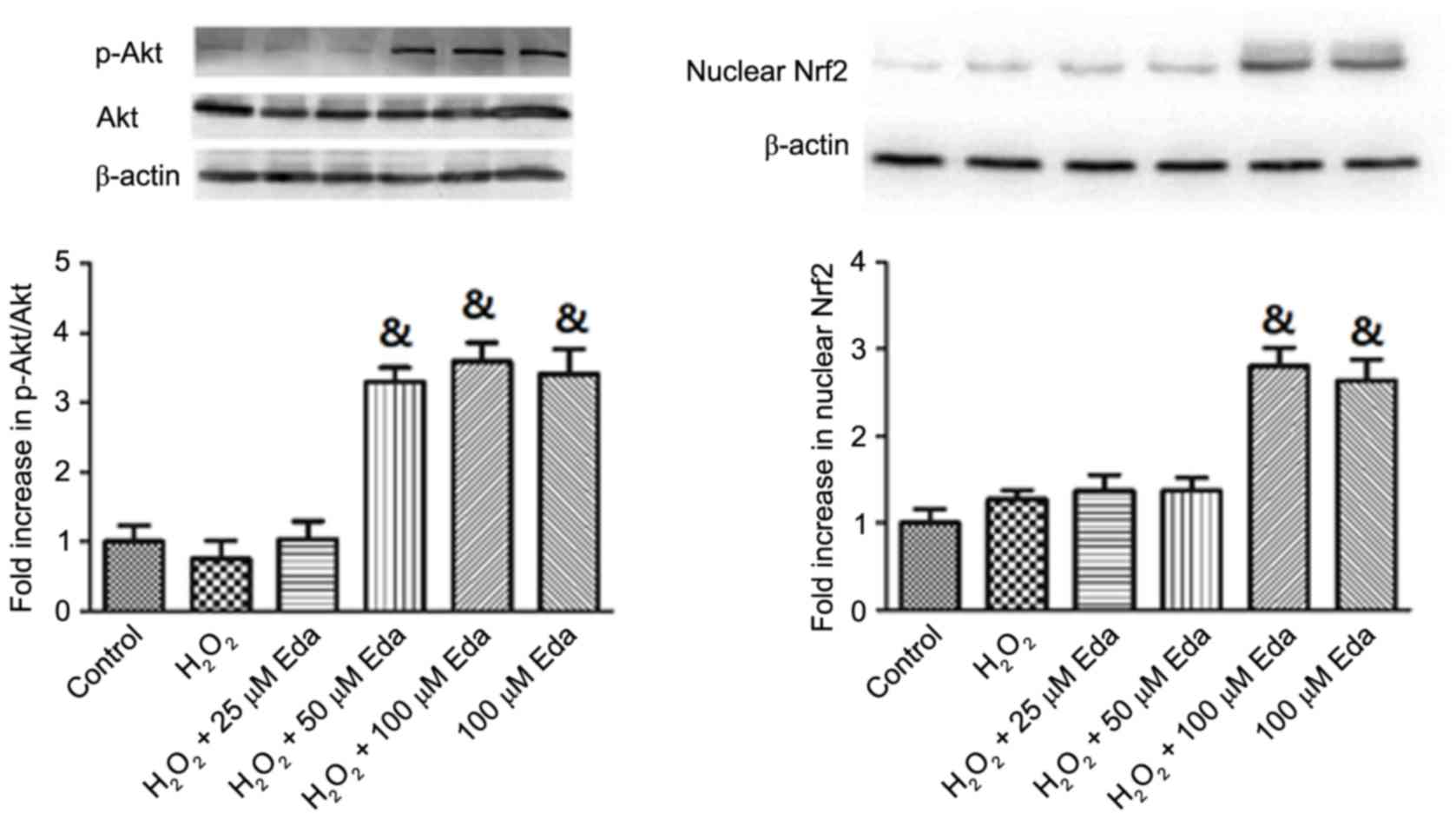
Expression levels of p-Akt, Akt and
nuclear Nrf2 in 661W cells are increased by edaravone
The expression levels of p-Akt, Akt and nuclear Nrf2
in cells were altered by oxidative stress and edaravone, as shown
in Fig. 2. It was demonstrated
that, following exposure of the 661W cells to
H2O2 for 2 h, no significant changes were
observed in the protein expression levels of p-Akt, Akt or nuclear
Nrf2 (P>0.05). In the presence of 25 µM edaravone, the
expression of p-Akt, Akt and nuclear Nrf2 remained unchanged,
however, treatment with 50 and 100 µM edaravone led to significant
increases in p-Akt/Akt and nuclear Nrf2 (P<0.05). Pretreatment
with 100 µM edaravone alone also significantly increased the
expression levels of p-Akt/Akt and nuclear Nrf2 in cells, compared
with those in the Control (P<0.05).
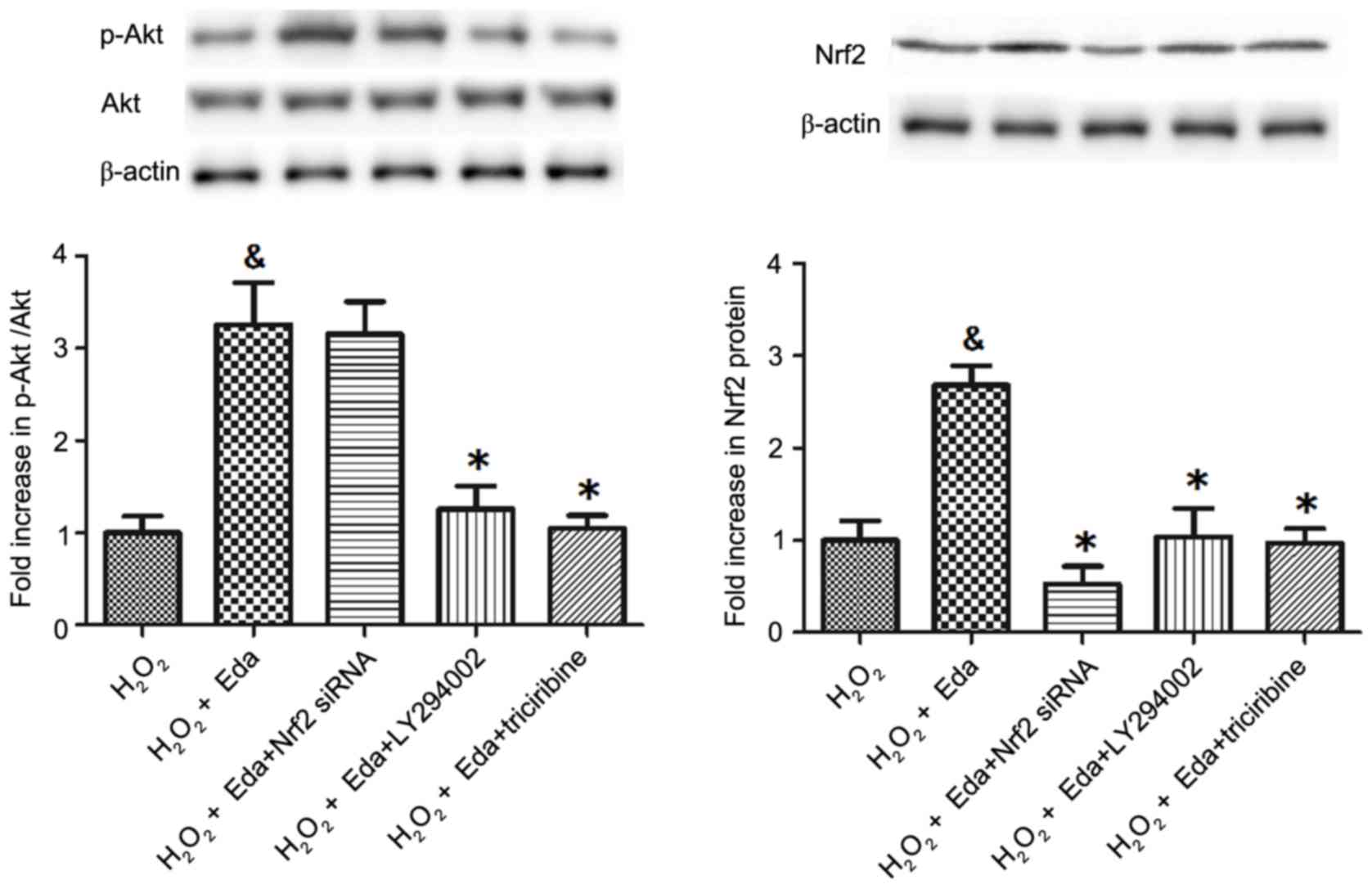
Expression levels of p-Akt, Akt and
Nrf2 in 661W cells are inhibited by Nrf2 siRNA or PI3K/Akt
inhibitors
The expression of p-Akt, Akt (Fig. 3A) and Nrf2 (Fig. 3B) in cells were altered by Nrf2
siRNA and the PI3K/Akt inhibitors. As shown in Fig. 3A, treatment with Nrf2 siRNA had no
significant effect on the p-Akt/Akt ratio (P>0.05), however, the
PI3K inhibitor (LY294002) and Akt inhibitor (triciribine)
significantly decreased the p-Akt/Akt ratio (P<0.05). For Nrf2
(Fig. 3B), the Nrf2 siRNA and the
PI3K/Akt inhibitor significantly decreased its expression
(P<0.05).
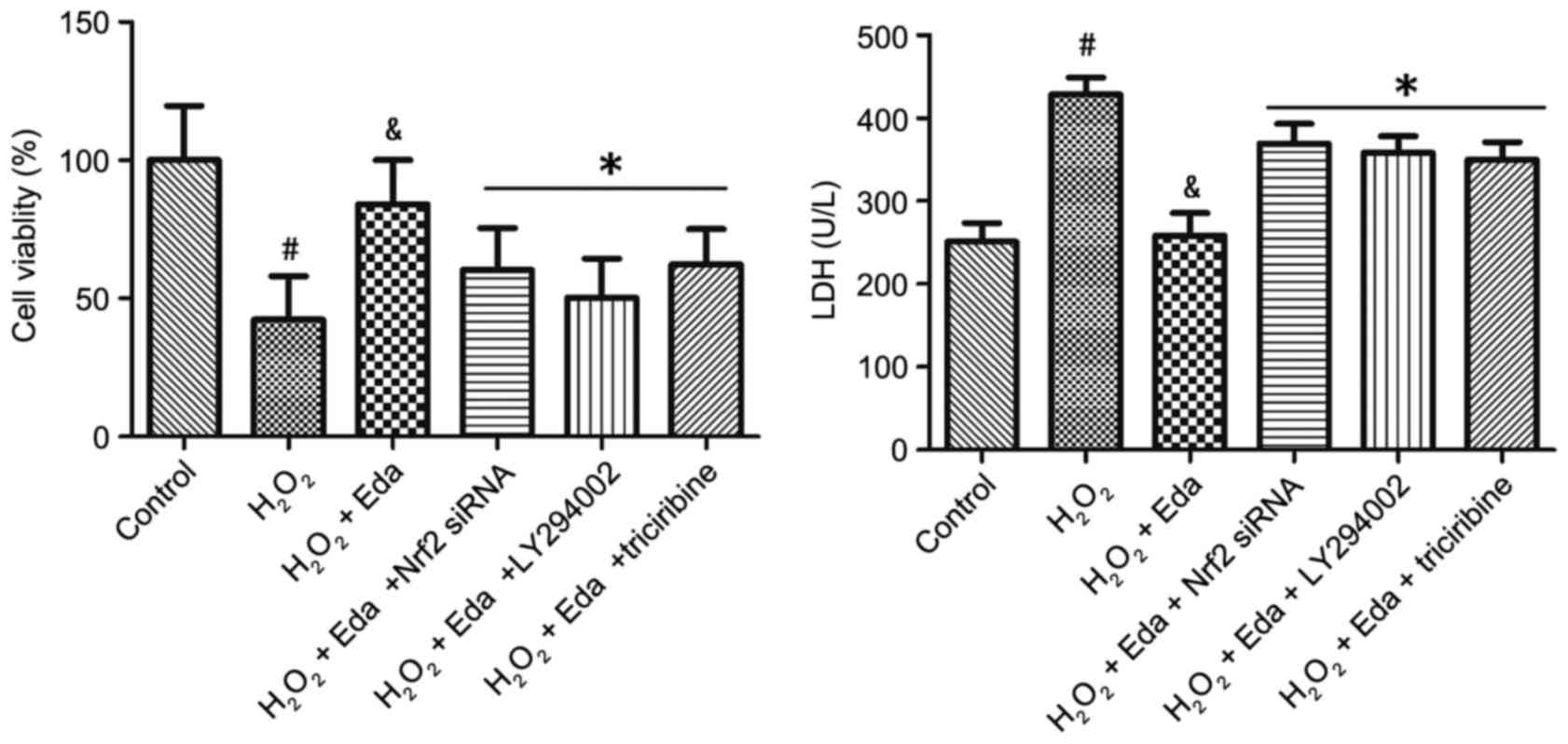
Nrf2 siRNA or PI3K/Akt inhibitors
counter the protective effect of edaravone on cell viability and
membrane integrity of 661W cells
The viabilities of 661W cells in the presence of
PI3K/Akt inhibitors or Nrf2 siRNA are exhibited in Fig. 4A. Treatment of cells with Nrf2
siRNA, PI3K inhibitor (LY294002) or Akt inhibitor (triciribine) all
eliminated the effect of edaravone on cell viability (P<0.05).
The membrane integrities of 661W cells in the presence of Nrf2
siRNA or PI3K/Akt inhibitors are shown in Fig. 4B. Treatment with Nrf2 siRNA,
LY294002 and triciribine all eliminated the effect of edaravone on
LDH leakage (P<0.05).
Discussion
Retinal I/R injury is associated with various
conditions, which can culminate in blindness if effective treatment
is not provided (22). The retina
consists of neurons, vasculature and glia, and each of these
compartments can be affected in retinal I/R injury (23–25).
The exact mechanism of cell death due to retinal I/R injury remains
to be fully elucidated, however, it has been previously
demonstrated that, in conditions of oxidative stress, retinal
ganglion cells are damaged as a result of increased intracellular
ROS and calcium influx (26). In
the process of I/R, ROS are extensively generated in the early
stage of reperfusion and can cause serious damage to various
organs, including the brain and heart (27,28).
It has also been demonstrated in multiple studies that the
oxidative stress induced by ROS is key in the pathophysiological
mechanisms involved in retinal I/R injury (29,30).
As demonstrated in the results of the present study, the rats
exposed to retinal I/R exhibited a significant increase in MDA and
DNA fragmentation, suggesting that ROS caused the peroxidation of
cellular lipid in addition to DNA oxidative damage. It also
significantly decreased the levels of TAS, SOD and GSH, indicating
that the balance of oxidative/anti-oxidative in the retina was
disturbed by I/R procedure. The production and accumulation of
excessive ROS is considered to be important in the mechanism of I/R
injury. ROS are the major free radicals in human body, including
O2−, OH and H2O2. The
nicotinamide adenine dinucleotide phosphate oxidase system, in
conjunction with mitochondria, is a major site of ROS generation
under H2O2 (31).
The overproduction of ROS can induce several
inflammatory mediators, including interleukin 1-β and tumor
necrosis factor-α, and apoptosis in the retina (32–34).
As a result, inflammation and cell apoptosis are considered to be
major causes of the pathological changes following I/R injury. As
exhibited in Table II, the rats
exposed to retinal I/R procedure exhibited significant increases in
retinal thickness and apoptotic indices in the GCL and INL. The
increase of retinal thickness indicated that the I/R injury caused
retinal inflammation, whereas the increase of AI in the GCL and INL
demonstrated cell apoptosis was induced by I/R.
Edaravone, a novel free radical scavenger, has been
approved for the treatment of ischemic stroke in China and Japan.
It is widely accepted that it produces neuroprotective effects by
scavenging free radicals, and inhibiting lipid peroxidation and
oxidative damage to cells (13).
In the animal experiments performed in the present study, it was
found that edaravone effectively attenuated the disruption of
oxidative/anti-oxidative balance induced by retinal I/R injury. The
oxidative parameters following I/R injury were all significantly
inhibited by edaravone: MDA and DNA fragmentation were decreased;
TAS, SOD and GSH were increased. Furthermore, edaravone
significantly decreased retinal thickness and AI in the GCL and
INL, indicating its protection against the retinal inflammation and
cell apoptosis induced by I/R. These results were consistent with
those in a study by Song et al (15). In this previous study, rats were
injected with edaravone at 30 min prior to ischemia, following
which retinal ischemia was induced by elevating intraocular
pressure to 110 mmHg for 60 min and then treated with edaravone
twice daily for 1 or 5 days post-I/R. An electroretinogram was
recorded 5 days following reperfusion. It was concluded that
edaravone lowered levels of MDA, increased SOD activity, and
attenuated I/R-induced apoptosis of retinal neurons and suppressed
I/R-induced reduction in a- and b-wave amplitudes of ERG. However,
the molecular events following I/R-induced retinal damage were not
examined. Understanding the underlying mechanisms may facilitate
the development of relevant treatments.
To further examine the underlying mechanism of the
protective effect of edaravone, the present study treated 661W
cells, a mouse photoreceptor cell line, with
H2O2 to produce oxidative stress, and examine
the effect of edaravone on cell viability and injury. The results
showed that edaravone dose-dependently enhanced cell viability,
which was decreased by H2O2 treatment; it
also dose-dependently protected cell integrity, which was impaired
by H2O2, demonstrated by LDH leakage.
Subsequently, the present study determined the effects of
H2O2 and edaravone on the activation of Akt
and expression of Nrf2. The results of the western blot analysis
showed no significant changes in the protein expression levels of
p-Akt, Akt or Nrf2 in the presence of H2O2,
however, edaravone significantly increased p-Akt/Akt and Nrf2. The
p-Akt/Akt ratio was not affected by treatment with Nrf2 siRNA, but
was significantly decreased by the PI3K inhibitor (LY294002) or Akt
inhibitor (triciribine). By contrast, the expression of Nrf2 was
inhibited by Nrf2 siRNA and the PI3K/Akt inhibitors.
It has been revealed that the PI3K-mediated
generation of 3′-phosphorylated phosphoinositide leads to the
recruitment of Akt to the cell membrane, where it is phosphorylated
by kinases, including phosphoinositide-dependent kinase-1, leading
to the activation of Akt (35).
Nrf2 is an important transcription factor in the coordinated
expression of stress-inducible genes. Several studies have shown
that Nrf2 can regulate the expression of phase-II detoxification
and antioxidant response element, including glutathione synthase,
hemeoxygenase (HO)-1 and catalase, under oxidative stress, and can
be activated by the PI3K/Akt pathway. For example, PI3K/Akt can
facilitate the release of Nrf2 from the Keap1-Nrf2 complex, and
enables it to translocate into the nucleus and induce phase II
defense enzymes (36). A study by
Hua et al (37)
demonstrated that edaravone significantly induced the translocation
of Nrf2 and HO-1 to the nucleus, and markedly increased components
of the cellular antioxidant defense system, including GSH, SOD and
HO-1, consistent with the present study. It is possible that the
rearrangement of actin microfilaments and the increase of cellular
Ca2+ are involved in the regulation of Nrf2 via the
PI3K/Akt pathway (38).
In conclusion, the present study revealed that
edaravone inhibited oxidative injury in the retina induced by
retinal I/R, and reduced the increases of retinal inflammation and
apoptosis. In vitro experiments demonstrated that edaravone
effectively protected cell viability and the membrane integrity of
the H2O2-treated 661W cells via the
PI3K/Akt/Nrf2 pathway. These results indicate the potential
protective effect of edaravone against retinal I/R injury and
provide a novel explanation for the protective effects of
edaravone.
References
|
1
|
Kaur C, Foulds WS and Ling EA:
Blood-retinal barrier in hypoxic ischaemic conditions: Basic
concepts, clinical features and management. Prog Retin Eye Res.
27:622–647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Archer DB: Tributary vein obstruction:
Pathogenesis and treatment of sequelae. Doc Ophthalmol. 40:339–360.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hayreh SS: Ischemic optic neuropathy. Int
Ophthalmol. 1:9–18. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Verma D: Pathogenesis of diabetic
retinopathy-the missing link? Med Hypotheses. 41:205–210. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nickells RW: Retinal ganglion cell death
in glaucoma: The how, the why, and the maybe. J Glaucoma.
5:345–356. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hayreh SS, Zimmerman MB, Kimura A and
Sanon A: Central retinal artery occlusion. Retinal survival time.
Exp Eye Res. 78:723–736. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mukaida Y, Machida S, Masuda T and Tazawa
Y: Correlation of retinal function with retinal histopathology
following ischemia-reperfusion in rat eyes. Curr Eye Res.
28:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aydemir O, Celebi S, Yilmaz T, Yekeler H
and Kükner AS: Protective effects of vitamin E forms
(alpha-tocopherol, gamma-tocopherol and d-alpha-tocopherol
polyethylene glycol 1000 succinate) on retinal edema during
ischemia-reperfusion injury in the guinea pig retina. Int
Ophthalmol. 25:283–289. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pace PW, Yao LJ, Wilson JX, Possmayer F,
Veldhuizen RA and Lewis JF: The effects of hyperoxia exposure on
lung function and pulmonary surfactant in a rat model of acute lung
injury. Exp Lung Res. 35:380–398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reddy NM, Kleeberger SR, Kensler TW,
Yamamoto M, Hassoun PM and Reddy SP: Disruption of Nrf2 impairs the
resolution of hyperoxia-induced acute lung injury and inflammation
in mice. J Immunol. 182:7264–7271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Watanabe T, Tanaka M, Watanabe K,
Takamatsu Y and Tobe A: Research and development of the free
radical scavenger edaravone as a neuroprotectant. Yakugaku Zasshi.
124:99–111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaur C and Ling EA: Antioxidants and
neuroprotection in the adult and developing central nervous system.
Curr Med Chem. 15:3068–3080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshida H, Yanai H, Namiki Y,
Fukatsu-Sasaki K, Furutani N and Tada N: Neuroprotective effects of
edaravone: A novel free radical scavenger in cerebrovascular
injury. CNS Drug Rev. 12:9–20. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu S, Li R, Ni X, Cai Z, Zhang R, Sun X,
Quock RM and Xu W: Perfluorocarbon-facilitated CNS oxygen toxicity
in rats: Reversal by edaravone. Brain Res. 1471:56–65. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song Y, Gong YY, Xie ZG, Li CH, Gu Q and
Wu XW: Edaravone (MCI-186), a free radical scavenger, attenuates
retinal ischemia/reperfusion injury in rats. Acta Pharmacol Sin.
29:823–828. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Institute of Laboratory Animal Resources,
Commission on Life Sciences, National Research Council, . Guide for
the Care and Use of Laboratory Animals. National Academy Press;
Washington, DC: 1996
|
|
17
|
Tong N, Zhang Z, Gong Y, Yin L and Wu X:
Diosmin protects rat retina from ischemia/reperfusion injury. J
Ocul Pharmacol Ther. 28:459–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohkawa H, Ohishi W and Yagi K:
Determination of lipid peroxidation by MDA. Anal Biochem.
95:351–358. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pathak R, Suke SG, Ahmed T, Ahmed RS,
Tripathi AK, Guleria K, Sharma CS, Makhijani SD and Banerjee BD:
Organochlorine pesticide residue levels and oxidative stress in
preterm delivery cases. Hum Exp Toxicol. 29:351–358. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bai L, Pang WJ, Yang YJ and Yang GS:
Modulation of Sirt1 by resveratrol and nicotinamide alters
proliferation and differentiation of pig preadipocytes. Mol Cell
Biochem. 307:129–140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang Y, Yang ST, Liu JH, Dong E, Wang Y,
Cao A, Liu Y and Wang H: In vitro toxicity evaluation of graphene
oxide on A549 cells. Toxicol Lett. 200:201–210. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Osborne NN, Casson RJ, Wood JP, Chidlow G,
Graham M and Melena J: Retinal ischemia: Mechanisms of damage and
potential therapeutic strategies. Prog Retin Eye Res. 23:91–147.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng L, Gong B, Hatala DA and Kern TS:
Retinal ischemia and reperfusion causes capillary degeneration:
Similarities to diabetes. Invest Ophthalmol Vis Sci. 48:361–367.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fernandez DC, Bordone MP, Chianelli MS and
Rosenstein RE: Retinal neuroprotection against ischemia-reperfusion
damage induced by postconditioning. Invest Ophthalmol Vis Sci.
50:3922–3930. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li C, Wang L, Huang K and Zheng L:
Endoplasmic reticulum stress in retinal vascular degeneration:
Protective role of resveratrol. Invest Ophthalmol Vis Sci.
53:3241–3249. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maher P and Hanneken A: Flavonoids protect
retinal ganglion cells from oxidative stress-induced death. Invest
Ophthalmol Vis Sci. 46:4796–4803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peters O, Back T, Lindauer U, Busch C,
Megow D, Dreier J and Dirnagl U: Increased formation of reactive
oxygen species after permanent and reversible middle cerebral
artery occlusion in the rat. J Cereb Blood Flow Metab. 18:196–205.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kevin LG, Camara AK, Riess ML, Novalija E
and Stowe DF: Ischemic preconditioning alters real-time measure of
O2 radicals in intact hearts with ischemia and reperfusion. Am J
Physiol Heart Circ Physiol. 284:H566–H574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen YQ, Pan WH, Liu JH, Chen MM, Liu CM,
Yeh MY, Tsai SK, Young MS, Zhang XM and Chao HM: The effects and
underlying mechanisms of S-allyl l-cysteine treatment of the retina
after ischemia/reperfusion. J Ocul Pharmacol Ther. 28:110–117.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Valko M, Leibfritz D, Moncol J, Cronin MT,
Mazur M and Telser J: Free radicals and antioxidants in normal
physiological functions and human disease. Int J Biochem Cell Biol.
39:44–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parinandi NL, Kleinberg MA, Usatyuk PV,
Cummings RJ, Pennathur A, Cardounel AJ, Zweier JL, Garcia JG and
Natarajan V: Hyperoxia-induced NAD(P)H oxidase activation and
regulation by MAP kinases in human lung endothelial cells. Am J
Physiol Lung Cell Mol Physiol. 284:L26–L38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schwartz M: Neuroprotection as a treatment
for glaucoma: Pharmacological and immunological approaches. Eur J
Ophthalmol. 13 Suppl 3:S27–S31. 2003.PubMed/NCBI
|
|
33
|
Clutton S: The importance of oxidative
stress in apoptosis. Br Med Bull. 53:662–668. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meldrum DR, Dinarello CA, Cleveland JC Jr,
Cain BS, Shames BD, Meng X and Harken AH: Hydrogen peroxide induces
tumor necrosis factor alpha-mediated cardiac injury by a P38
mitogen-activated protein kinase-dependent mechanism. Surgery.
124:291–297. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Toker A and Cantley LC: Signalling through
the lipid products of phosphoinositide-3-OH kinase. Nature.
387:673–676. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ryu MJ, Kang KA, Piao MJ, Kim KC, Zheng J,
Yao CW, Cha JW, Chung HS, Kim SC, Jung E, et al:
7,8-Dihydroxyflavone protects human keratinocytes against oxidative
stress-induced cell damage via the ERK and PI3K/Akt-mediated
Nrf2/HO-1 signaling pathways. Int J Mol Med. 33:964–970. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hua K, Sheng X, Li TT, Wang LN, Zhang YH,
Huang ZJ and Ji H: The edaravone and 3-n-butylphthalide
ring-opening derivative 10b effectively attenuates cerebral
ischemia injury in rats. Acta Pharmacol Sin. 36:917–927. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kang KW, Lee SJ and Kim SG: Molecular
mechanism of nrf2 activation by oxidative stress. Antioxid Redox
Signal. 7:1664–1673. 2005. View Article : Google Scholar : PubMed/NCBI
|