Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an
autosomal dominant disorder characterized by the occurrence of
tumors of the parathyroids, anterior pituitary and neuroendocrine
tumors of the pancreas and duodenum. It is furthermore associated
with germline mutations of the MEN1 tumor suppressor gene.
Parathyroid tumors, which lead to hypercalcemia are the most common
feature of MEN1 occurring in about 95% of patients. Pancreatic
islet cell tumors occur in approximately 40% of patients, while
anterior pituitary tumors occur in around 30% of patients (1). The MEN1 gene, which was
identified in 1997 by Chandrasekharappa et al (2), consists of 10 exons that encode the
protein referred to as menin. A comprehensive review and analysis
of 38 years of data related to 613 MEN1 mutations found that
frameshift mutations represent the highest rate (42%), while
missense mutations show a frequency of 25.5%. Nonsense mutations
represent 14%, splice-site mutations 10.5%; in-frame del/ins 5.5%
and gross deletions represent the remaining 2.5% (3). Furthermore, the most mutations
resulted in the 2, 9 and 10 exons, but thus far the
genotype-phenotype correlation has not been established (4). Therefore, DNA testing for MEN1
gene is an established and useful tool for the diagnosis of MEN1.
In the present study we report a familial MEN1 case with
prolactinoma and parathyroid adenoma, and a missense mutation
c.482G>A in exon 3 of MEN1 gene, which has been shown to
change the 161st amino acid from glycine to aspartate.
Case report
A 46-year-old woman was admitted to our hospital
because of malaise and gastrointestinal discomfort. For the past 6
years she has been receiving bromocriptine treatment for
prolactinoma, and she had a month old fracture in the left leg. Her
mother died of ‘gastric cancer’ 6 years ago, and her brother was
diagnosed with multiple kidney calculus 4 years ago. The rest of
the family history was noncontributory. Her body mass index (BMI)
was 31.1 kg/m2. The patient's initial laboratory
examinations are listed in Table
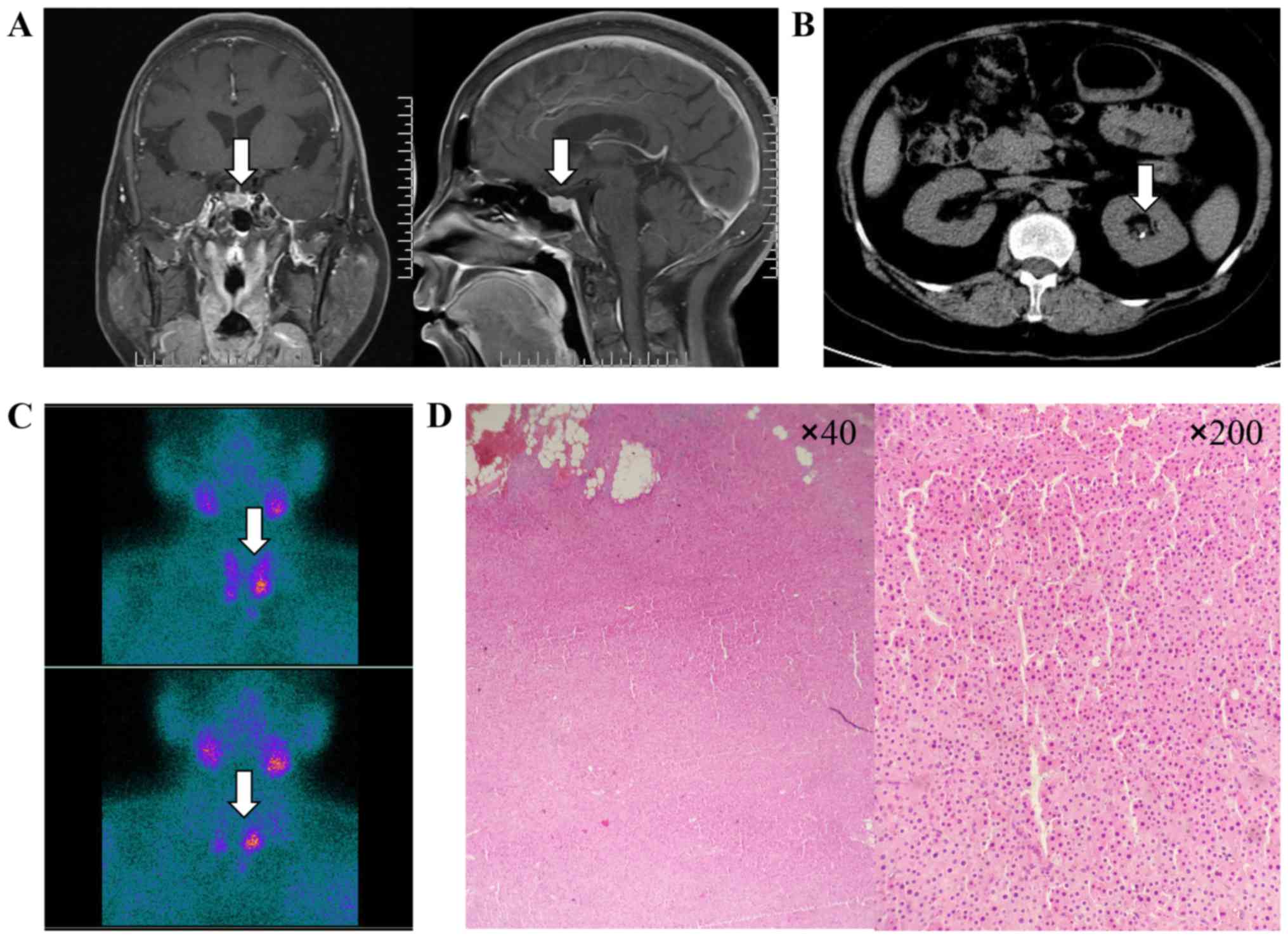
I. Pituitary magnetic resonance imaging (MRI) scanning showed
an adenoma in the pituitary (Fig.
1A). Abdominal computed tomography (CT) revealed multiple small
calculi in the left kidney (Fig.
1B), while bone mineral density showed severe osteoporosis
(Hologic Inc., Waltham, MA, USA). Dual-phase
99mTechnetium-Sestamibi (99mTc-MIBI)
scintigraphy revealed a significant uptake in the parathyroid gland
located in the inferior side of bilateral thyroid, which was
suggestive of parathyroid adenoma (Fig. 1C). The blood calcium decreased
gradually after using bisphosphonate and salmon calcitonin. The
postoperative pathology confirmed the bilateral parathyroid adenoma
(Fig. 1D), and on day after
parathyroidectomy, the levels of serum calcium (2.4 mmol/l) and
parathyroid hormone (PTH) (92.2 ng/ml) were immediately
reduced.
 | Table I.Laboratory results of the proband. |
Table I.
Laboratory results of the proband.
| Variable | Laboratory data | Normal range |
|---|
| PTH (ng/ml) | 426.6 | 15–65 |
| Calcium (mmol/l) | 3.08 | 2.2–2.7 |
| 24-h urine Ca
(mmol/day) | 8.9 | 2.5–7.5 |
| Phosphate
(mmol/l) | 0.68 | 0.81–1.9 |
| PRL (ng/ml) | 35.7 | 5.18–26.53 |
| GH (ng/ml) | 0.5 | 0.06–5 |
| IGF-1 (ng/ml) | 166 | 94–252 |
| HbA1c (%) | 5.1 | 4.0–6.5 |
| ACTH (8:00 am)
(pg/ml) | 45.6 | 7.2–63.3 |
| Cortisol (8:00 am)
(ng/ml) | 511 | 171–536 |
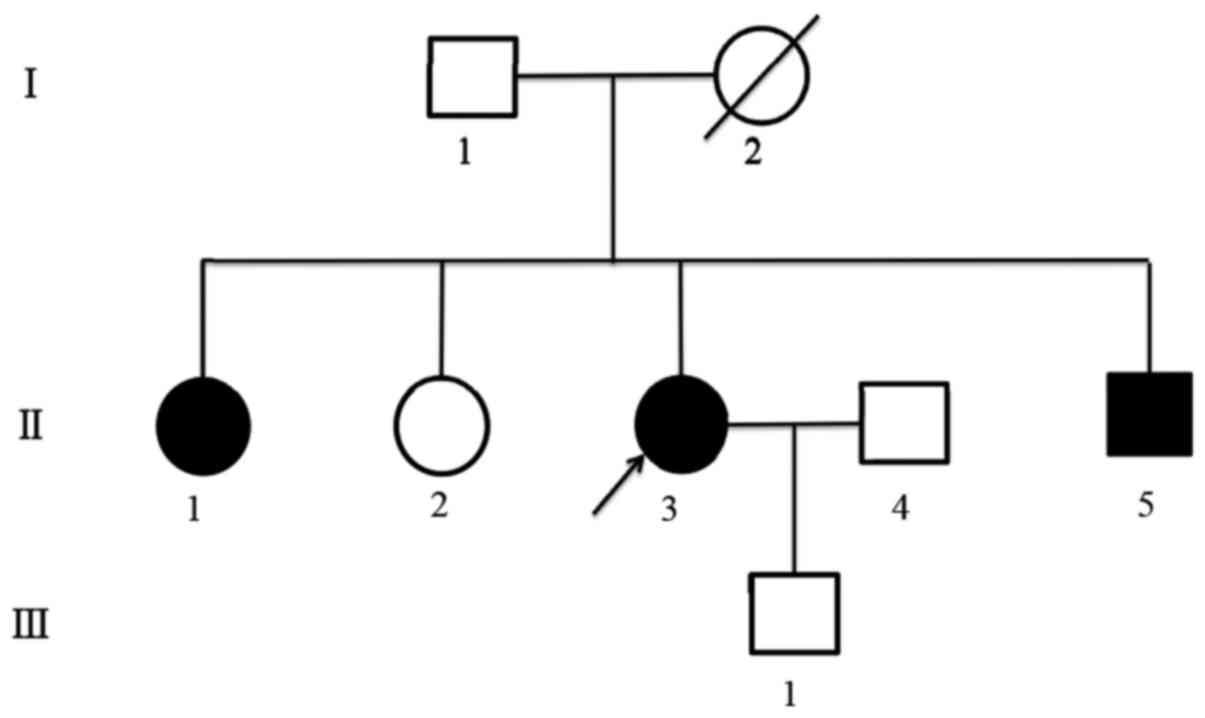
DNA was extracted from peripheral blood and resected
parathyroid tissue samples of the proband. The samples were taken
from her father, two elder sisters, brother and son after they
signed a written informed consent. Simultaneously, their serum
calcium, PTH and prolactin (PRL) were tested. We found the mutation
c.482G>A (p.Gly161Asp) in the third exon of MEN1 gene in
both peripheral blood and resected parathyroid tissue of the
proband (Fig. 2A). Patient's
brother and one of the elder sisters revealed the same mutation
(Fig. 2B), as well as high blood
calcium, PTH and PRL. Other family members tested normal, in both
gene testing and related laboratory examination (Fig. 2C and D). The identified mutation
has previously been reported (5);
nevertheless previous studies did not clearly illustrate
pathogenicity of this mutation and the function of the mutant
protein. Polyphen2, SNPs3D and SIFT were used to predict the
function of the 161st amino acid substitute from glycine to
aspartate. Polyphen2 predicted result score was 1.00 (i.e., score
closer to 1.00 was interpreted as the greater mutation damage;
score closer to 0 was interpreted as the smaller damage); SNPs3D
predicted result score was −1.97 (positive score was interpreted as
harmless; negative score was interpreted as harmful); SIFT
predicted result score was 0.00 (<0.05 was interpreted as having
the ability to affect protein function). All the obtained results
revealed that mutation may be harmful for the menin protein. Gene
sequencing was performed by Sangon Biotech Co., Ltd. (Shanghai,
China). The seqman of DNAstar package was used for alignment of
sequenced and normal genes. The name of mutation referenced the
cDNA sequence NM_00244.3 of Genbank. The tertiary structure of
menin protein was predicted using Swiss-model (6).
Discussion
The clinical diagnosis of MEN1 depends on the
identification of neoplastic disease in at least two of the
commonly affected organs, i.e., the parathyroid gland, endocrine
pancreas, and anterior pituitary (7). In the present case, the diagnosis of
MEN1 was demonstrated by prolactinoma and primary
hyperparathyroidism with bilateral parathyroid adenoma. Genetic
testing of the patient and family members revealed a missense
mutation c.482G>A (p.Gly161Asp) in patients and patient's elder
sister and brother. The sequencing peak pattern revealed additional
peak confirming the heterozygous mutation, and suggesting the
patient's mother might had the same mutation in the pedigree, while
other familiars tested normal (Fig.
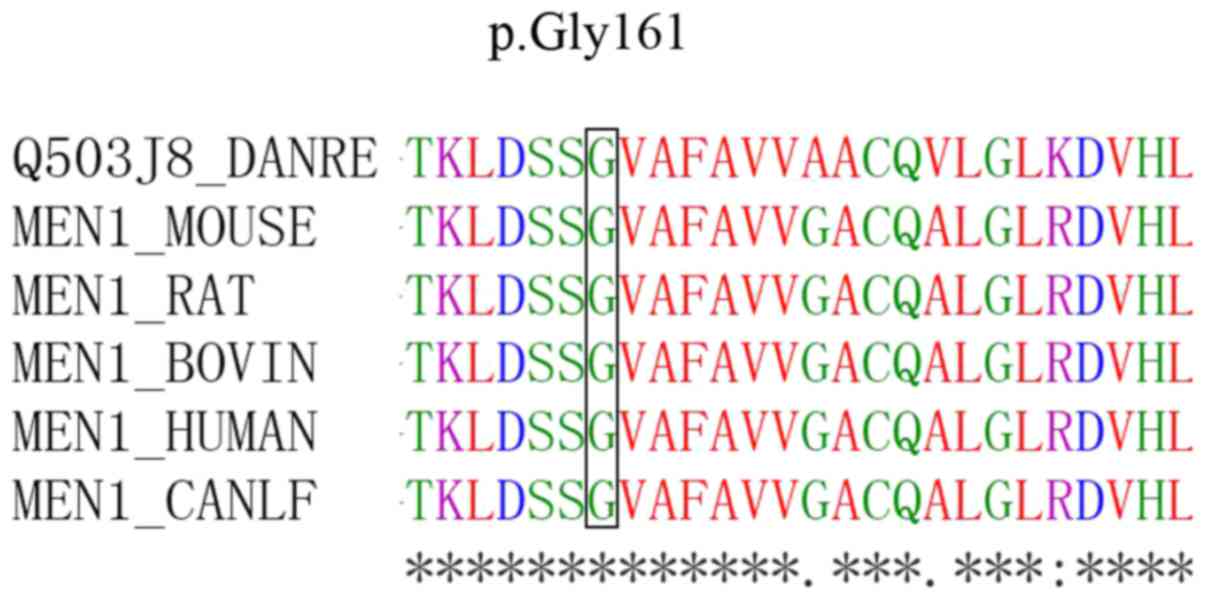
3). Sequence alignments revealed that the glycine residues were
highly conserved among menin proteins from different species
(Fig. 4).
The product of the MEN1 gene, menin, is a nuclear
protein (8) and it is believed to
function as a tumor suppressor having a number of potential roles
in maintaining normal cell function. Some of the more researched
menin functions indicated by direct interactions with many proteins
and other molecules are: 3 JunD-interacting domains at codons 1–40,
139–242, and 323–428, Smad family at codons 40–278, and 477–610,
and 3 nuclear localization signals at codons 479–497, 546–572, and
588–608 (1). The missense mutation
c.482G>A (p.Gly161Asp) was identified at codon 161 which
involved menin-JunD or menin-Smad3 interactions regions. Menin
contains a deep pocket that can bind mixed-lineage leukemia 1 (MLL1
or KMT2A) protein or the transcription factor (TF) JunD, with
opposite effects on gene transcription (9). In transcriptional regulation, menin
has been shown to interact with the activating protein-1 (AP-1) and
TF JunD (10), and to suppress
Jun-mediated transcriptional activation (11). Thevenon et al (12) reported that patients with mutations
affecting the JunD interacting domain had a higher risk of death
secondary to a MEN1 tumor. In addition, members of the Smad family,
Smad3 and the Smad 1/5 complex have been shown to be involved in
the transforming growth factor-β (TGF-β) (13) and the bone morphogenetic protein-2
(BMP-2) signaling pathways (14),
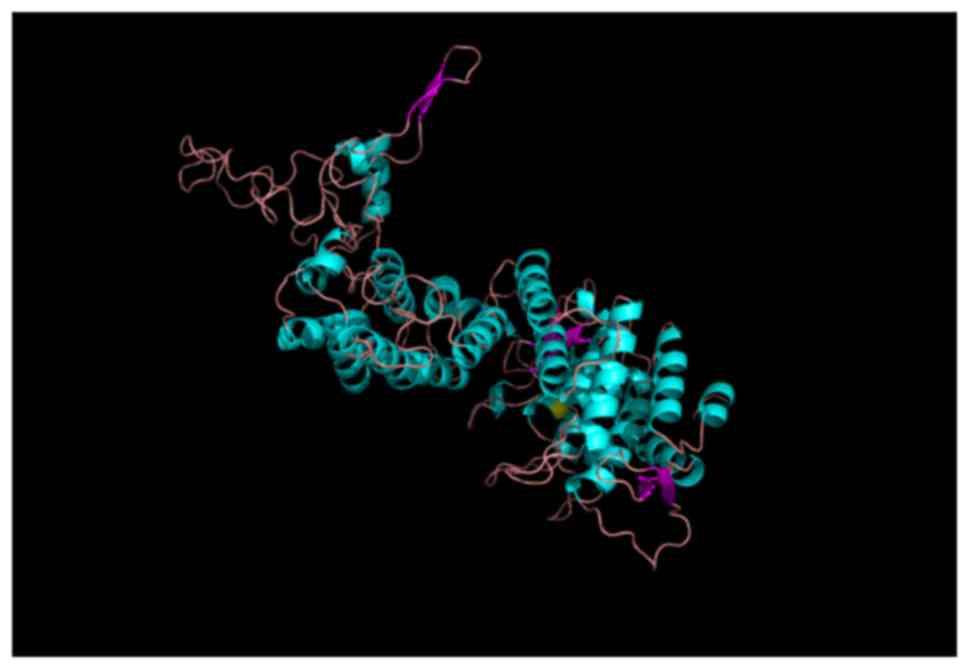
respectively. For the tertiary structure, the mutation p.Gly161Asp
changes the location of hydrogen bands (Fig. 5). The substitution of glycine, a
neutral amino acid for aspartate, an acidic amino acid, might
interfere with the menin-JunD or menin-Smad3 interactions. In the
present case of the Gly161Asp mutation, the Polyphen2, SNPs3D and
SIFT results, all supported the damage function of menin. Further
studies are necessary to explore the function of mutant protein
in vivo and in vitro.
Knudson's two-hit theory with a loss of
heterozygosity (LOH) in the MEN1 gene is commonly used to
explain tumorigenesis in the MEN1 patients (15). In the present study, the identified
miscellaneous peak of the proband's resected parathyroid tissue
confirmed a heterozygous missense mutation without LOH, implying
LOH might not be a unique condition. Luzi et al (16) found that microRNA24-1 (miR-24-1)
has ability to bind to the untranslated region (3′UTR) of MEN1
mRNA. Also, the negative feedback-loop between the oncomir miR-24-1
and menin, existing in MEN1 parathyroid adenoma without LOH,
modulates the MEN1 tumorigenesis by mimicking the ‘Knudson's second
hit’.
In summary, we herein reported a case of MEN1 with a
missense mutation c.482G>A (p.Gly161Asp) of the MEN1
gene, which appeared to be responsible for MEN1. The proband and
two family members had the same mutation and abnormal laboratory
examinations. This suggests that identification of mutations might
be very important for prompt diagnosis and monitoring of at-risk
family members in MEN1 pedigrees (17).
Acknowledgements
This study was supported by the the Joint Project of
Medical Science and Technology of Henan Province, China (grant no.
201201015).
Glossary
Abbreviations
Abbreviations:
|
MEN1
|
multiple endocrine neoplasia type
1
|
|
BMI
|
body mass index
|
|
MRI
|
magnetic resonance imaging
|
|
CT
|
computed tomography
|
|
BMD
|
bone mineral density
|
References
|
1
|
Lemos MC and Thakker RV: Multiple
endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations
reported in the first decade following identification of the gene.
Hum Mutat. 29:22–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chandrasekharappa SC, Guru SC, Manickam P,
Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z,
Lubensky IA, Liotta LA, et al: Positional cloning of the gene for
multiple endocrine neoplasia-type 1. Science. 276:404–407. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Concolino P, Costella A and Capoluongo E:
Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new
germline variants reported in the last nine years. Cancer Genet.
209:36–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stewart C, Parente F, Piehl F, Farnebo F,
Quincey D, Silins G, Bergman L, Carle GF, Lemmens I, Grimmond S, et
al: Characterization of the mouse Men1 gene and its expression
during development. Oncogene. 17:2485–2493. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mutch MG, Dilley WG, Sanjurjo F,
DeBenedetti MK, Doherty GM, Wells SA Jr, Goodfellow PJ and Lairmore
TC: Germline mutations in the multiple endocrine neoplasia type 1
gene: Evidence for frequent splicing defects. Hum Mutat.
13:175–185. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guex N, Peitsch MC and Schwede T:
Automated comparative protein structure modeling with SWISS-MODEL
and Swiss-PdbViewer: A historical perspective. Electrophoresis. 30
Suppl 1:S162–S173. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thakker RV, Newey PJ, Walls GV, Bilezikian
J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F and Brandi
ML: Endocrine Society: Clinical practice guidelines for multiple
endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab.
97:2990–3011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guru SC, Goldsmith PK, Burns AL, Marx SJ,
Spiegel AM, Collins FS and Chandrasekharappa SC: Menin, the product
of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci USA.
95:pp. 1630–1634. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang J, Gurung B, Wan B, Matkar S,
Veniaminova NA, Wan K, Merchant JL, Hua X and Lei M: The same
pocket in menin binds both MLL and JUND but has opposite effects on
transcription. Nature. 482:542–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Agarwal SK, Guru SC, Heppner C, Erdos MR,
Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS,
Spiegel AM, et al: Menin interacts with the AP1 transcription
factor JunD and represses JunD-activated transcription. Cell.
96:143–152. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pfarr CM, Mechta F, Spyrou G, Lallemand D,
Carillo S and Yaniv M: Mouse JunD negatively regulates fibroblast
growth and antagonizes transformation by ras. Cell. 76:747–760.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thevenon J, Bourredjem A, Faivre L,
Cardot-Bauters C, Calender A, Murat A, Giraud S, Niccoli P, Odou
MF, Borson-Chazot F, et al: Higher risk of death among MEN1
patients with mutations in the JunD interacting domain: A Groupe
d'Εtude des Tumeurs Endocrines (GTE) cohort study. Hum Mol Genet.
22:1940–1948. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaji H, Canaff L, Lebrun JJ, Goltzman D
and Hendy GN: Inactivation of menin, a Smad3-interacting protein,
blocks transforming growth factor type beta signaling. Proc Natl
Acad Sci USA. 98:pp. 3837–3842. 2001; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sowa H, Kaji H, Canaff L, Hendy GN,
Tsukamoto T, Yamaguchi T, Miyazono K, Sugimoto T and Chihara K:
Inactivation of menin, the product of the multiple endocrine
neoplasia type 1 gene, inhibits the commitment of multipotential
mesenchymal stem cells into the osteoblast lineage. J Biol Chem.
278:21058–21069. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA. 68:pp.
820–823. 1971; View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luzi E, Marini F, Giusti F, Galli G,
Cavalli L and Brandi ML: The negative feedback-loop between the
oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by
mimicking the ‘Knudson's second hit’. PLoS One. 7:e397672012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Falchetti A: Genetic screening for
multiple endocrine neoplasia syndrome type 1 (MEN-1): When and how.
F1000 Med Rep. 2:pii: 142010.
|