Introduction
Ischemic heart disease (IHD) is the main cause of
death and disability in the world. The incidence of acute
myocardial infarction (MI), a consequence of an imbalance between
oxygen supply and demand, is growing in the global burden of IHD
(1). Autophagy is a highly
conserved intracellular degradation process that is an adaptive
role to protect organisms against diverse pathologies, including
infections, cancer, aging, and heart disease (2). Recent studies have showed that
cardiomyocytes autophagy was activated in ischemic heart disease
(3). The increase of autophagy is
associated with the protective effect of ischemic preconditioning
(4). However, the mechanistic
process remains to be fully elucidated between myocardial ischemia
and autophagy. Hence, cardiomyocytes autophagy may be a promising
approach for the prevention of myocardial ischemia and progression
to heart failure.
3,3′-Diindolylmethane (DIM) is extracted from
natural product, derived from the acid-catalyzed condensation of
indole-3-carbinol in cruciferous vegetables. Numerous studies have
suggested that DIM has various pharmacological effects, including
antioxidant, antitumor, anti-angiogenic, anti-inflammatory and
anti-apoptotic (5–8). Meanwhile DIM could affect AMPKα and
JNK signaling pathway (9,10). DIM could inhibit the proliferation
of cancer cells through modulating autophagy (11). The previous studies showed that DIM
attenuated TGFβ1-induced myofibroblast differentiation through
down-regulated AKT/GSK-3β signaling pathways (12). The cardioprotective effects of DIM
on cardiac hypertrophy are partly mediated through AMPKa signaling
(13). It also improved myocardial
energy metabolism imbalance induced by pressure overload via AMPKα
in mice (14). All these studies
indicate the protective effect of DIM on cardiovascular disease.
However, it remains unknow whether DIM would protect cardiomyocyte
from hypoxia injury. The purpose of the study was to evaluate the
effect of DIM on hypoxia injury in cardiomyocytes and the
underlying mechanism.
Materials and methods
Materials
The primary antibodies included phosphorylated
(p)-AMPKα (sc101631; Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA), T-AMPKα (BS4046, BS1061, BS6271, respectively; Bioworld
Technology Inc., St. Louis Park, MN, USA) and p-c-Jun N-terminal
kinase (JNK) (4668P), T-JNK (9258), Bax (2722), Bcl-xl (2764P),
C-caspase3 (9664) and GAPDH (2118) (all purchased from Cell
Signaling Technology, Inc., Danvers, MA, USA). DIM was purchased
from Sigma-Aldrich (St. Louis, MO, USA; D9568-5G) and dissolved in
dimethyl sulfoxide (RNBC0311; Sigma-Aldrich) for the in
vitro bioassay.
Cell culture
Rat cardiac H9c2 cells (Cell Bank of the Chinese
Academy of Sciences, Shanghai, China) were cultured in Dulbecco's
modified Eagle's medium (DMEM, C11995; Gibco-BRL, Carlsbad, CA,
USA) supplemented with 10% fetal bovine serum (FBS; 10099-133;
Gibco-BRL), 100 U/ml penicillin/100 mg/ml streptomycin (15140;
Gibco-BRL) and 5% CO2 at 37°C. The media was exchanged
every 2 days and subcultured to 70–80% confluency. Cells were
plated at an appropriate density according to each experimental
design. H9c2 cells were seeded in 6-well plates at a density of
0.25×106 cells/well. Cells were pretreated with DIM (1,
5, and 10 µM) in serum-free DMEM at 37°C for 12 h prior to the
experiment. Cells were divided into four groups (Normoxia-PBS
group, Normoxia-DIM group, Hypoxia-PBS group, Hypoxia-DIM group).
Cell hypoxia used AnaeroPack-Anaero (Mitsubishi Gas Chemical
Company, Inc., Tokyo, Japan) and anaerobic jar. The Pack could
consume all the oxygen and produce equivalent carbon dioxide. H9c2
cells, anaerobic indicator and AnaeroPack were placed in anaerobic
jar and immediately closed the jar lid. After approximately 30 min,
the oxygen concentration decreased to less than 0.1%. The whole
anoxic process was conducted at 37°C.
Quantitative polymerase chain reaction
(qPCR)
qPCR was used to detect RNA expression levels of
autophagy and fibrotic markers. Following 12 h pre-treatment with
DIM, H9c2 cells were incubated with hypoxia for 12 h prior to the
extraction of total RNA using TRIzol, and their yields and purities
were spectrophoto metrically estimated using the A260/A280 and
A230/260 ratios via a SmartSpec Plus Spectrophotometer (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). RNA (2 µg from each sample)
was reverse-transcribed into cDNA using oligo (DT) primers (Sangon
Biotech Co., Ltd., Shanghai, China) and the Transcriptor First
Strand cDNA Synthesis kit (04896866001; Roche Diagnostics). PCR
amplifications were quantified using the LightCycler 480 SYBR-Green
I Master mix. GAPDH was used as the internal control. The PCR
cycling conditions were as follows: Initial activation at 95°C for
10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1
min. The sequences of the oligonucleotide primers (Sangon Biotech
Co., Ltd.) were as follows: GAPDH, forward:
5′-GACATGCCGCCTGGAGAAAC-3′ and reverse: 5′-AGCCCAGGATGCCCTTTAGT-3′;
IL-1β, forward: 5′-GGGATGATGACGACCTGCTAG-3′ and reverse:
5′-ACCACTTGTTGGCTTATGTTCTG-3′; IL-6, forward:
5′-GTTGCCTTCTTGGGACTGATG-3′ and reverse:
5′-ATACTGGTCTGTTGTGGGTGGT-3′; TNF-α, forward:
5′-AGCATGATCCGAGATGTGGAA-3′ and reverse:
5′-TAGACAGAAGAGCGTGGTGGC-3′; Beclin1, forward:
5′-AGCTTTTCTGGACTGTGTGC-3′ and reverse: 5′-TGAACTTGAGCGCCTTTGTC-3′;
Atg7, forward: 5′-CACCAAAACAGATCCAGGCC-3′ and reverse:
5′-AGGGTGCTGGGTTAGGTTAC-3′; Atg5, forward:
5′-CAAGGATGCAGTTGAGGCTC-3′ and reverse: 5′-AGTTTCCGGTTGATGGTCCA-3′;
P62 forward: 5′-AAGAGGCTCCATCACCAGAG-3′ and reverse:
5′-CCCCTTGACTCTGGCTGTAA-3′.
Anti-oxidative assessments
Anti-oxidative was assessed by the release of
nitrite oxide (NO) and the activity of superoxide dismutase (SOD)
according to the instructions (Beyotime Institute of Biotechnology,
Shanghai, China). For NO detection, the supernatant was collected
and transferred to another 96-well plate. Then NO released from
H9c2 cells was measured at 540 nm using spectrophotometer according
to the manufacturer's instructions (Beyotime Institute of
Biotechnology). For SOD assessment, proteins were extracted and
quantified before activity of SOD measurement at 450 nm using the
commercial kit (Beyotime Institute of Biotechnology).
TUNEL staining
A TUNEL assay was performed to label apoptotic
nuclei according to the manufacturer's instructions (ApopTag Plus
Fluorescein In Situ Apoptosis Detection kit). Briefly, following 12
h pre-treatment with DIM (10 µM), cells were incubated with hypoxia
for 12 h and subsequently fixed on coverslips in 1%
paraformaldehyde in phosphate-buffered saline (both from Sinopharm
Chemical Reagent Co., Ltd., Shanghai, China), stained with TUNEL
reagents (EMD Millipore, Billerica, MA, USA) and DAPI (Invitrogen
Life Technologies, Carlsbad, CA, USA) and observed under a
microscope (BX51; Olympus Corp., Tokyo, Japan). The index of cell
apoptosis was calculated as the percentage of apoptotic
nuclei/total number of nuclei.
LC3 immunofluorescent staining
The effect of DIM on LC3 expression of
cardiomyocytes hypoxia was examined by immunofluorescence method.
Cells were grown on slides in 24-well plates and were pretreated
with or without DIM. The cells were fixed in 4% paraformaldehyde
and then permeabilised with 0.1% Triton X-100. After blocking with
8% goat serum for 60 min, the slides were incubated with rabbit
polyclonal anti-LC3 in a 1:50 dilution. After 2 h of incubation in
a chamber at 37°C, the slides were washed with phosphate-buffered
saline (PBS) and incubated at 37°C for 1 h with a secondary goat
anti-rabbit antibody conjugated to Alexa Fluor® 488. The
cells on coverslips were mounted onto glass slides with Slow Fade
Gold antifade reagent with DAPI (S36939; Invitrogen Life
Technologies). The cells were observed using a DX51 microscope
(Olympus Corp.) and images were taken with a camera.
Western blot analysis
Cultured cardiac H9c2 cells were lysed in
radioimmuno precipitation (RIPA) lysis buffer and 50 µg cell lysate
was used for protein separation by 10% SDS-PAGE. The proteins were
then transferred to polyvinylidene difluoride (PVDF) membranes
(Millipore Corp., Billerica, MA, USA). Specific protein expression
levels were normalized to the GAPDH protein levels of the total
cell lysate and cytosolic proteins on the same PVDF membranes. The
following primary antibodies were used: p-AMPKα, T-AMPKα, p-JNK,
T-JNK, LC3, Bax, Bcl-xl, C-caspase3 and GAPDH. Antibody incubation
was performed overnight with gentle shaking at 4°C. Quantification
of the western blots was performed using an Odyssey infrared
imaging system (LI-COR Biosciences, Lincoln, NE, USA). Then the
membranes were incubated with IRDye® 800CW conjugated
secondary antibodies for 1 h. The blots were scanned using an
infrared Li-Cor scanner, allowing for simultaneous detection of two
targets (phosphorylated and total protein) within the same
experiment.
Statistical analysis
Values are expressed as the mean ± standard error of
the mean. Differences among groups were determined by one-way
analysis of variance followed by Tukey's post hoc test. Comparisons
between two groups were performed using an unpaired Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
DIM attenuates the inflammation in
response to hypoxia in H9c2 cells
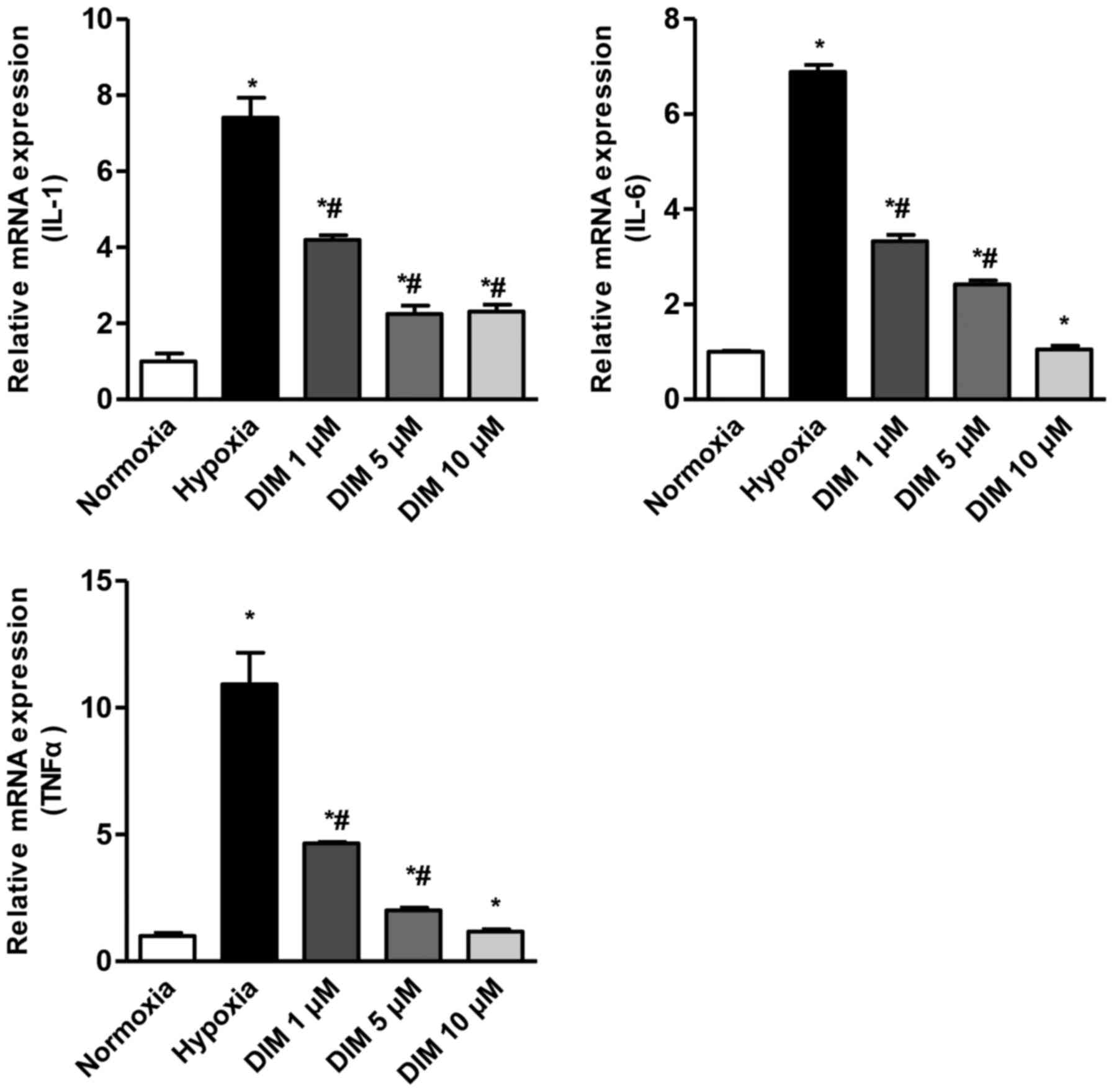
After H9c2 cardiomyocytes being expose to hypoxia
for 12 h, the mRNA levels of IL-1β, IL-6 and TNF-α were
significantly increased. We measured the effect of DIM with
different concentration (1, 5 and 10 µM) in reduction of IL-1β,
IL-6 and TNF-α in response to hypoxia (Fig. 1). As a result, DIM pretreatment
markedly attenuated this increase of mRNA expression in H9c2 cells
in a dose dependent maner.
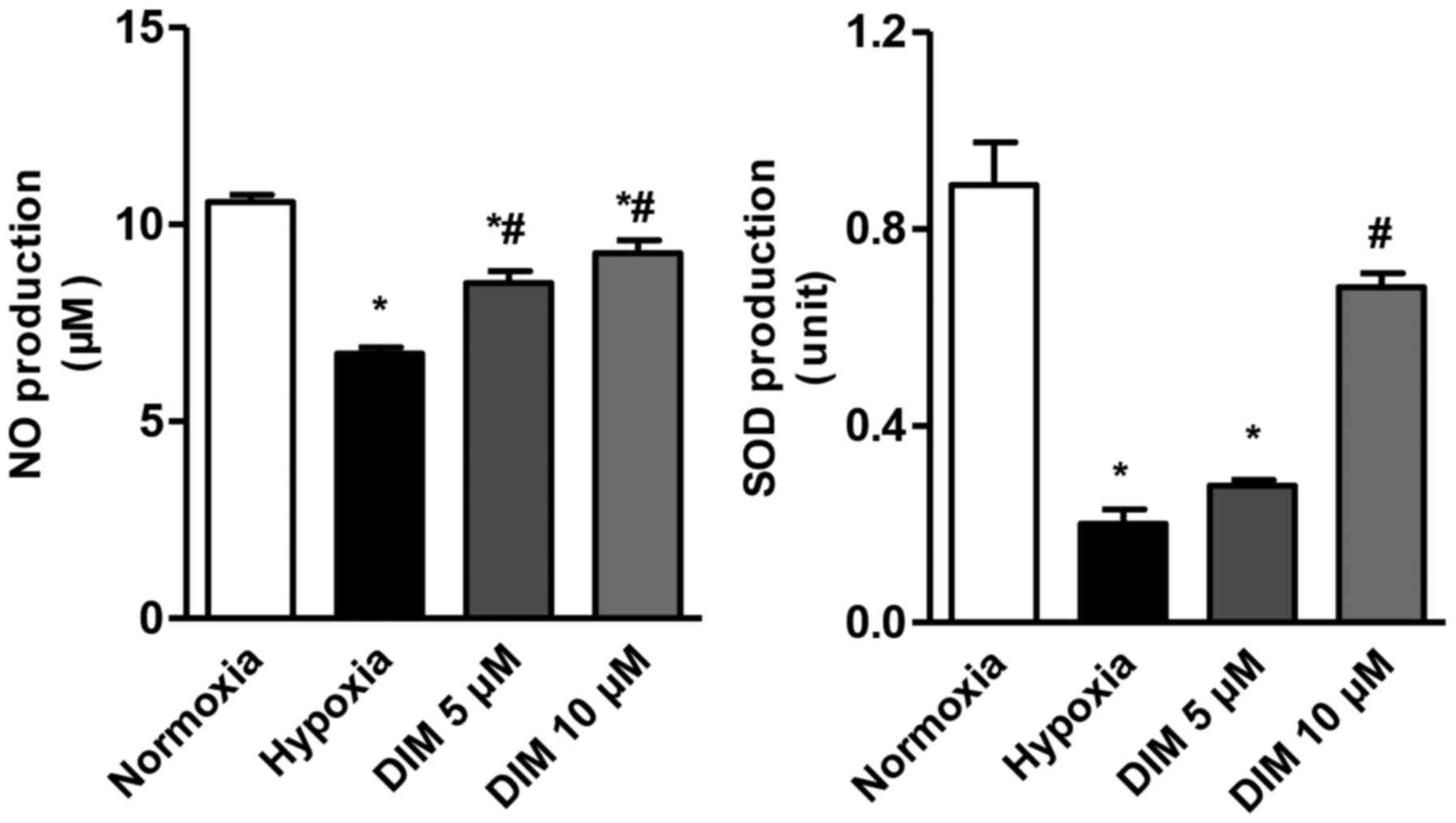
Anti-oxidative ability of DIM in
hypoxia-induced H9c2 cells
The effect of DIM at different concentrations (5 and
10 µM) on the production of NO and SOD in cardiomocytes exposed to
hypoxia was measured. Compared with hypoxia group, the production
of NO and SOD was increased after DIM pretreatment (Fig. 2). This result indicates that DIM
protects against the oxidative injury induced by hypoxia. Since 10
µM DIM has a better performance, it was chosen for the further
study.
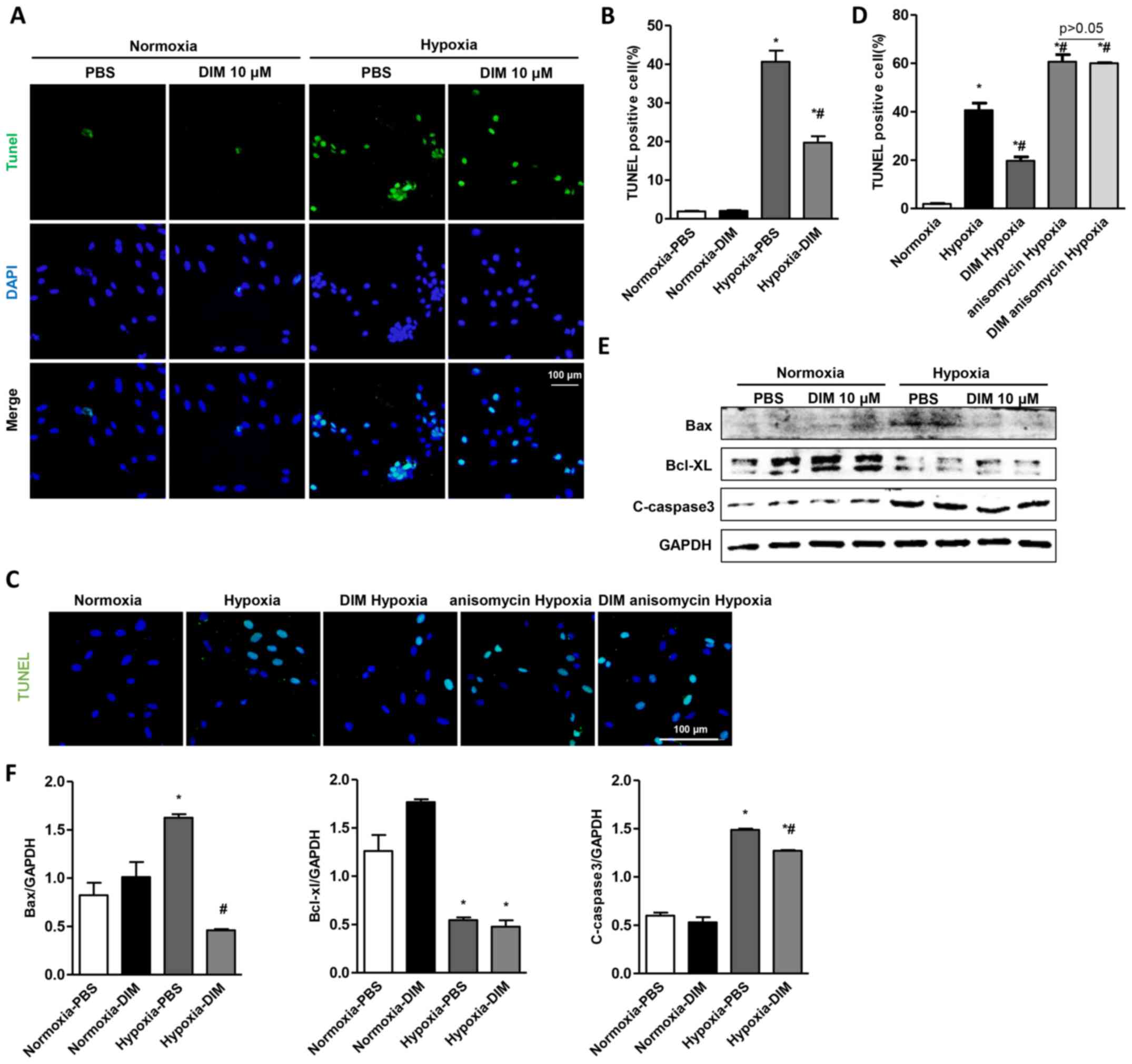
DIM attenuates apoptosis in
hypoxia-induced H9c2 cells
To demonstrate the protective role of DIM in
hypoxia-induced apoptosis in H9c2 cells, TUNEL staining was used to
detect the apoptotic nuclei. A significant increase in the number
of TUNEL-positive nuclei was observed in H9c2 cells in the hypoxia
group, whereas DIM pretreatment markedly reduced the percentage of
TUNEL-positive cells induced by hypoxia (Fig. 3A and B). TUNEL staining also showed
that the effect of DIM on hypoxia induced apoptosis was abolished
after being pretreated with JNK activator (anisomycin, 40 ng/ml)
(Fig. 3C and D). In addition, DIM
decreased the protein expression of Bax and C-caspase3 comparable
to hypoxia group, while increasing the protein Bcl-xl in H9c2 cells
(Fig. 3E and F).
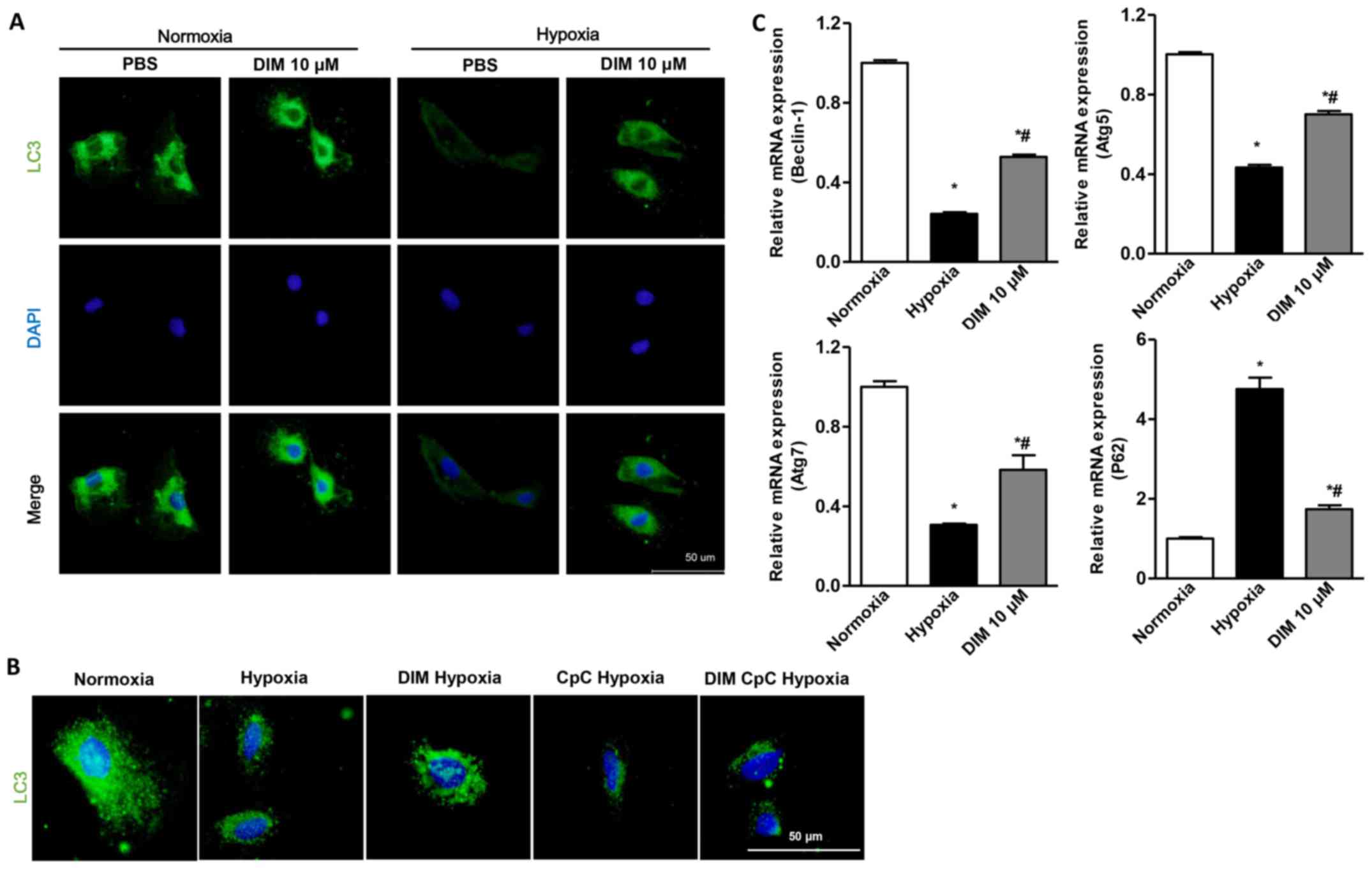
DIM activates autophagy in response to
hypoxia in H9c2 cells
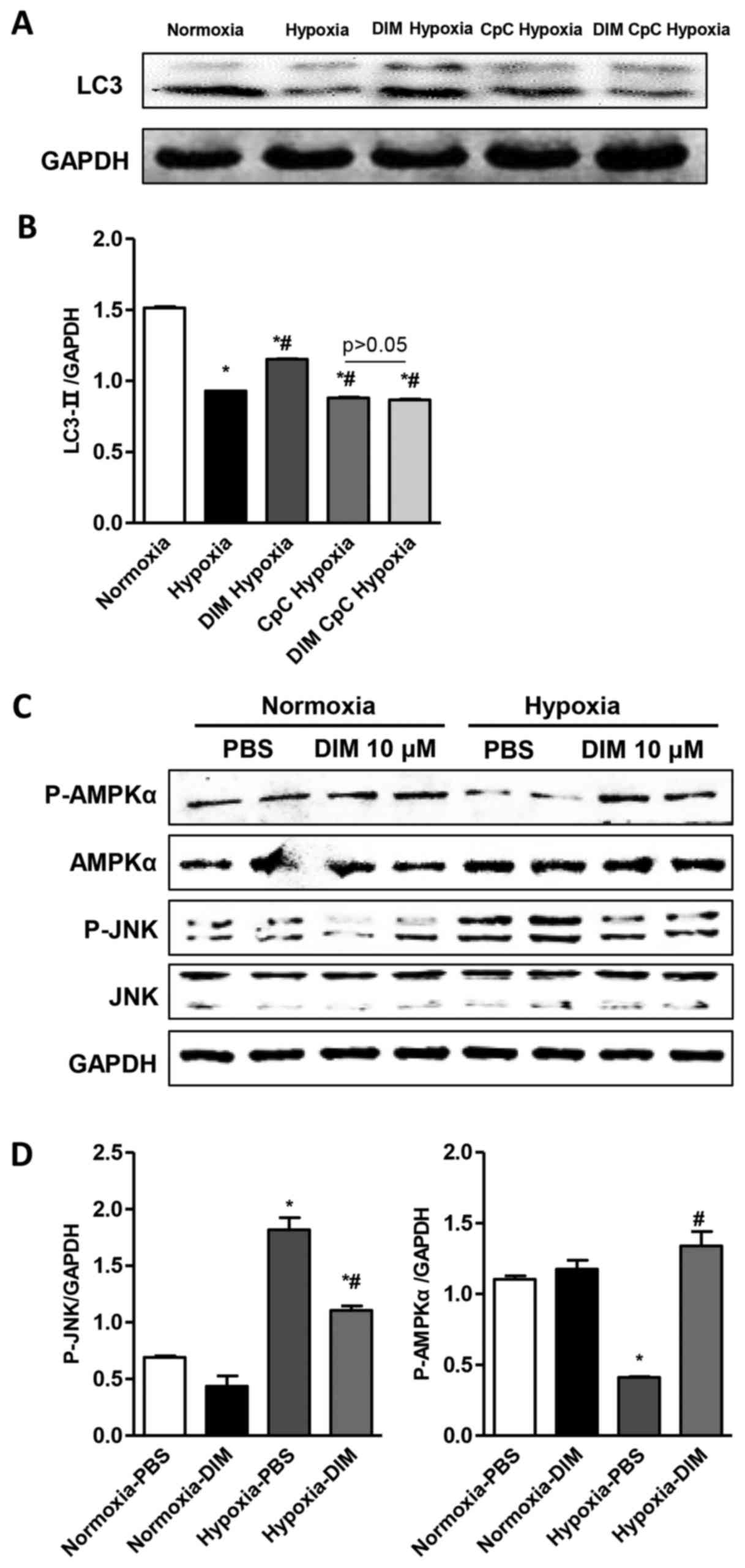
A significant decrease of LC3 expression through LC3
immunofluorescent staining was observed in H9c2 cells in the
hypoxia group, whereas DIM pretreatment markedly increased LC3
expression induced by hypoxia (Fig.
4A). LC3 immunofluorescent staining also showed that the effect
of DIM on autophagy was vanished after being pretreated with AMPKa
inhibitor (CpC, 20 µM) following stimulation with hypoxia (Fig. 4B). Beclin1 plays a central role in
autophagy induction. When autophagy occurs, Atg7 and Atg5 are
located in the isolated membrane, thus facilitating the extension
of the isolated membrane and forming the phagocytic vesicles. P62
is localized to the autophagosome formation site on the endoplasmic
reticulum (ER). It is deposited in the cells when autophagy is
reduced. After H9c2 cardiomyocytes being expose to hypoxia, the
relative mRNA levels of Beclin1, Atg5, Atg7 decreased, and P62
increased. With the pretreatment of DIM, the Beclin1, Atg5, Atg7
increased, and P62 decreased in response to hypoxia (Fig. 4C). The results indicated that DIM
(10 µM) ameliorated cardiomyocytes hypoxia injury via activating
autophagy.
DIM promotes p-AMPKa activity and
inhibits JNK signaling pathway in response to hypoxia in H9c2
cells
The mechanisms underlying the anti-inflmmatory,
anti-apoptotic and pro-autophagy effects of DIM on hypoxia induced
H9c2 cells injury were investigated via Western blot analysis. The
Western blot analysis showed that DIM pretreatment increased LC3
expression induced by hypoxia. While the effect of DIM on LC3
expression was vanished after being pretreated with AMPKa inhibitor
(CpC, 20 µM) following stimulation with hypoxia (Fig. 5A and B). The results showed that
the phosphorylation and activation of AMPKα was inhibited and the
phosphorylation and activation of JNK was increased after
cardiomyocytes expose to hypoxia. In comparison, 10 µM DIM
pretreatment increased the activation of AMPKα and decreased the
phosphorylation of JNK (Fig. 5C and
D). These findings indicates that DIM protects against
cardiomyocytes hypoxia injury, may through stimulating p-AMPKα
activity and inhibiting JNK signaling.
Discussion
The present study showed that DIM reduced the
inflammation and apoptosis in response to hypoxia and increased the
production of antioxidant NO and SOD. It also activated autophagy
to exert anti-hypoxia effects in cardiomyocytes by regulating AMPKa
and JNK signaling pathway.
Previous studies have showed that the inflammatory
response induced by hypoxia in cardiomyocytes subsequently induces
the induction of inflammatory mediators, including TNF-α, IL-1β and
IL-6 (15,16). From cardiac injury to failure,
inflammatory cytokines have been throughout the whole process
(17). Therefore, it is very
meaningful for myocardial ischemia to control inflammation
(18). DIM is a possible option
for inflammation in certain tissues and cells (19,20).
The present study showed that DIM could decrease the expression of
TNF-α, IL-1β and IL-6 in hypoxia-induced H9c2 cells injury model,
which indicated the effect of DIM on inflammation. Anti-oxidative
ability of DIM was performed by the detection of intracellular ROS,
the release of intracellular NO and the activity of intracellular
SOD (21). NO, the main effect of
the expansion of blood vessels, attenuates myocardial ischemia
(22). SOD can resist cell damage
caused by oxygen free radicals and timely repair of damaged cells,
reflecting the body's ability to clear free radicals, in
particular, to reflect the capacity of clearing superoxide anion
(23). Increased levels of NO and
SOD partly contribute towards the action of DIM to attenuate
cardiomyocyte hypoxia injury.
Myocardial apoptosis is involved in many
cardiovascular diseases, including myocardial hypertrophy, heart
failure, diabetic cardiomyopathy, ischemia/reperfusion injury
(24). The present study indicated
that DIM attenuated hypoxia-induced cardiomyocyte apoptosis. The
balance between regulation of apoptotic protein Bax and
anti-apoptotic protein Bcl-2 determines cells undergo apoptosis or
survive (25). The results showed
that DIM pretreatment downregulated the Bax and C-caspase3,
meanwhile the levels of Bcl-xl remained unchanged in cardiomyocyte
with hypoxia injury on DIM pretreatment. Activation of JNK, members
of the mitogen-activated protein kinase family, has been reported
to be induced by hypoxia (26).
Following activation, JNK phosphorylated a wide array of
intracellular targets, resulting in the reprogramming of cardiac
gene expression, the inflammation and apoptosis of cardiomyocytes
(27). Inhibition of the JNK
pathways in cardiomyocytes results in attenuated hypertrophic
growth induced by agonist stimulation (28). In addition, previous studies have
demonstrated that the activation of JNK is involved in the
induction of the inflammatory response and apoptosis (29,30).
The present study revealed that DIM inhibited the phosphorylation
of JNK in hypoxia-induced H9c2 cells, which indicated that the
anti-apoptosis effect of DIM may mediated by blocking JNK
pathway.
Autophagy is a conserved metabolic process, and
cellular components are transported to the lysosomal degradation
(31). Many studies have showed
that autophagy is involved in the pathogenesis of acute and chronic
ischemia heart injury and heart failure (32,33).
Emerging studies have demonstrated that autophagy is significantly
upregulated in cardiomyocytes ischemia, suggesting a role of
autophagy in the process of ischemia injury (34). Autophagy is stimulated by
myocardial ischemia and exert protective effect during myocardial
ischemia (35). The autophagy
pathway was involved in the cardio-protective effect induced by
ischemic preconditioning (IPC) (36). The cardiomyocyte-specific knockout
of Atg7 aggravates the Myocardial ischemia injury (37). AMPK-mediated autophagy has been
shown to play a possible protective role during myocardial ischemia
(38). AMPKα can not only regulate
autophagy through mTOR-TSC2, but also leads to autophagy by direct
phosphorylation of ULK1 (39,40).
Beclin1 (mammalian ortholog of yeast Atg6) has a central role in
autophagy and it is a key autophagic protein regulating both
autophagosome formation and processing (41). When autophagy occurs, Atg16 is
combined with Atg12-Atg5 to form an Atg16-Atg12-Atg5 complex. The
complex is located on the isolation membrane, which promotes the
extension of the isolation film and forms a phagocytic bubble
(42). P62, a multifunctional
ubiquitin-binding protein, localizes to the autophagosome formation
site on the ER, where autophagosomes are nucleated (43). It plays a critical role in cell's
selective autophagy and oxidative stress response (44). In the current study, we found that
pretreatment with DIM could activate autophagy, as revealed by the
upregulated expression of Atg5, Atg7 and Beclin1 and decreased
level of P62 in hypoxia-induced H9c2 cells. And the pro-autophagy
effect of DIM may through activation of AMPKα.
In conclusion, the present study indicated that DIM
attenuated the inflammation and apoptosis in response to hypoxia in
H9c2 cells. DIM also activated autophagy in response to hypoxia in
H9c2 cells. DIM protected against hypoxia induced cardiomyocyte
injury. The effect may be mediated by activation of AMPKα and
inhibition of JNK pathways. These observations may provide
experimental evidence for the application of DIM in the protective
effect of myocardial ischemia in cardiovascular diseases.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81400178) and the
Natural Science Foundation of Jiangsu Province (grant nos.
BK20140226 and BK20160231).
References
|
1
|
Frangogiannis NG: Pathophysiology of
myocardial infarction. Compr Physiol. 5:1841–1875. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rothermel BA and Hill JA: Myocyte
autophagy in heart disease: Friend or foe? Autophagy. 3:632–634.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yan WJ, Dong HL and Xiong LZ: The
protective roles of autophagy in ischemic preconditioning. Acta
Pharmacol Sin. 34:636–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jayakumar P, Pugalendi KV and Sankaran M:
Attenuation of hyperglycemia-mediated oxidative stress by
indole-3-carbinol and its metabolite 3,3′-diindolylmethane in
C57BL/6J mice. J Physiol Biochem. 70:525–534. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kunimasa K, Kobayashi T, Kaji K and Ohta
T: Antiangiogenic effects of indole-3-carbinol and
3,3′-diindolylmethane are associated with their differential
regulation of ERK1/2 and Akt in tube-forming HUVEC. J Nutr.
140:1–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cho HJ, Seon MR, Lee YM, Kim J, Kim JK,
Kim SG and Park JH: 3,3′-Diindolylmethane suppresses the
inflammatory response to lipopolysaccharide in murine macrophages.
J Nutr. 138:17–23. 2008.PubMed/NCBI
|
|
8
|
Azmi AS, Ahmad A, Banerjee S, Rangnekar
VM, Mohammad RM and Sarkar FH: Chemoprevention of pancreatic
cancer: Characterization of Par-4 and its modulation by 3,3′
diindolylmethane (DIM). Pharm Res. 25:2117–2124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen D, Banerjee S, Cui QC, Kong D, Sarkar
FH and Dou Q: Activation of AMP-activated protein kinase by
3,3′-Diindolylmethane (DIM) is associated with human prostate
cancer cell death in vitro and in vivo. PloS One. 7:e471862012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xue L, Firestone GL and Bjeldanes LF: DIM
stimulates IFNgamma gene expression in human breast cancer cells
via the specific activation of JNK and p38 pathways. Oncogene.
24:2343–2353. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ye Y, Fang Y, Xu W, Wang Q, Zhou J and Lu
R: 3,3′-Diindolylmethane induces anti-human gastric cancer cells by
the miR-30e-ATG5 modulating autophagy. Biochem Pharmacol.
115:77–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Zhang W, Jiao R, Yang Z, Yuan Y, Wu
Q, Hu Z, Xiang S and Tang Q: DIM attenuates TGF-β1-induced
myofibroblast differentiation in neonatal rat cardiac fibroblasts.
Int J Clin Exp Pathol. 8:5121–5128. 2015.PubMed/NCBI
|
|
13
|
Zong J, Wu QQ, Zhou H, Zhang JY, Yuan Y,
Bian ZY, Deng W, Dai J, Li FF, Xu M, et al: 3,3′-Diindolylmethane
attenuates cardiac H9c2 cell hypertrophy through 5′-adenosine
monophosphate-activatedprotein kinase-α. Mol Med Rep. 12:1247–1252.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng W, Zong J, Wei L, Guo H, Cheng Z,
Zhang R, Lin Y and Tang Q: 3,3′-Diindolylmethane improves
myocardial energy metabolism imbalance induced by pressure overload
via AMPKα in mice. Int J Cardiol. 177:235–237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu S, Mao J, Wang T and Fu X:
Downregulation of aquaporin-4 protects brain against hypoxia
ischemia via anti-inflammatory mechanism. Mol Neurobiol. Oct
10–2016.(Epub ahead of print).
|
|
16
|
Cheng CY: Anti-inflammatory effects of
traditional Chinese medicines against ischemic injury in in vivo
models of cerebral ischemia. Evid Based Complement Alternat Med.
2016:57394342016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
González A, Ravassa S, Beaumont J, López B
and Díez J: New targets to treat the structural remodeling of the
myocardium. J Am Coll Cardiol. 58:1833–1843. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cuenca-López MD, Brea D, Galindo MF,
Antón-Martínez D, Sanz MJ, Agulla J, Castillo J and Jordán J:
Inflammatory response during ischaemic processes: Adhesion
molecules and immunomodulation. Rev Neurol. 51:30–40. 2010.(In
Spanish). PubMed/NCBI
|
|
19
|
Jeon EJ, Davaatseren M, Hwang JT, Park JH,
Hur HJ, Lee AS and Sung MJ: Effect of oral administration of
3,3′-diindolylmethane on dextran sodium sulfate-induced acute
colitis in mice. J Agric Food Chem. Oct 4–2016.(Epub ahead of
print). View Article : Google Scholar
|
|
20
|
Rzemieniec J, Litwa E, Wnuk A, Lason W,
Krzeptowski W and Kajta M: Selective aryl hydrocarbon receptor
modulator 3,3′-diindolylmethane impairs AhR and ARNT signaling and
protects mouse neuronal cells against hypoxia. Mol Neurobiol.
53:5591–5606. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun HJ, Wang Y, Hao T, Wang CY, Wang QY
and Jiang XX: Efficient GSH delivery using PAMAM-GSH into
MPP-induced PC12 cellular model for Parkinson's disease. Regen
Biomater. 3:299–307. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rong B, Xie F, Sun T, Hao L, Lin MJ and
Zhong JQ: Nitric oxide, PKC-ε, and connexin43 are crucial for
ischemic preconditioning-induced chemical gap junction uncoupling.
Oncotarget. 7:69243–69255. 2016.PubMed/NCBI
|
|
23
|
Zhang WP, Zong QF, Gao Q, Yu Y, Gu XY,
Wang Y, Li ZH and Ge M: Effects of endomorphin-1 postconditioning
on myocardial ischemia/reperfusion injury and myocardial cell
apoptosis in a rat model. Mol Med Rep. 14:3992–3998. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corbalan J Jose, Vatner DE and Vatner SF:
Myocardial apoptosis in heart disease: Does the emperor have
clothes? Basic Res Cardiol. 111:312016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tichý A: Apoptotic machinery: The Bcl-2
family proteins in the role of inspectors and superintendents. Acta
Medica (Hradec Kralove). 49:13–18. 2006.PubMed/NCBI
|
|
26
|
Wu X, Gu W, Lu H, Liu C, Yu B, Xu H, Tang
Y, Li S, Zhou J and Shao C: Soluble receptor for advanced glycation
end product ameliorates chronic intermittent hypoxia induced renal
injury, inflammation and apoptosis via P38/JNK signaling pathways.
Oxid Med Cell Longev. 2016:10153902016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Balakumar P and Jagadeesh G: Multifarious
molecular signaling cascades of cardiac hypertrophy: Can the muddy
waters be cleared? Pharmacol Res. 62:365–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang Q and Molkentin JD: Redefining the
roles of p38 and JNK signaling in cardiac hypertrophy: Dichotomy
between cultured myocytes and animal models. J Mol Cell Cardiol.
35:1385–1394. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu T, Wang G, Tao H, Yang Z, Wang Y, Meng
Z, Cao R, Xiao Y, Wang X and Zhou J: Capsaicin mediates caspases
activation and induces apoptosis through P38 and JNK MAPK pathways
in human renal carcinoma. BMC Cancer. 16:7902016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tien YC, Lin JY, Lai CH, Kuo CH, Lin WY,
Tsai CH, Tsai FJ, Cheng YC, Peng WH and Huang CY: Carthamus
tinctorius L. prevents LPS-induced TNFalpha signaling activation
and cell apoptosis through JNK1/2-NFkappaB pathway inhibition in
H9c2 cardiomyoblast cells. J Ethnopharmacol. 130:505–513. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Costa R, Morrison A, Wang J, Manithody C,
Li J and Rezaie AR: Activated protein C modulates cardiac
metabolism and augments autophagy in the ischemic heart. J Thromb
Haemost. 10:1736–1744. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yan L, Vatner DE, Kim SJ, Ge H, Masurekar
M, Massover WH, Yang G, Matsui Y, Sadoshima J and Vanter SF:
Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci
USA. 102:pp. 13807–13812. 2005; View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: Enhancing macroautophagy protects against ischemia/reperfusion
injury in cardiac myocytes. J Biol Chem. 281:29776–29787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peng W, Liu Y, Xu WJ and Xia QH: Role of
Beclin 1-dependent autophagy in cardioprotection of ischemic
preconditioning. J Huazhong Univ Sci Technolog Med Sci. 33:51–56.
2007. View Article : Google Scholar
|
|
37
|
Li S, Liu C, Gu L, Wang L, Shang Y, Liu Q,
Wan J, Shi J, Wang F, Xu Z, et al: Autophagy protects
cardiomyocytes from the myocardial ischaemia-reperfusion injury
through the clearance of CLP36. Open Biol. 6:1601772016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nishida K, Kyoi S, Yamaguchi O, Sadoshima
J and Otsu K: The role of autophagy in the heart. Cell Death
Differ. 16:31–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: Metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2009. View
Article : Google Scholar
|
|
41
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Itakura E and Mizushima N: p62 targeting
to the autophagosome formation site requires self-oligomerization
but not LC3 binding. J Cell Biol. 192:17–27. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu SM, Dong YJ and Liu B: Progress of
study on p62 and protein degradation pathways. Sheng Li Xue Bao.
67:48–58. 2015.PubMed/NCBI
|