Introduction
Ovarian cancer is the sixth most common cancer and
the seventh leading cause of cancer-associated mortality in women
worldwide (1). Traditionally,
patients suffering from the disease are treated by cytoreductive
surgery and/or chemotherapy (2).
Unfortunately, the past decades have seen little improvement in the
survival rate among ovarian cancer patients (3). Challenges in the surgical treatment
of ovarian cancer include late detection, tumor metastasis within
the peritoneal cavity, drug resistance and cancer recurrence even
after initial therapy (4).
Therefore, increasing attention has been drawn to the use of
chemotherapeutic agents.
As the first-line agent to treat ovarian cancer,
cisplatin (cis-diamminedichloroplatinum, DDP) and its platinum
derivatives cause DNA interstrand cross-links, which then induces
DNA damage responses and the initiation of apoptotic mechanism, and
ultimately triggers the death of cancer cells (5,6).
However, the continued use of these DNA cross-linking agents fuels
the rise of drug resistance, which is difficult to overcome. This
issue becomes more troublesome with the presence of glucose
tolerance or type II diabetes among cancer patients receiving
chemotherapy (7–10). In other words, the sensitivity of
cancer patients with diabetes to chemotherapy is reduced (8). As is well known, insulin (INS) is the
most effective hypoglycemic drug, which binds to cell surface INS
receptor, phosphorylates INS receptor substrates, and in turn
mediates a broad spectrum of specific pathways controlling cellular
proliferation (10). Recent
studies have determined the overexpression of insulin receptor
substrates in malignant cells (7,8),
while INS can promote the sensitivity of cancer cells to paclitaxel
(10). Therefore, possibilities
exist that a combined treatment of INS and DPP produce a superior
inhibitory effect on the proliferation of ovarian cancer cells,
when compared with DPP treatment alone.
Most anti-cancer drugs are designed to perturb cell
cycle by inducing/damaging cell cycle events, which activate
checkpoints, arrest cancer cells and causes apoptosis (11). The cell cycle is a set of
coordinated events that take place in a cell and function to
integrate various signaling cascades with cell growth and
proliferation (12). Cancer cells
usually deregulate the cell cycle and undergo unscheduled cell
division. Therefore, inhibition of the cell cycle constitutes a
potential strategy for therapeutic intervention in cancer therapy
(13,14). c-Jun N-terminal kinases (JNKs) are
multifunctional kinases involved in different physiological
processes. The JNK signal transduction pathway has been
demonstrated to serve a crucial role in apoptosis in many cell
death paradigms. This kinase, together with tumor suppressor p53,
are two apoptosis-regulatory factors frequently involved in the
modulation of cancer cell death (15), which may also be signaling
components participating in the inhibition of ovarian cancer cell
proliferation after the combined therapy of INS and DPP.
The present study demonstrated that the combination
of INS and DDP facilitated DDP-induced apoptosis in A2780 cells.
The combined therapy resulted in a dramatic decrease in the
percentage of G0/G1 phase cells, but a corresponding increase in
the proportion of S phase cells. These changes could be associated
with the activation of the JNK signaling pathway and the
involvement of p53 at both mRNA and protein levels.
Materials and methods
Materials
The A2780 human ovarian cell line was obtained from
the China Center for Type Culture Collection (Wuhan, China).
Insulin, Dulbecco's modified Eagle's medium (DMEM) and fetal bovine
serum (FBS) were purchased from Life Technologies; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Annexin V-fluorescein
isothiocyanate (FITC), penicillin, streptomycin,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI),
propidium iodide (PI), RNase A and dimethyl sulfoxide (DMSO) were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Cis-platinum (DDP) was from Anji Haosen Pharmaceutical Co., Ltd.
(Huzhou, China). Antibodies against phosphorylated (p)-JNK (#9255)
and JNK (#9252) were bought from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Antibodies against β-actin (sc-47778) and p53
protein (sc-47698) were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Horseradish peroxidase (HRP)-conjugated
secondary antibodies for detection of rabbit (A0208) and mouse
(A0216) primary antibodies and an enhanced chemiluminescence (ECL)
western blotting detection system were purchased from Beyotime
Institute of Biotechnology (Nantong, China).
Cell culture
A2780 cells were cultured in DMEM supplemented with
10% FBS, 1% penicillin and 1% streptomycin at 37°C in a humidified
atmosphere of 95% air/5% CO2. During the experiments,
the cells used were in the exponential growth phase, while culture
media were replaced every 2 or 3 days.
Colorimetric MTT assay
The compound MTT was adopted to assess cell
proliferation as previously described (16). Briefly, A2780 cells were seeded
into 96-well plates at a density of 2×103 cells per well
in quintuplicate for 24 h before exposure to different
concentrations of DDP (0, 5, 10, 15, 20, 25 or 30 µg/ml), INS (0,
1, 2, 5, 10, or 15 mU/ml) or both as described above. After
incubation for another 24 h, 20 µl 5 mg/ml MTT was added to each
well and the cells were incubated at 37°C for another 4 h.
Subsequently, the media was discarded followed by the addition of
150 µl DMSO per well to dissolve purple precipitate. The absorbance
(A) was measured at 570 nm using a plate microreader. The cell
growth inhibitory rate (I%) was calculated according to the
following equation: inhibitory rate% = (Acontrol -
Asample) / (Acontrol - Ablank) ×
100%, where Acontrol is the absorbance of the untreated
cells, Asample is the absorbance of the cells exposed to
DDP, INS or both, and A blank is the absorbance of the
media.
DAPI staining
A2780 cells were seeded into 6-well plates at a
density of 5×104 cells per well and incubated at 37°C
for 24 h. Subsequently, the cells were fixed with 4% formaldehyde
in PBS containing 4% sucrose at room temperature for 15 min. After
washing with PBS three times, the cells were stained using 10 µg/ml
DAPI in the dark at room temperature for 15 min. The solution was
then removed and the cells were washed with PBS twice. Images were
observed under an Olympus X81 microscope (Olympus Corporation,
Tokyo, Japan) and analyzed by MetaMorph® Microscopy
Automation & Image Analysis Software (Molecular Devices, LLC,
Sunnyvale, CA, USA).
Apoptosis assay
A2780 cells (104/well) were incubated in
24-well plates. After treatment, the cells were collected and
resuspended in 500 µl binding buffer to reach a density of
2×106 cells/ml. The resultant samples were treated with
5 µl Annexin V-FITC and 5 µl PI in the dark at room temperature for
15 min before flow cytometry analysis (Miltenyi Biotec GmbH,
Bergisch Gladbach, Germany) using CellQuest™ version 3.3 (BD
Biosciences, San Jose, CA, USA).
Cell cycle analysis
Cell cycle stages were determined by PI staining
with flow cytometry. Briefly, after treatment, the cells (at
~1×106 cells per group) in triplicate were harvested
through centrifugation at 300 × g for 5 min at 4°C, followed by
washing with ice-cold PBS. Following this, the cells were
resuspended in 70% pre-cooled ethanol and fixed at 4°C overnight.
Next, the fixed cells were washed with PBS and incubated with RNase
A and PI at 37°C in the dark for 30 min. DNA content was quantified
by flow cytometry analysis.
Western blotting analysis
After measurement of protein concentrations using an
Enhanced BCA Protein Assay kit (Beyotime Institute of
Biotechnology), the obtained lysates (20 µg/well) were subjected to
4–10% SDS-PAGE and subsequently blotted to polyvinylidene
difluoride membranes (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) before incubation at 4°C overnight using the following primary
antibodies: p-JNK (1:500), JNK (1:1,000), β-actin (1:3,000) and p53
(1:500). The membranes were then incubated with HRP-conjugated
secondary antibodies (1:1,000, 1 h, room temperature) and
visualized using an ECL detection system. Optical densities of the
blots were measured using ImageJ version 1.47 (National Institutes
of Health, Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
According to the manufacturer's protocol, total RNA
was isolated from cells and reverse transcribed with oligo (dT)
primers by Takara Mini BEST Universal RNA Extraction kits (Takara
Biotechnology Co., Ltd., Dalian, China) with on column RNase-free
DNase I treatment (Takara Biotechnology Co., Ltd.). Samples were
run on the Light Cycler R 480 system (Roche Diagnostics GmbH,
Mannheim, Germany) using SYBR Premix Ex Taq (Takara Biotechnology,
Co., Ltd.) according to the manufacturer's protocol. The cycling
conditions were as follows: 95°C for 2 min, followed by 40 cycles
of 95°C for 20 sec, 56°C for 1 min and 72°C for 30 sec. All values
were normalized to GAPDH expression and data were analyzed using
the 2−∆∆Cq method (16). Primer sequences (Sangon Biotech
Co., Ltd., Shanghai, China) were as follows: p53
5′-CCTGTCATCTTCTGTCCCTT-3′ (forward) and 5′-GGGAGTAGGTGCAAGTCA-3′
(reverse); p21 5′-GCGGAACAAGGAGTCAGACAT-3′ (forward) and
5′-CCCAATACTCCAAGTACACTAAGCA-3′ (reverse); GAPDH
5′-TCAACTACATGGTTTACATGTTC-3′ (forward) and
5′-GATCTCGCTCCTGGAAGAT-3′ (reverse).
Statistical analysis
All data in the different experimental groups are
expressed as the mean ± standard deviation. Differences between the
groups were assessed by one-way analysis of variance followed by
the Student-Newman-Keuls, least-significant difference (for equal
variances) or Dunnett T3 (unequal variances) multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of DDP and INS on the
proliferation of A2780 cells
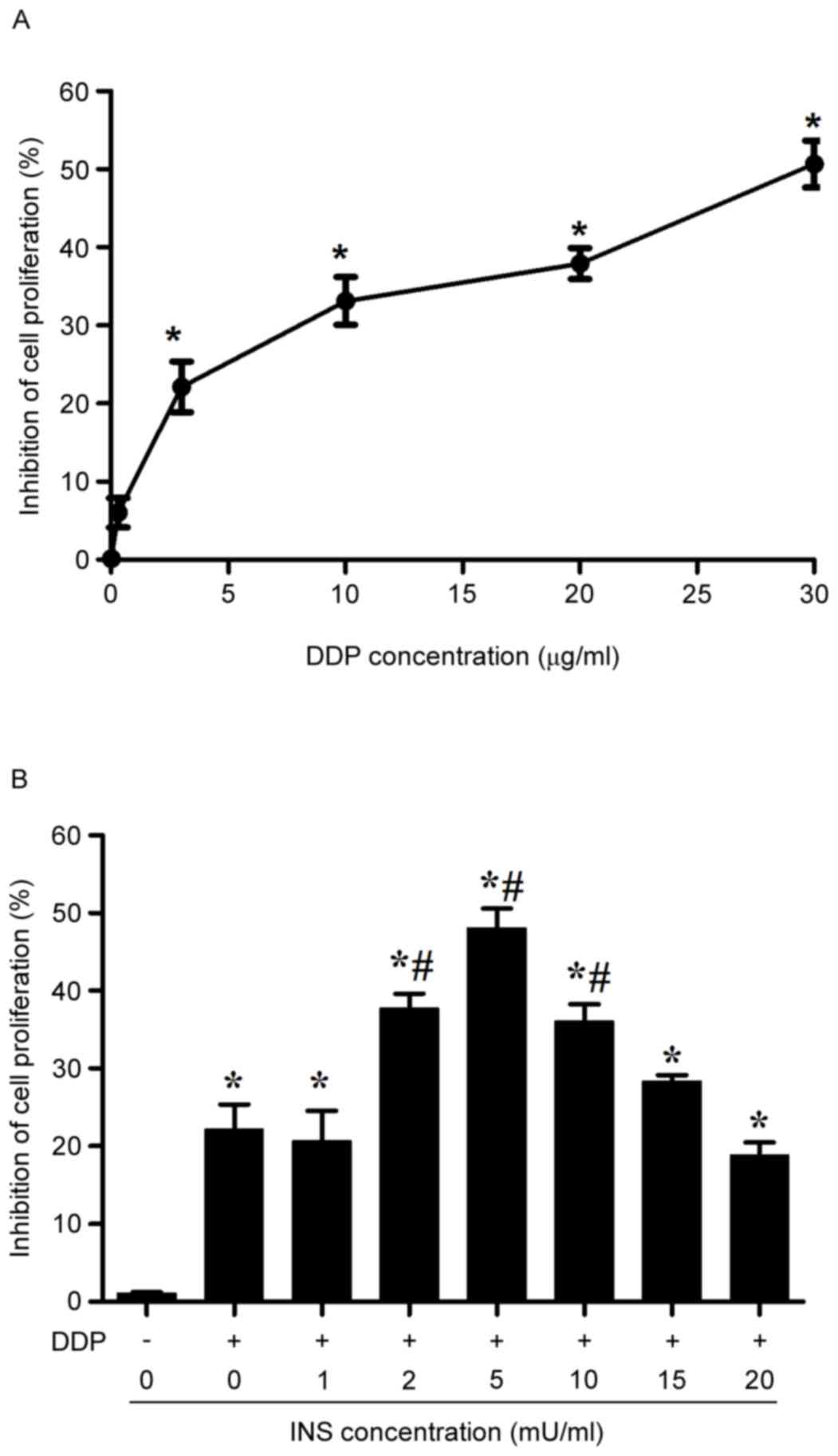
MTT assay demonstrated that the proliferation of
A2780 cells was systemically inhibited with the rise of DDP
concentrations from 0.3–30 µg/ml after 24 h of treatment (Fig. 1A). Notably, the inhibitory effect
became significant when the cells were exposed to 3 µg/ml DDP.
Therefore, such a concentration was used throughout the following
experiments. With respect to the presence of impaired glucose
tolerance or type II diabetes in patients receiving DDP-based
chemotherapy who may also receive INS, the anti-proliferative
effects of INS in combination with DDP on A2780 cells were measured
by MMT. In the assay, the cells were administrated with various
concentrations of INS (0, 1, 2, 5, 10, 15 and 20 mU/ml) for 6 h
before treatment with 3 µg/ml DDP for 24 h. The results suggested
that pre-exposure to INS could cause an increase in the inhibitory
rate of A2780 cell proliferation due to administration of 3 µg/ml
DDP, with the most substantial impact at 5 mU/ml (Fig. 1B).
INS facilitates DDP-induced
apoptosis
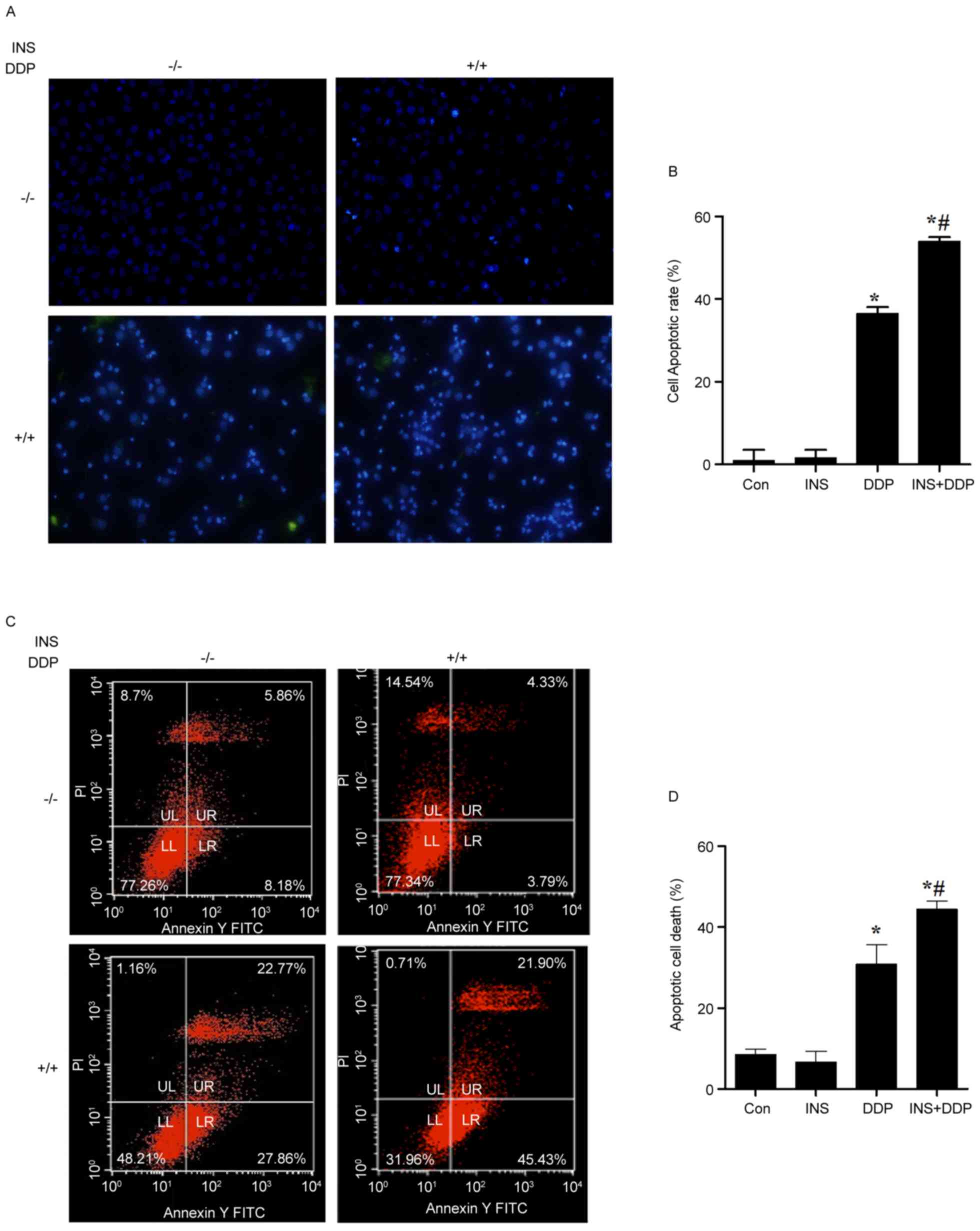
To investigate the possible influence of INS on
DDP-induced apoptosis, nuclear changes of A2780 cells were observed
using DAPI stating under a fluorescence microscope. As presented in
Fig. 2A, fewer normal nuclei but
more ‘ghost nuclei’ were observed in the INS and DDP combination
group, compared with the cells treated with INS or DDP alone. The
cell apoptotic rate was increased in the DDP and INS+DDP groups,
compared with the control and INS groups (Fig. 2B). The same results were determined
by flow cytometry analysis (Fig. 2C
and D).
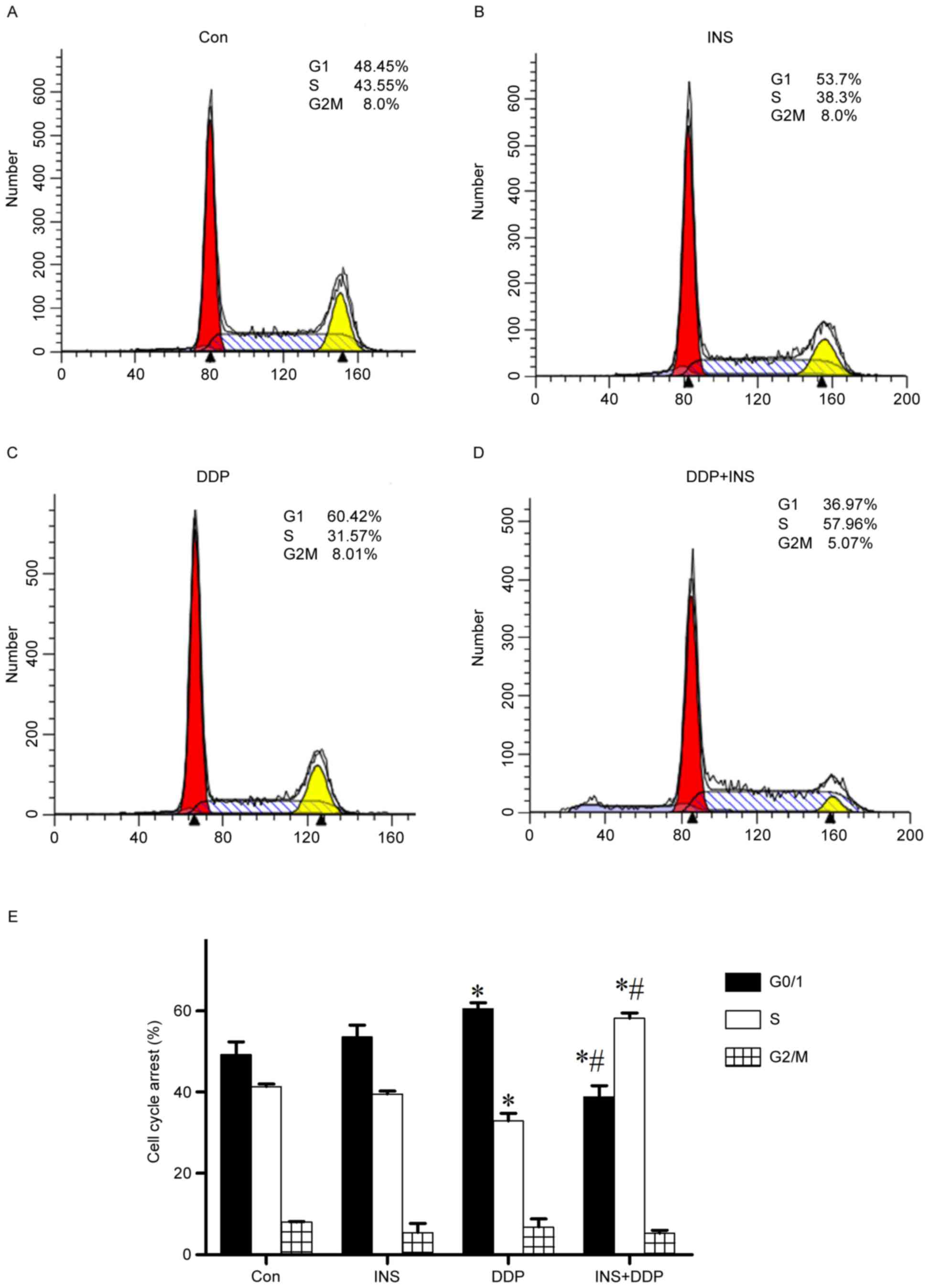
Cell cycle arrest
To further determine the mechanism by which INS
heightens the sensitivity of A2780 cells to DDP, the cell cycle
stages of both treated and normal cells were examined by flow
cytometry. The results demonstrated that compared with the control
group, administration of DDP alone significantly induced
G0/G1-phase accumulation, with a visible reduction in the number of
S phase cells (Fig. 3). However,
INS-DDP treatment brought in a marked decrease in the percentage of
G0/G1 phase cells, but a corresponding increase in the proportion
of S phase cells (Fig. 3).
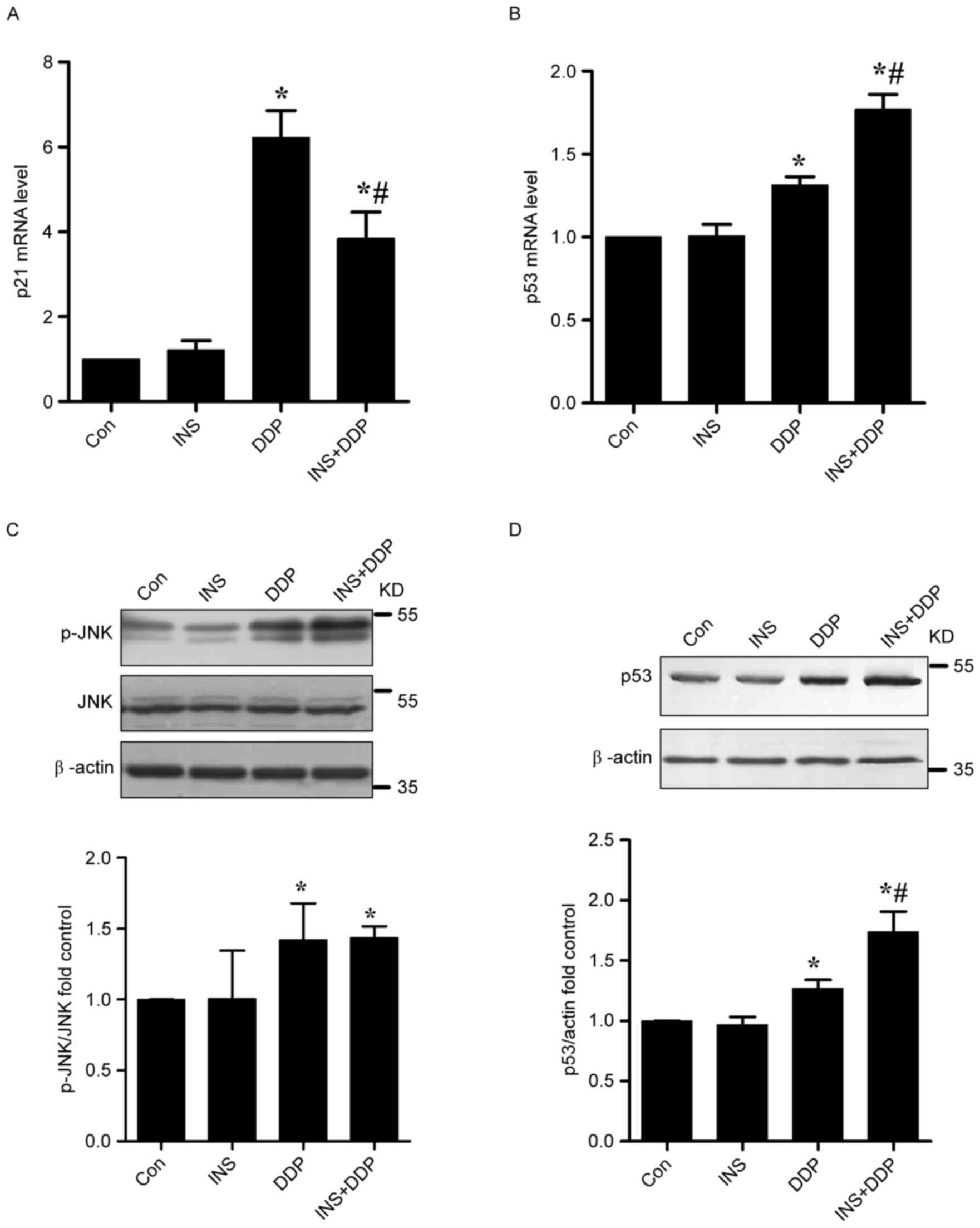
To further verify the impact of the INS on cell
cycle, the expression of the cell cycle-associated p21 gene was
examined. As presented in Fig. 4A,
increased expression levels of p21 mRNA were examined in the DDP
groups. In contrast, reduced amounts of p21 mRNA were observed when
the cells were pre-exposed to INS followed by DDP administration,
compared with DDP treatment alone. In other words, INS-DDP
administration could cause cell cycle redistribution compared with
DDP treatment alone.
Effects of DDP and INS on the JNK-p53
signaling pathway in A2780 cells
The role of JNK signaling in cancer progression
remains unclear. This signaling cascade can be attributed to both
tumor-promoting and tumor-suppressing functions. Previous studies
have reported that, under a stress state, JNK phosphorylates p53
protein, inhibiting its degradation and consequently promoting cell
apoptosis (4,17). The expression of p53 mRNA in A2780
cells was measured by RT-qPCR. The results indicated that DDP
treatment could significantly up-regulate the levels of p53 mRNA
compared with the control. In contrast, increased amounts of p53
mRNA were determined when the cells were pre-exposed to INS
followed by DDP administration, compared with DDP treatment alone
(Fig. 4B).
As presented in Fig.
4C, increased quantities of p-JNK were observed in both the DDP
and INS-DDP groups compared with the control and INS groups. The
p53 protein functions in the checkpoints that arrest human cells in
G1/S, S or G2/M phases. According to western blotting, in
comparison with the control, the expression levels of p53 in A2780
cells exposed to DDP were substantially increased, which were
further elevated after INS-DDP administration (Fig. 4D).
Discussion
In the current study, INS in combination with DDP
facilitated DDP-induced apoptosis in A2780 cells through activation
of JNK, which was in accordance with previous reports (17–19).
This combined administration also upregulated the mRNA and protein
expression levels of p53 compared with DDP treatment alone, which
may then activate other cell cycle checkpoint factors and result in
cell cycle redistribution.
Apoptosis is the key to understanding some malignant
diseases. A failure in apoptosis can lead uncontrolled triggering
of programmed cell death, which is considered as one of the
hallmarks of cancer (20).
Therefore, activation of apoptosis signaling pathways is a
potential mechanism for cancer therapeutic targets, including
chemotherapy (3,4,19).
DDP, one of the chemotherapy drugs commonly used to treat ovarian
cancer, can kill tumor cells mainly through inducing DNA damage and
activating apoptosis-associated signaling pathways, which finally
leads to cell apoptosis (6,7). In
the present study, administration of DDP could significantly
inhibit the proliferation of A2780 cells in a dose-dependent manner
and cause cell apoptosis. In contrast, exposure to both INS and DDP
resulted in a higher inhibitory rate of A2780 cell proliferation
and facilitated apoptosis. Treating with higher concentrations of
INS results in a lower inhibitory rate. This kind of insulin
resistance may be because insulin increases the bioactivity of
insulin-like growth factor 1 (IGF-I) which appear to have a role in
tumor initiation (21).
Circulating IGF-1 and IGF-2 bind to the IGF-1 receptor and trigger
a signal transduction cascade that leads to increased proliferation
and enhanced survival of IGF-responsive cells (22).
The cell cycle is a series of events that take place
in a cell leading to its division and duplication (8). Regulation of the cell cycle involves
processes crucial to the survival of a cell (9,10).
According to the present study, a significant accumulation of cells
in G0/G1 phase and a visible reduction in S phase were observed in
DDP-treated A2780 cells. However, INS-DDP treatment brought in a
dramatic decrease in the percentage of G0/G1 phase cells, but a
corresponding increase in the proportion of S phase cells, which
means DDP-INS administration may redistribute the cell cycle
compared with DDP treatment alone.
Many anticancer drugs target the cell cycle through
different signaling pathways and function through activating cell
cycle checkpoints and blocking cell division (8). Among these signaling components
involved is JNK, which under a stress state can phosphorylate p53
protein, inhibiting its degradation and consequently promoting cell
apoptosis (4,17), especially arresting human cells in
G1/S, S or G2/M phases (11,23,24).
Increased amounts of p-JNK were measured both the DDP and INS-DDP
groups. The quantities of p53 mRNA and protein in A2780 cells
exposed to DDP were substantially increased, which were further
elevated after INS-DDP administration. Although a drug may not
possess a particular effect, it may induce the tissues or receptors
to respond to a different drug; a process that is known as
sensitization. In the present study, INS served a role in
chemotherapeutic sensitization within A2780 cells; this effect may
be related to appropriate dosage and action time. Therefore, INS
may be useful as an adjuvant chemotherapy to reduce DDP dosage,
this would result in the ability to alleviate the toxicity and side
effects, and to improve the life quality of the patients.
References
|
1
|
Seeber LM and Van Diest PJ: Epigenetics in
ovarian cancer. Methods Mol Biol. 863:253–269. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meyerhardt JA, Catalano PJ, Haller DG,
Mayer RJ, Macdonald JS, Benson AB III and Fuchs CS: Impact of
diabetes mellitus on outcomes in patients with colon cancer. J Clin
Oncol. 21:433–440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Papa V, Pezzino V, Costantino A, Belfiore
A, Giuffrida D, Frittitta L, Vannelli GB, Brand R, Goldfine ID and
Vigneri R: Elevated insulin receptor content in human breast
cancer. J Clin Invest. 86:1503–1510. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Belfiore A: The role of insulin receptor
isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr
Pharm Des. 13:671–686. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartucci M, Morelli C, Mauro L, Andò S and
Surmacz E: Differential insulin-like growth factor I receptor
signaling and function in estrogen receptor (ER)-positive MCF-7 and
ER-negative MDA-MB-231 breast cancer cells. Cancer Res.
61:6747–6754. 2001.PubMed/NCBI
|
|
6
|
Miglietta A, Panno ML, Bozzo F, Gabriel L
and Bocca C: Insulin can modulate MCF-7 cell response to
paclitaxel. Cancer Lett. 209:139–145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zou K, Ju JH and Xie H: Pretreatment with
insulin enhances anticancer functions of 5-fluorouracil in human
esophageal and colonic cancer cells. Acta Pharmacol Sin.
28:721–730. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hellawell GO, Turner GD, Davies DR,
Poulsom R, Brewster SF and Macaulay VM: Expression of the type 1
insulin like growth factor receptor is up-regulated in primary
prostate cancer and commonly persists in metastatic disease. Cancer
Res. 62:2942–2950. 2002.PubMed/NCBI
|
|
9
|
Cox ME, Gleave ME, Zakikhani M, Bell RH,
Piura E, Vickers E, Cunningham M, Larsson O, Fazli L and Pollak M:
Insulin receptor expression by human prostate cancers. Prostate.
96:33–40. 2009. View Article : Google Scholar
|
|
10
|
Law JH, Habibi G, Hu K, Masoudi H, Wang
MY, Stratford AL, Park E, Gee JM, Finlay P, Jones HE, et al:
Phosphorylated insulin-like growth factor-i/insulin receptor is
present in all breast cancer subtypes and is related to poor
survival. Cancer Res. 68:10238–10246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
DeVita VT: Dose-response is alive and
well. J Clin Oncol. 4:1157–1159. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Minden A and Karin M: Regulation and
function of the JNK subgroup of MAP kinases. Biochim Biophys Acta.
1333:F85–104. 1997.PubMed/NCBI
|
|
13
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kyriakis JM, Banerjee P, Nikolakaki E, Dai
T, Rubie EA, Ahmad MF, Avruch J and Woodgett JR: The
stress-activated protein kinase subfamily of c-Jun kinase. Nature.
369:156–160. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li-Weber M: New therapeutic aspects of
flavones: The anticancer properties of Scutellaria and its main
active constituents Wogonin, Baicalein and Baicalin. Cancer Treat
Rev. 35:57–68. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Osborne CK, Bolan G, Monaco ME and Lippman
ME: Hormone responsive human breast cancer in long-term tissue
culture: Effect of insulin. Proc Natl Acad Sci USA. 73:pp.
4536–4540. 1976; View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Crescenzi E, Chiaviello A, Canti G, Reddi
E, Veneziani BM and Palumbo G: Low doses of cisplatin or
gemcitabine plus Photofrin/photodynamic therapy: Disjointed cell
cycle phase-related activity accounts for synergistic outcome in
metastatic non-small cell lung cancer cells (H1299). Mol Cancer
Ther. 5:776–785. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Genet Dev. 12:14–21. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arcidiacono B, Iiritano S, Nocera A,
Possidente K, Nevolo MT, Ventura V, Foti D, Chiefari E and Brunetti
A: Insulin resistance and cancer risk: An overview of the
pathogenetic mechanisms. Exp Diabetes Res. 2012:7891742012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Weroha SJ and Haluska P: The insulin-like
growth factor system in cancer. Endocrinol Metab Clin North Am.
41:335–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ichijo H: From receptors to
stress-activated MAP kinase. Oncogene. 18:6087–6093. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bogoyevitch MA: The isoform-specific
functions of the c-Jun N-terminal Kinases (JNKs): Differences
revealed by gene targeting. Bioessays. 28:923–934. 2006. View Article : Google Scholar : PubMed/NCBI
|