Introduction
Lung cancer is a type of malignant tumor, which is
one of the most life-threatening to humans. The morbidity and
mortality rates in men with lung cancer are the highest of all
types of malignancy, and lung cancer has the second highest
following breast cancer in women (1). Smoking is the major cause of lung
cancer (2) and accounts for 85% of
lung cancer cases in the world (3). Lung cancer includes small-cell lung
carcinoma and non-small-cell lung carcinoma (NSCLC) (4), and lung adenocarcinoma is a type of
NSCLC, which is found in peripheral lung tissue (1). It is the most common type of lung
cancer in smokers and in those who have never smoked (5). Lung adenocarcinoma grows slowly and
usually forms small masses, however, they readily metastasize at
the early stage (6).
Cancer is caused by genetic and environmental
factors, and ~8% of lung cancer cases are caused by genetic factors
(7). Genetic and environmental
factors can damage DNA to alter the epigenetics, which affects the
normal functions of cells, including DNA repair, cell proliferation
and apoptosis (8). There have been
numerous studies investigating the association between gene
variation and lung adenocarcinoma. The epidermal growth factor
receptor and Kristen rat sarcoma viral oncogene homolog have been
identified as mutations in lung adenocarcinoma as important driver
genes (9). In addition, single
nucleotide polymorphisms (SNPs) have been found to be associated
with lung cancer, including colony-stimulating factor 1 receptor,
tumor protein p63 and co-repressor interacting with RBPJ1 (10). Successful mapping of the human
genome and the emergence of next-generation sequencing technology
have assisted in the identification of specific variations
associated with disease comprising SNPs, insertions and deletions
(InDels), structure variations (SVs) and copy number variations
(CNVs). This has already assisted in understanding and
investigating several diseases, including melanoma (11), lung cancer (12), breast cancer (13) and acute myelogenous leukemia
(14), and has also assisted in
disease diagnosis, screening of drug targets and prediction of
disease risk.
In the present study, the genome variations of four
tumor tissues from different patients with lung adenocarcinoma were
detected, using a whole genome re-sequencing method performed on
the Illumina HiSeq Xten platform. From this, the SNPs, InDels, SVs
and CNVs from these four samples were identified, and the SNPs and
InDels were annotated, respectively.
Patients and methods
Patient samples and DNA
extraction
Tumor samples were collected from four patients with
lung adenocarcinoma. The four patients were all diagnosed with lung
adenocarcinoma using pathological methods and were classified to
have stage IV tumors, as bony metastases had occurred prior to
diagnosis. All were treated with chemotherapy only (Table I). Written informed consent was
obtained from all patients. The study was approved by the ethics
committee of Shanghai Tongji Hospital (Shanghai, China). Genomic
DNA was extracted from tumor samples using standard
phenol/chloroform extraction methods (15). Agarose gel electrophoresis was used
to confirm that the DNA samples were not degraded and that RNA was
not contaminated. The quality and quantity of DNA were assessed
using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and Qubit® 2.0 Fluorometer
(Thermo Fisher Scientific, Inc.), following which the DNA samples
with optical density values between 1.8 and 2.0, and a content
>1.5 µg were used for library construction.
 | Table I.Patient information. |
Table I.
Patient information.
| Patient | Sex | Age (years) | Cancer | Stage | Tumor sample |
|---|
| YJY | Female | 67 | Lung
adenocarcinoma | IV | Primary tumor |
| GMY | Female | 75 | Lung
adenocarcinoma | IV | Primary tumor |
| ZCG | Male | 65 | Lung
adenocarcinoma | IV | Primary tumor |
| JLY2 | Male | 52 | Lung
adenocarcinoma | IV | Primary tumor |
Library construction and
sequencing
The present study was performed by Beijing Novogene
Bioinformatics Technology Co., Ltd, Beijing, China (www.novogene.com/). Briefly, the qualified DNA samples
were randomly sheared into DNA fragment sizes of 350 bp using the
Covaris S220 Focused UItrasonicator (Covaris, Woburn, MA, USA), and
library construction was performed according to the manufacturer's
protocol of the TruSeqDNA Library Construction kit (Illumina, Inc.,
San Diego, CA, USA). The Qubit® 2.0 Fluorometer was used
first for preliminary quantification following the completion of
library construction, following which the Agilent 2100 Bioanalyzer
(Agilent Technologies, Inc., Waldbronn, Germany) was used for
determining the insert size of the library. This was followed by
accurate quantification using the quantitative polymerase chain
reaction method to ensure the library effective concentration was
>2 nM. Sequencing was performed using the Illumina HiSeq Xten
platform (Illumina, Inc.) to obtain the raw reads. Reads with
adapter sequences or of low quality were filtered out to obtain
clean reads, which were used in the following analysis.
Read mapping and identification of
SNPs, InDels, SVs and CNVs
The high quality filtered reads were mapped to the
reference genome UCSC hg19 (16)
using BWA 0.7.8-r455 software (17). The initial alignment results
underwent duplicate removal, local realignment and base quality
recalibration processing using Picard 1.111 (sourceforge.net/projects/picard/), GATK v3.1
(software.broadinstitute.org/gatk/) (18) and SAMtools 1.0 (samtools.sourceforge.net) (19), respectively. The effective data
were evaluated for the sequence read depth and coverage of the
clean reads mapping to the reference genome. SAMtools 1.0 software
(19) was used to identify SNPs
and InDels (fragment size of insertion or deletion <50 bp),
estimate the accuracy of SNP data with the transition/transversion
ratio (ts/tv), with the whole genome ratio being ~2.2 (20), and matching these SNPs and InDels
to the SNP database (dbSNP) (www.ncbi.nlm.nih.gov/projects/SNP/) (21). The identification of SVs was
performed using Breakdancer 1.4.4 software (genome.ustl.edu/tools/cancer-genomics/) (22), containing large fragments of
deletions, insertions, duplication and copy number variants,
inversion and translocation. CNVs were identified using
Control-FREEC v6.7 software (bioinfo.curie.fr/projects/freec/tutorial.html)
(23), containing deletions and
duplications.
Results
Genome sequencing and mapping to the
reference genome hg19
Genome DNA was extracted from tumor tissues of four
patients with lung adenocarcinoma and genome sequencing was
performed with the Illumina HiSeq Xten platform. A total of 415.98
G raw data was generated on 25 paired-end lanes, and an average of
693,316,202 raw reads were obtained for each sample, clean reads
accounted for ~99.88% of the original data. A Phred quality score
is a measure of the quality of the identification of the
nucleobases generated by automated DNA sequencing. Phred quality
scores Q are defined as a property which is logarithmically related
to the base-calling error probabilities P, Q=-10 log10 P
(24). The proportion of four
bases was 86–91% when the Phred score was >30, amongst which GC
content accounted for 40.92–43.71% (Table II).
| Table II.Quality of sequencing data. |
Table II.
Quality of sequencing data.
| Sample | Raw reads (n) | Raw data (G) | Clean reads (n) | Effective (%) | Q20 (%) | Q30 (%) | GC (%) |
|---|
| YJY | 621,362,127 | 93.20 | 620,492,220 | 99.86 | 96.27; 93.27 | 90.88; 85.32 | 42.05; 41.99 |
| GMY | 695,467,557 | 104.32 | 694,563,450 | 99.87 | 96.53; 93.07 | 91.35; 86.04 | 40.93; 40.93 |
| ZCG | 680,810,250 | 102.12 | 679,993,278 | 99.88 | 96.43; 94.08 | 91.26; 86.94 | 43.71; 43.66 |
| JLY2 | 775,624,875 | 116.34 | 774,771,688 | 99.89 | 96.55; 93.63 | 91.38; 86.22 | 40.95; 40.92 |
The numbers of filtered clean reads were
620,492,220, 694,563,450, 679,993,278 and 774,771,688 for YJY, GMY,
ZCG and JLY2, of which 96.38, 97.20, 97.71 and 97.25%,
respectively, were properly mapped to the reference genome of UCSC
hg19. The average sequencing depths were 28.31-, 31.08-, 30.55- and
34.47-fold, and 99.66, 98.96, 99.62, 98.93 of the reference genomes
were covered by the clean reads, respectively. Coverage at least
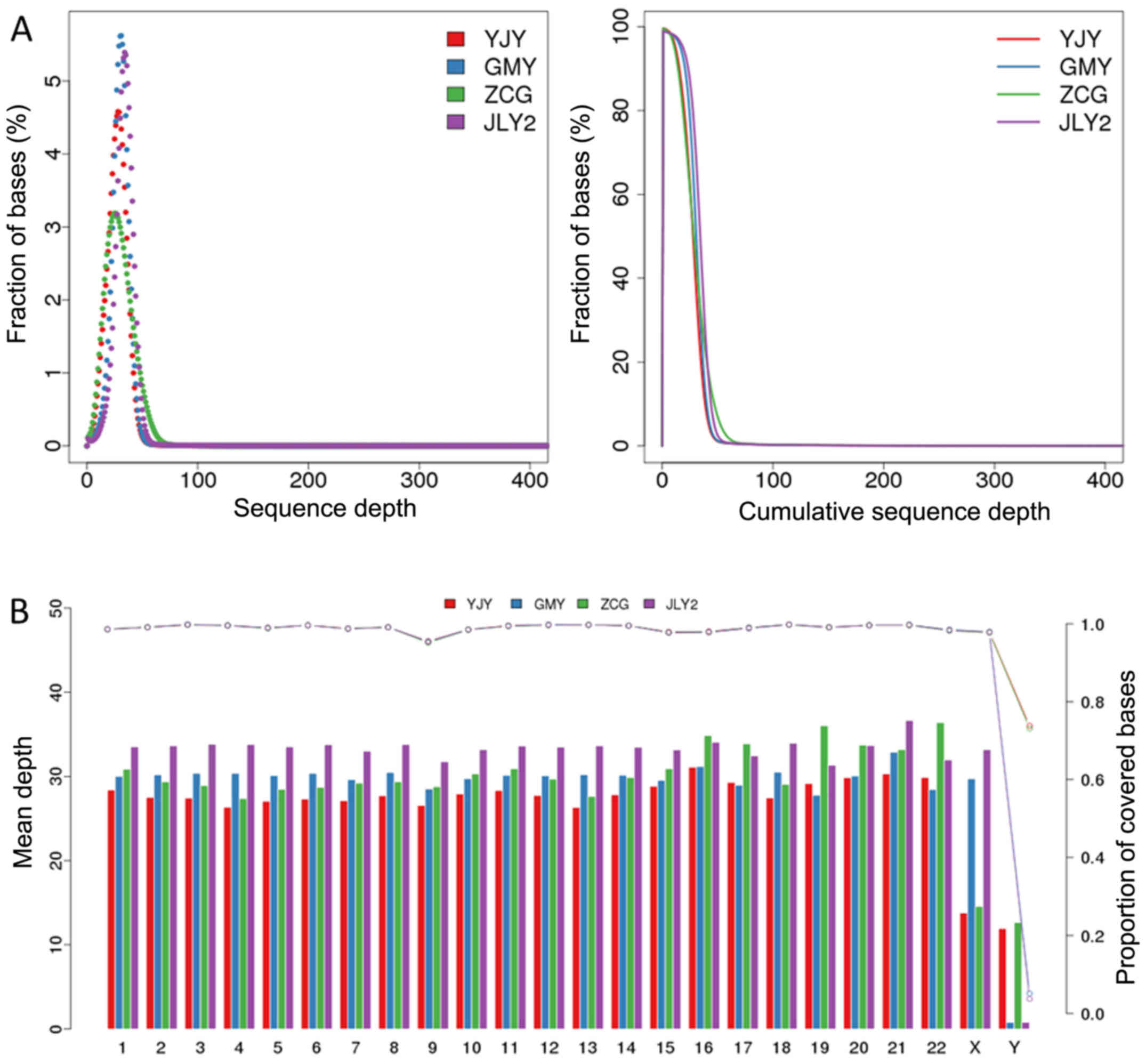
4-fold were 99.32, 98.69, 99.19 and 98.70%, respectively (Table III). As shown in Fig. 1A, the association between the ratio
of bases in each sample and the sequence depth was in accordance
with the Poisson distribution. The mean depths and coverage of each
chromosome of the reference genome are shown in Fig. 1B, showing a mean depth of ~47 and
coverage of ~97%, with the exception of sex chromosomes.
| Table III.Summary of sequenced reads aligned to
the reference genome of hg19. |
Table III.
Summary of sequenced reads aligned to
the reference genome of hg19.
| Sample | YJY | GMY | ZCG | JLY2 |
|---|
| Total | 620,492,220
(100%) | 694,563,450
(100%) | 67,9993,278
(100%) | 774771688 (100%) |
| Duplicate | 62,418,141
(10.10%) | 83,850,807
(12.12%) | 78,946,241
(11.66%) | 98027674
(12.70%) |
| Mapped | 617,787,854
(99.56%) | 691,799,013
(99.60%) | 677,069,702
(99.57%) | 771,860,901
(99.62%) |
| Properly mapped | 598,003,088
(96.38%) | 675,101,840
(97.20%) | 664,436,740
(97.71%) | 753,442,342
(97.25%) |
| PE mapped | 615,682036
(99.22%) | 689,578,896
(99.28%) | 674,885,134
(99.25%) | 769,555,296
(99.33%) |
| SE mapped | 4,211,636
(0.68%) | 4,440,234
(0.64%) | 4,369,136
(0.64%) | 4,611,210
(0.60%) |
| With mate mapped to
a different chromosome | 3,958,804
(0.64%) | 2,711,738
(0.39%) | 3,465,530
(0.51%) | 2,738,078
(0.35%) |
| With mate mapped to
a different chromosome [(mapQ ≥5)] | 2,839,756
(0.46%) | 1,788,585
(0.26%) | 2,347,772
(0.35%) | 1,691,563
(0.22%) |
| Average sequencing
depth | 28.31 | 31.08 | 30.55 | 34.47 |
| Coverage | 99.66% | 98.96% | 99.62% | 98.93% |
| Coverage at least
4X | 99.32% | 98.69% | 99.19% | 98.70% |
| Coverage at least
10X | 97.00% | 97.93% | 96.07% | 98.15% |
| Coverage at least
20X | 79.91% | 91.11% | 76.60% | 94.19% |
Detection of SNPs and InDels
The detection of SNPs and InDels were performed
using SAMtools, and matched to the dbSNP. There were 3,346,792,
3,387,147, 3,334,068 and 3,389,071 SNPs, respectively for YJY, GMY,
ZCG and JLY2, and ~6% (191,998, 193,563, 189,761 and 193,356) of
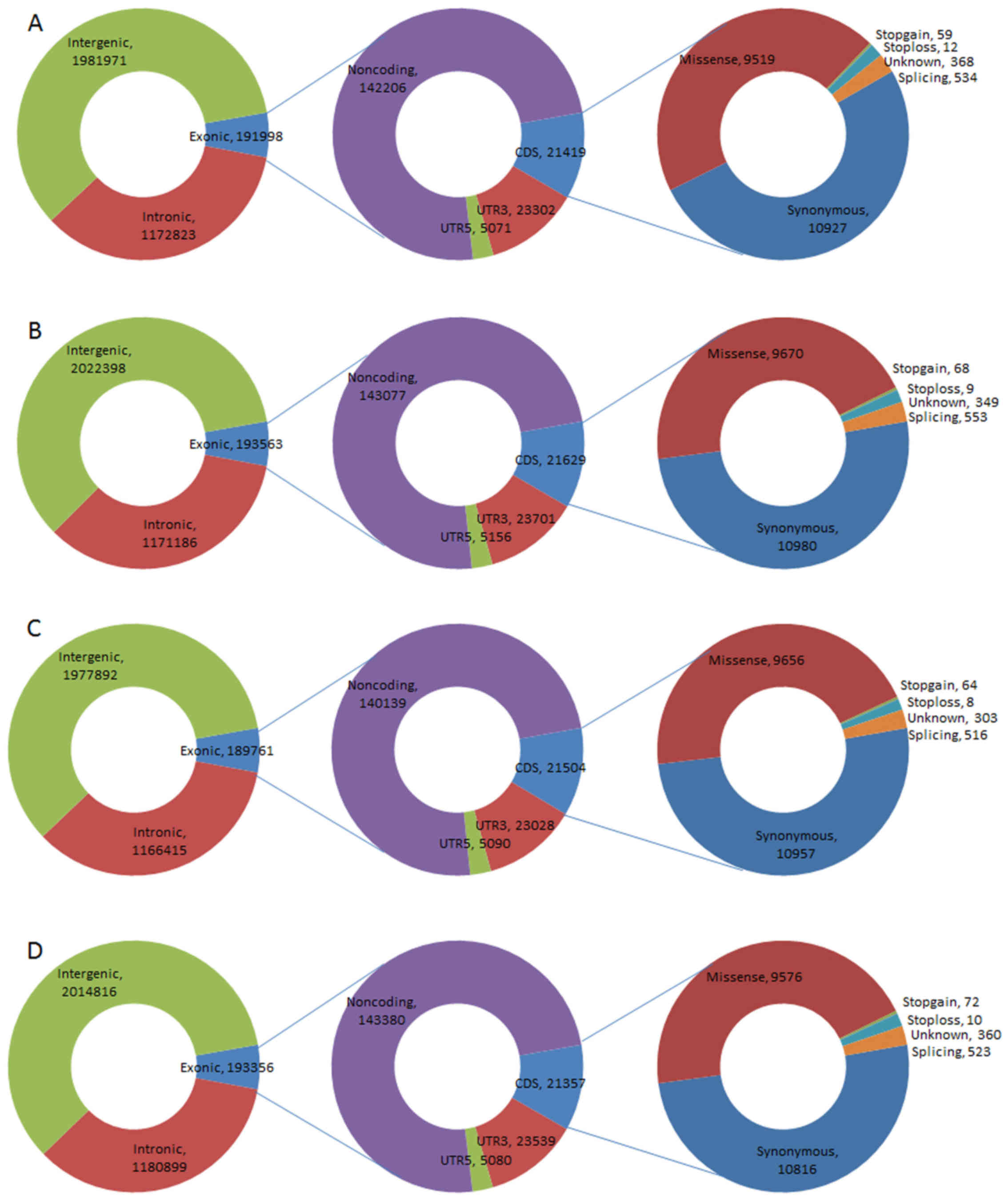
these were located in exonic regions. SNPs identified as being
locating in coding sequence (CDS) regions constituted ~11, and ~44%
were missense mutations (9,519 in 21,419, 9,670 in 21,629, 9,656 in
21,504, and 9,576 in 21,357; Fig.
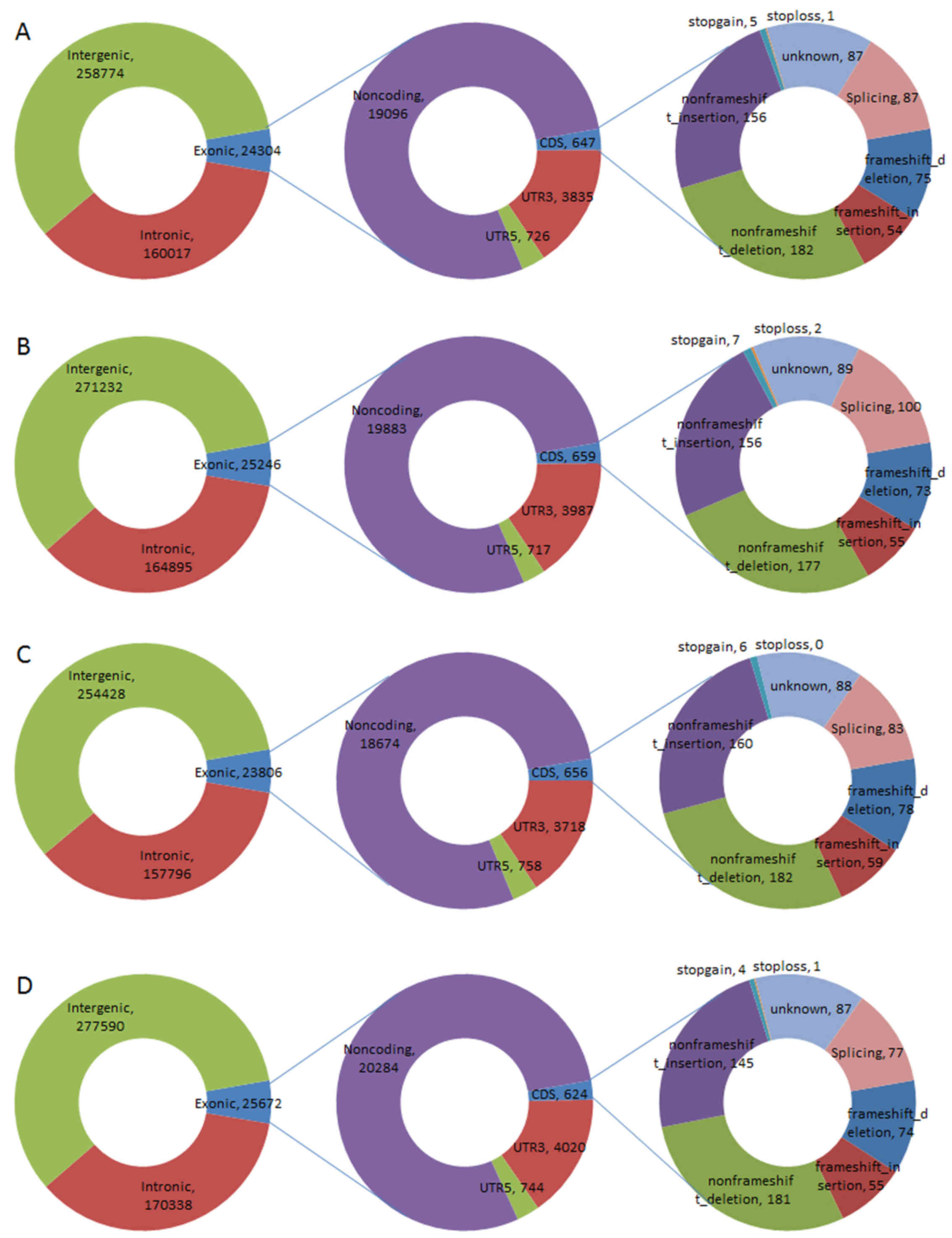
2). Similarly, of the InDels, ~6% were located in exonic
regions (24,304 in 443,118, 2,5246 in 461,393, 23,806 in 436,058,
25,672 in 473,617), an average of 647 InDels were located in the
CDS, ~12% (75, 73, 78, and 74) were identified as frame shift
deletions, and 9% (54, 55, 59, and 55) were frame shift insertions
(Fig. 3).
Annotation of SNPs and InDels
The ts/tv ratios of all samples were ~2.2 (YJY,
2.11; GMY, 2.10; ZCG, 2.11; JLY2, 2.10), the numbers of TS and TV
types of SNPs were 2,269,999 and 1,076,793, 2,296,037 and
1,091,110, 2,263,437 and 1,070,631, 2,297,128 and 1,091,943 for
each sample. Compared with the dbSNP, an average of 98.76% of the
SNP sites were matched and an average of 41,563 SNPs were novel in
each sequence data (40,762, 41,619, 41,694, 42,176; Table IV). For the InDels, there were
443,118, 461,393, 436,058 and 473,617 in YJY, GMY, ZCG and JLY2,
respectively, the majority of which were heterozygote and
homozygote; ~28.28% (126,222, 130,260, 124,274 and 132,201) were
found in the dbSNP, and the remaining were novel (Table V).
| Table IV.Statistics of single nucleotide
polymorphisms for high quality reads from YJY, GMY, ZCG and JLY2
mapped onto the reference genome of hg19. |
Table IV.
Statistics of single nucleotide
polymorphisms for high quality reads from YJY, GMY, ZCG and JLY2
mapped onto the reference genome of hg19.
| Sample | YJY | GMY | ZCG | JLY2 |
|---|
| Total | 3,346,792 | 3,387,147 | 3,334,068 | 3,389,071 |
| Heterozygote | 1,890,012 | 1,951,091 | 1,881,111 | 1,961,901 |
| Homozygote | 1,456,780 | 1,436,056 | 1,452,957 | 1,427,170 |
| Transition | 2,269,999 | 2,296,037 | 2,263,437 | 2,297,128 |
| Transversion | 1,076,793 | 1,091,110 | 1,070,631 | 1,091,943 |
| ts/tv | 2.11 | 2.10 | 2.11 | 2.10 |
| dbSNP
percentage | 3,306,030
(98.78%) | 3,345,528
(98.77%) | 3,292,374
(98.75%) | 3,346,895
(98.76%) |
| Novel | 40,762 | 41,619 | 41,694 | 42,176 |
| Novel ts | 26,861 | 27,531 | 27,471 | 27,834 |
| Novel tv | 13,901 | 14,088 | 14,223 | 14,342 |
| Novel ts/tv | 1.93 | 1.95 | 1.93 | 1.94 |
| Table V.Statistics of insertions and
deletions for high quality reads from YJY, GMY, ZCG and JLY2 mapped
onto the reference genome of hg19. |
Table V.
Statistics of insertions and
deletions for high quality reads from YJY, GMY, ZCG and JLY2 mapped
onto the reference genome of hg19.
| Sample | YJY | GMY | ZCG | JLY2 |
|---|
| Total | 443,118 | 461,393 | 436,058 | 473,617 |
| Heterozygote | 191,092 | 202,768 | 187,294 | 207,839 |
| Homozygote | 252,026 | 258,625 | 248,764 | 265,778 |
| dbSNP
percentage | 126,222
(28.48%) | 130,260
(28.23%) | 124,274
(28.50%) | 132,201
(27.91%) |
| Novel | 316,896 | 331,133 | 311,784 | 341,416 |
Analysis of SVs and CNVs
SV identification was performed using Breakdancer
1.4.4 software, the results showed that the largest number of
structural variations were deletions with average of 2,422
variations (YJY, 2,193; GMY, 2,390; ZCG, 2,220 and JLY2, 2,886).
The majority of the variations were located at intergenic (~57%)
and intron (~35%) regions. By contrast, the least common structural
variation was inversions, with an average of 138 (YJY, 150; GMY,
130; ZCG, 133; JLY2, 138), which were predominantly located in the
intergenic, intron and CDS regions (Table VI). CNVs were identified using
Control-FREEC v6.7 software, which contained deletions and
duplications, the majority of which were in intergenic regions (92,
174, 98 and 159) and CDS regions (41, 35, 43 and 42). The frequency
of deletion occurrence was higher, compared with that of
duplication occurrence in the four samples (Table VII).
| Table VI.Statistics of structural variations
for high quality reads from YJY, GMY, ZCG and JLY2 mapped onto the
reference genome of hg19. |
Table VI.
Statistics of structural variations
for high quality reads from YJY, GMY, ZCG and JLY2 mapped onto the
reference genome of hg19.
| Sample | VarType | Total | CDS | Splicing | UTR5 | UTR3 | Intron | Upstream | Downstream | ncRNA | Intergenic | Unknown |
|---|
| YJY | Insertion | 295 | 5 | 0 | 1 | 1 | 114 | 2 | 3 | 16 | 153 | 0 |
|
| Inversion | 150 | 33 | 0 | 0 | 0 | 32 | 1 | 0 | 12 | 72 | 0 |
|
| Deletion | 2,193 | 49 | 2 | 2 | 5 | 748 | 12 | 20 | 79 | 1,276 | 0 |
|
| Translocation | 298 | 3 | 0 | 1 | 4 | 83 | 3 | 0 | 14 | 190 | 0 |
| GMY | Inversion | 130 | 30 | 0 | 0 | 0 | 24 | 1 | 0 | 13 | 62 | 0 |
|
| Deletion | 2,390 | 51 | 0 | 2 | 4 | 809 | 14 | 15 | 92 | 1,403 | 0 |
|
| Insertion | 176 | 4 | 0 | 0 | 1 | 71 | 3 | 3 | 8 | 86 | 0 |
|
| Translocation | 300 | 1 | 0 | 0 | 10 | 92 | 3 | 0 | 17 | 177 | 0 |
| ZCG | Deletion | 2,220 | 52 | 2 | 3 | 5 | 807 | 24 | 20 | 77 | 1,230 | 0 |
|
| Inversion | 133 | 49 | 0 | 0 | 0 | 29 | 1 | 1 | 11 | 42 | 0 |
|
| Insertion | 376 | 7 | 0 | 1 | 0 | 156 | 1 | 4 | 19 | 188 | 0 |
|
| Translocation | 378 | 4 | 0 | 1 | 5 | 110 | 3 | 0 | 19 | 236 | 0 |
| JLY2 | Deletion | 2,886 | 57 | 1 | 3 | 8 | 1,002 | 13 | 24 | 94 | 1,684 | 0 |
|
| Insertion | 631 | 9 | 0 | 1 | 2 | 243 | 6 | 7 | 28 | 335 | 0 |
|
| Inversion | 138 | 29 | 0 | 1 | 0 | 36 | 1 | 0 | 10 | 61 | 0 |
|
| Translocation | 356 | 3 | 0 | 1 | 6 | 101 | 2 | 0 | 18 | 225 | 0 |
| Table VII.Statistics of copy number variations
for high quality reads from YJY, GMY, ZCG and JLY2 mapped onto the
reference genome of hg19. |
Table VII.
Statistics of copy number variations
for high quality reads from YJY, GMY, ZCG and JLY2 mapped onto the
reference genome of hg19.
| Sample | VarType | Total | CDS | Splicing | UTR5 | UTR3 | Intron | Upstream | Downstream | ncRNA | Intergenic | Unknown |
|---|
| YJY | Loss | 96 | 17 | 0 | 2 | 1 | 16 | 0 | 0 | 4 | 56 | 0 |
|
| Gain | 67 | 24 | 0 | 0 | 0 | 2 | 0 | 1 | 4 | 36 | 0 |
| GMY | Gain | 82 | 22 | 0 | 0 | 0 | 7 | 3 | 1 | 8 | 41 | 0 |
|
| Loss | 195 | 13 | 0 | 1 | 1 | 35 | 2 | 1 | 9 | 133 | 0 |
| ZCG | Gain | 74 | 21 | 0 | 1 | 1 | 4 | 1 | 0 | 9 | 37 | 0 |
|
| Loss | 106 | 22 | 0 | 1 | 1 | 13 | 2 | 1 | 5 | 61 | 0 |
| JLY2 | Gain | 88 | 22 | 0 | 0 | 0 | 7 | 2 | 1 | 9 | 47 | 0 |
|
| Loss | 178 | 20 | 0 | 1 | 1 | 33 | 1 | 3 | 7 | 112 | 0 |
Discussion
The characteristics of tumor cells include infinite
proliferation and growth, evasion of the body's immune
surveillance, energy metabolism on the basis of glycolysis
metabolism, dedifferentiation, and invasion or migration in a clone
growth manner. These biological characteristics of tumor cells
differ from the basic features and evolution processes of normal
cells, and are considered a result of decisive factors and
genetics. Therefore, the investigation of cancer from the
perspective of molecular genetics has become the mainstream. A
study by Boveri (25) suggested
that cancer was caused by abnormal genetic material, and that the
damage and induced mutation of DNA leading to cancer also indicated
the importance of genetic material in cancer. Rather than
explaining the role of single genes or single mutations in cancer
cells, investigations are now primarily aimed at clarifying the
nature of carcinogenesis through identifying whole genome
variation. This includes the identification of novel mutation sites
and gene mutations associated with tumors, the molecular regulatory
network and the cellular signaling pathways, which these mutations
are involved in, for determining the cause of cancer from the gene
variation profile (26). Several
cancer genes and pathways have been identified in cancer genome
investigations, including the stromal antigen 2 mutation in
transitional cell carcinoma of the bladder (27), FAT atypical cadherin 1, FAT
atypical cadherin 2 and zinc finger protein 750 mutations in
esophageal squamous cell carcinoma (28) and AT-rich interactive domain-1A,
vascular cell adhesion molecule 1 and cyclin-dependent kinase 14
mutations in liver cancer (29).
In the present study, re-sequencing of four tumor
genomes was performed from different patients with lung
adenocarcinoma, and these sequence data were aligned to reference
genome hg19 to obtain information regarding the numbers of SNPs,
InDels, SVs and CNVs in each sample, respectively. The alignment
results showed that changes of base ratio with sequence depth in
each sample were in accordance with the Poisson distribution, and
this suggested that the sequencing results were of high quality and
coverage.
SNPs can occur anywhere in the genome, including
CDS, untranslated region, splicing, non-coding RNA and intergenic
regions; they can occur as a synonymous SNP, missense SNP, stopgain
and stoploss in the CDS region, which can lead to errors in the
amino acid sequence of proteins or in the regulation of
translation, thus affecting cell features and function. In the
present study, an average of 3,364,269 SNPs was detected in each
sample, of which 98.77% were matched to the dbSNP. The ts/tv ratios
were all ~2.1, indicating a high level of accuracy of the SNP data
(whole genome ratio is ~2.2). InDels of the CDS region and splice
site are likely to alter protein translation. Frameshift mutations,
in which the base lengths of insertion or deletions are not a
multiple of three bases, may lead to changes in the reading frame.
The present study also identified numerous InDels in each sample,
the majority of which were located in intergenic regions and
noncoding regions, and a large proportion of InDels (~82%) were
novel. However, changes in frame coding proteins, including
frameshift deletions, frameshift insertions, stopgain and stoploss
were present in CDS regions.
SV is widespread in the human genome, and is the
source of individual differences and susceptibility to certain
diseases. SV also exists in cancer cells, compared with the genome
of normal tissue cells and may lead to the occurrence of fusion
genes, which may be associated with cancer (30). The results of the present study
showed that the most common SV type was deletions, with an average
of 2,422 variations. CNV may be an important cause of certain
diseases. Deletions and duplications at the chromosome level have
become a focus of investigations of several diseases. The results
of the present study indicated that deletions and duplications were
present in all four samples, predominantly in intergenic regions
(92, 174, 98 and 159) and CDS regions (41, 35, 43 and 42).
In conclusion, the present study described the
genome re-sequencing results of four patients with lung
adenocarcinoma and the alignment results of these sequence data.
The SNPs, InDels, SVs and CNVs of each sample were identified,
which aligned to the reference genome of hg19, and a simple
annotation of SNPs and InDels was performed. Increasing evidence
indicates that genetic variation is closely associated with
diseases, including cancer. SNPs, InDels, SVs and CNVs may affect
gene expression or signaling pathways, which may lead to changes in
cell viability and metastasis. The occurrence and progression of
lung adenocarcinoma is a complicated process with specific gene
expression profile and gene functions, which are the result of
genetic variation and/or environmental factors. The results of the
present study showed that investigating genome variation in
patients with lung adenocarcinoma assists in understanding the
mechanism of lung adenocarcinoma oncogenesis. More samples and
investigations of specific genetic analysis and functional
annotations are required to further examine of the associations
between gene variation and lung adenocarcinoma.
Acknowledgements
The present study was supported by the ‘12th Five
Year’ National Science and Technology Supporting Program (grant no.
2011BAI11B16).
References
|
1
|
Stewart BW and Wild C: International
Agency for Research on Cancer and World Health Organization. World
cancer report. 2014.
|
|
2
|
Biesalski HK, de Mesquita B Bueno, Chesson
A, Chytil F, Grimble R, Hermus RJ, Köhrle J, Lotan R, Norpoth K,
Pastorino U and Thurnham D: European consensus statement on lung
cancer: Risk factors and prevention. Lung Cancer Panel. CA Cancer J
Clin. 48:164–176. 1998. View Article : Google Scholar
|
|
3
|
Yu YH, Liao CC, Hsu WH, Chen HJ, Liao WC,
Muo CH, Sung FC and Chen CY: Increased lung cancer risk among
patients with pulmonary tuberculosis: A population cohort study. J
Thorac Oncol. 6:32–37. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Devesa SS, Bray F, Vizcaino AP and Parkin
DM: International lung cancer trends by histologic type: Male:
Female differences diminishing and adenocarcinoma rates rising. Int
J Cancer. 117:294–299. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Travis WD: World Health Organization,
International Agency for Research on Cancer., International
Association for the Study of Lung Cancer. And International Academy
of Pathology.: Pathology and genetics of tumours of the lung,
pleura, thymus and heart. IARC Press Oxford University Press.
(distributor). LyonOxford: 2004
|
|
6
|
Kumar V and Robbins SL: Robbins basic
pathology. Saunders/Elsevier; Philadelphia, PA: 2007
|
|
7
|
Yang IA, Holloway JW and Fong KM: Genetic
susceptibility to lung cancer and co-morbidities. J Thorac Dis. 5
Suppl 5:S454–S462. 2013.PubMed/NCBI
|
|
8
|
Hong WK; American Association for Cancer
Research, : Holland Frei cancer medicine. 8. People's Medical Pub.
House; Shelton, Conn: 2010
|
|
9
|
Yatabe Y, Koga T, Mitsudomi T and
Takahashi T: CK20 expression, CDX2 expression, K-ras mutation and
goblet cell morphology in a subset of lung adenocarcinomas. J
Pathol. 203:645–652. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang HG, Lee SY, Jeon HS, Choi YY, Kim S,
Lee WK, Lee HC, Choi JE, Bae EY, Yoo SS, et al: A functional
polymorphism in CSF1R gene is a novel susceptibility marker for
lung cancer among never-smoking females. J Thorac Oncol.
9:1647–1655. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pleasance ED, Cheetham RK, Stephens PJ,
McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR,
Bignell GR, et al: A comprehensive catalogue of somatic mutations
from a human cancer genome. Nature. 463:191–196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee W, Jiang Z, Liu J, Haverty PM, Guan Y,
Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, et al: The mutation
spectrum revealed by paired genome sequences from a lung cancer
patient. Nature. 465:473–477. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shah SP, Morin RD, Khattra J, Prentice L,
Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, et al:
Mutational evolution in a lobular breast tumour profiled at single
nucleotide resolution. Nature. 461:809–813. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ley TJ, Mardis ER, Ding L, Fulton B,
McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S,
Hickenbotham M, et al: DNA sequencing of a cytogenetically normal
acute myeloid leukaemia genome. Nature. 456:66–72. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mamiatis T, Fritsch EF, Sambrook J and
Engel J: Molecular cloning? A laboratory manual. New York Cold
Spring Harbor Laboratory. 1982, 545 S., 42 Volume 5, Issue 1. Acta
Biotechnologica. 5:104. 1985. View Article : Google Scholar
|
|
16
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The Sequence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
1000 Genomes Project Consortium, ;
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker
RE, Kang HM, Marth GT and McVean GA: An integrated map of genetic
variation from 1,092 human genomes. Nature. 491:56–65. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen K, Wallis JW, McLellan MD, Larson DE,
Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et
al: BreakDancer: An algorithm for high-resolution mapping of
genomic structural variation. Nat Methods. 6:677–681. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boeva V, Popova T, Bleakley K, Chiche P,
Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O and
Barillot E: Control-FREEC: A tool for assessing copy number and
allelic content using next-generation sequencing data.
Bioinformatics. 28:423–425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ewing B and Green P: Base-calling of
automated sequencer traces using phred. II. Error probabilities.
Genome Res. 8:186–194. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boveri T: Concerning the origin of
malignant tumours by Theodor Boveri. Translated and annotated by
Henry Harris. J Cell Sci. 121 Suppl 1:S1–S84. 2008. View Article : Google Scholar
|
|
26
|
Garraway LA and Lander ES: Lessons from
the cancer genome. Cell. 153:17–37. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y,
Shi JY, Zhu YM, Tang L, Zhang XW, et al: Exome sequencing
identifies somatic mutations of DNA methyltransferase gene DNMT3A
in acute monocytic leukemia. Nat Genet. 43:309–315. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Song Y, Li L, Ou Y, Gao Z, Li E, Li X,
Zhang W, Wang J, Xu L, Zhou Y, et al: Identification of genomic
alterations in oesophageal squamous cell cancer. Nature. 509:91–95.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li
N, Zhu ZD, Zhou B, Liu XY, Liu RF, et al: Exome sequencing of
hepatitis B virus-associated hepatocellular carcinoma. Nat Genet.
44:1117–1121. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shao D, Lin Y, Liu J, Wan L, Liu Z, Cheng
S, Fei L, Deng R, Wang J, Chen X, et al: A targeted next-generation
sequencing method for identifying clinically relevant mutation
profiles in lung adenocarcinoma. Sci Rep. 6:223382016. View Article : Google Scholar : PubMed/NCBI
|