Introduction
Liver fibrosis is a complex pathophysiological
process characterized by the production and excessive deposition of
extracellular matrix (ECM) (1–3),
leading to loss of liver function, remodeling of liver blood
vessels and destruction of liver structures. Liver fibrosis is
generally considered a reversible wound healing response following
liver injury. This process occurs in all chronic liver diseases,
and can eventually lead to irreversible cirrhosis and liver failure
(4,5); therefore preventing and reversing
liver fibrosis is a primary measure for the treatment of liver
diseases (6).
Epithelial-mesenchymal transition (EMT) refers to a
process in which epithelial cells lose their typical epithelial
properties and become motile as mesenchymal cells (7). The EMT process is accompanied by a
loss of epithelial characteristics, including E-cadherin
expression, and an increase in mesenchymal cells and fibroblast
markers, including α-smooth muscle actin (α-SMA) (8,9). EMT
has been reported to be associated with liver fibrosis. Previous
studies have suggested that activation of collagen-producing
myofibroblasts can be enhanced by hepatocytes and biliary
epithelial cells via EMT during liver fibrosis (10,11),
which can result in excessive deposition of ECM in the liver
(12).
Transforming growth factor-β (TGF-β) is a principal
profibrotic cytokine (13), which
can induce EMT during liver fibrosis. It has previously been
suggested that EMT is regulated by various profibrotic cytokines,
with TGF-β1 considered the most important (14). TGF-β1 has been reported to initiate
and finalize EMT processes in animal disease models and patients
(15). Growing evidence has
indicated that the prevention of EMT development may control and
reverse liver fibrosis (16).
Blocking TGF-β signal transduction and understanding
the mechanism underlying EMT has great significance for the
development of novel effective therapies for the treatment of
EMT-associated fibrosis diseases (17). A novel truncated (27–123 residues)
type II TGF-β receptor (tTGFβRII) was designed and constructed,
which works as a dominant-negative receptor. A previous study
demonstrated that recombinant tTGFβRII (rtTGFβRII) can efficiently
trap TGF-β1 from access to wild-type receptors and can prevent
TGF-β1-triggered signals (18).
Therefore, the present study aimed to determine whether rtTGFβRII
ameliorates liver fibrosis by inhibiting EMT.
Materials and methods
Expression and purification of
rtTGFβRII protein
The tTGFβRII sequence was amplified by reverse
transcription-polymerase chain reaction (RT-PCR) and cloned into
the pET-28a vector prior to transformation into Escherichia
coli BL21 cells. Briefly, the tTGFβRII sequence was synthesized
from the RNA of human peripheral blood using the Catrimox-14
reagent (Takara Biotechnology, Co., Ltd., Dalian, China). Informed
consent was obtained for the collection of blood sample from a
healthy volunteer, and this study was approved by the Ethics
Committee of Mudanjiang Medical University (Mudanjiang, China). The
total RNA (1 µg) was reverse transcribed to cDNA using the
First-Strand cDNA Synthesis kit (Roche Diagnostics, Indianapolis,
IN, USA) according to the manufacturer's protocol. The 10X
polymerase chain reaction buffer and dNTP mixture were purchased
from Takara Biotechnology Co., Ltd. The primers (synthesized by
Sangon Biotech Co., Ltd., Shanghai, China), were as follows:
Forward, 5′-CCGGAATTCCACGTTCAGAAGTCGGTTAA-3′ and reverse,
5′-TTGCGGCCGCCATAATGCACTTTGGAGAAG-3′. The PCR conditions were as
follows: 94°C for 5 min, 30 cycles of 94°C for 30 sec, 55°C for 30
sec and 72°C for 30 sec, and a final extension of 72°C for 10
min.
The recombinant plasmid pET-28a/tTGFβRII contained 6
histidine tags, and was grown overnight in lysogeny broth (LB)
medium that contained 50 µg/ml kanamycin at 37°C, with agitation at
210 rpm. Subsequently, the culture was diluted 1:100 in fresh LB
medium that contained 50 µg/ml kanamycin and was cultured at 37°C
until optical density at 600 nm values of the media reached 0.6.
Afterwards, 1 mmol/l isopropyl β-D-thiogalactopyranoside was added
for induction at 37°C and the cells were harvested after 12 h.
Purification was completed using Ni-NTA agarose affinity
chromatography. The expression and purity of rtTGFβRII was analyzed
by 15% SDS-PAGE with Coomassie brilliant blue staining. The
purified recombinant protein was measured using a bicinchoninic
acid (BCA) protein assay kit according to the manufacturer's
protocol and stored at −80°C until further use.
Animals and treatment
All experimental protocols were approved by the
Mudanjiang Medical University Animal Care and Veterinary Services
Committee (Mudanjiang, China). The methods were carried out in
accordance with the Guidelines for the Care and Use of Laboratory
Animals (19). A rat model of
liver fibrosis was induced using carbon tetrachloride
(CCl4). A total of 24 male Sprague Dawley rats (age, 10
weeks; weight, 200–250 g) were obtained from the SLAC Laboratory
Animal Co., Ltd. (Shanghai, China). The animals were maintained
under a 12 h light/dark cycle at 22°C with free access to food and
water. After acclimation for 1 week, the rats were randomly divided
into three groups (n=8 rats/group): Normal control (NC), model
(CCl4), and treatment (CCl4 + rtTGFβRII)
groups. The NC group received an intraperitoneal (i.p.) injection
of 2 ml/kg body weight (b.w.) pure olive oil twice a week for 8
weeks and were then administered 2 ml/kg b.w. pure olive oil (i.p.)
once a week for 4 weeks. The CCl4 group received 2 ml/kg
b.w., CCl4 solution (40% CCl4 in pure olive
oil, i.p.) twice a week for 8 weeks and afterwards were
administered a CCl4 solution 2 ml/kg b.w. once a week
for 4 weeks. The treatment group (CCl4 + rtTGFβRII)
received 2 ml/kg b.w. CCl4 solution (40% CCl4
in pure olive oil, i.p.) twice a week for 8 weeks and once a week
for 4 weeks. Once the rats had been treated for 8 weeks they were
administered 1 mg/kg b.w. rtTGFβRII (i.p.), two times a week, over
a total of 4 weeks. All of the rats were sacrificed under
anesthesia with an i.p. injection of 10% chloral hydrate (3 ml/kg
b.w.), and blood and liver samples were collected. Blood samples
were centrifuged at 1,500 × g for 10 min at 4°C to separate serum;
the serum activities of ALT and AST were then detected by the
Hongqi Hospital of Mudanjiang Medical University using a fully
automatic biochemical analyzer (AU640; Olympus Corporation, Tokyo,
Japan). A portion of the liver was immediately fixed in 10%
formalin for histological analyses. The rest of the liver tissue
was rapidly frozen and maintained at −80°C until assessed. Liver
samples were acquired for analyses of liver function, and the
detection of mRNA and protein expression levels of
fibrosis-associated factors by RT-quantitative (q)PCR, histology,
immunofluorescence and western blot analysis.
RNA extraction and RT-qPCR
analysis
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the manufacturer's protocol. Total RNA (1 µg) was
reverse transcribed to cDNA using the First-Strand cDNA Synthesis
kit (Roche Diagnostics) according to the manufacturer's
instructions. Gene expression was measured by RT-qPCR using a SYBR
Green RT-qPCR Master Mix and a StepOne Real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: 95°C for 10 min, followed
by 40 cycles of 95°C for 15 sec and 60°C for 1 min. PCR primer
sequences were as follows: β-actin forward,
5′-GGAGATTACTGCCCTGGCTCCTA-3′ and reverse,
5′-GACTCATCGTACTCCTGCTTGCTG-3′; vimentin forward,
5′-TGACATTGAGATCGCCACCT-3 and reverse, 5′-TCATCGTGGTGCTGAGAAGT-3′;
fibroblast-specific protein (FSP-1) forward,
5′-ATGTAATAGTGTCCACCTTCC-3′ and reverse,
5′-ACTTCATTGTCCCTGTTGCT-3′; fibronectin forward,
5′-CCGAATCACAGTAGTTGCGG-3′ and reverse, 5′-GCATAGTGTCCGGACCGATA-3′;
E-cadherin forward, 5′-TCATCACAGACCCCAAGACC-3′ and reverse,
5′-GATCTCCAGACCCACACCAA-3′; collagen I forward,
5′-CTGCTGGTGAGAGAGGTGAA-3′ and reverse, 5′-GGAAACCTCTCTCGCCTCTT-3′;
and α-smooth muscle actin (α-SMA) forward,
5′-CATCATGCGTCTGGACTTGG-3′ and reverse, 5′-CCAGGGAAGAAGAGGAAGCA-3′.
β-actin was used as an internal control and relative quantification
of gene expression was performed using the 2−ΔΔCq method
(20).
Histological analysis
The liver tissues were fixed in 10% neutraformaline
for 24 h at room temperature and were embedded in paraffin. Tissue
sections (5 µm) underwent hematoxylin and eosin (H&E), Masson's
trichrome (MTS; cat no. D026; Nanjing Jiancheng Bioengineering
Institute, Nanjing, China) and Sirius red staining (cat no.
ab150681; Abcam, Cambridge, MA, USA) to study morphological
alterations. All histological analyses were performed according to
the manufacturer's instructions. For semi-quantitative morphometry,
fibrotic areas stained with MTS and Sirius red staining were
detected and calculated with ImageJ v1.42q software [National
Institutes of Health (NIH), Bethesda, MD, USA] in eight randomly
selected micrographs.
Immunofluorescence staining
Immunofluorescence analysis was performed on 5 µm
liver sections that were dewaxed with xylene and hydrated using
sequential ethanol and distilled water. The sections were
permeabilized in 0.1% Triton X-100/PBS for 10 min, blocked with 1%
bovine serum albumin (BSA)/PBS containing 0.1% Tween (both from
Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) for 60 min at room temperature and were incubated with
antibodies against E-cadherin (1:100 dilution; cat no. sc-7870),
fibronectin (1:100 dilution; cat no. sc-69682) (both from Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), α-SMA (1:200 dilution;
cat. no. ab5694; Abcam) and vimentin (1:100 dilution; cat. no.
sc-7558; Santa Cruz Biotechnology, Inc.) at 4°C overnight. Samples
were washed with PBS prior to incubation with fluorescein
isothiocyanate- (1:500 dilution; cat nos. ab97063 and ab6881;
Abcam) and Cy3- (1:500 dilution; cat no. ab97035; Abcam) labeled
secondary antibodies for 1 h at room temperature. Subsequently, 1
µg/ml DAPI was used to stain for 10 min in the dark. Images were
observed under a laser scanning confocal microscope (Olympus
Corporation). For semi-quantitative morphometry, immunofluorescence
staining was detected and calculated with ImageJ v1.42q software
(NIH) in eight randomly selected micrographs.
Western blot analysis
Liver tissues were lysed in radioimmunoprecipitation
assay buffer (P0013B; Beyotime Institute of Biotechnology,
Shanghai, China), supplemented with protease inhibitor 1 mM
phenylmethylsulfonyl fluoride for 30 min on ice and were
centrifuged at 7,000 × g at 4°C for 15 min. Protein concentration
was detected by a spectrophotometer using a BCA protein assay kit
according to the manufacturer's protocol. The protein samples (50
µg) were separated by 8–10% SDS-PAGE and were transferred to
polyvinylidene difluoride membranes. Membranes were blocked in 5%
BSA/PBS containing 0.1% Tween for 1 h at room temperature, and were
then incubated with primary antibodies against E-cadherin (1:500
dilution; cat no. sc-7870), fibronectin (1:200 dilution; cat no.
sc-69682) (both from Santa Cruz Biotechnology Inc.), collagen I
(1:5,000 dilution; cat no. ab34710), Smad2/3 (1:500 dilution; cat
no. ab217553), phosphorylated (p)-Smad2/3 (1:1,000 dilution; cat
no. ab63399) and GAPDH (1:5,000 dilution; cat no. ab181602) (all
from Abcam) at 4°C overnight. GAPDH was used as an internal
control. Horseradish peroxidase-conjugated goat anti-rabbit IgG
(1:5,000 dilution; cat no. ab97051) or goat anti-mouse IgG (1:5,000
dilution; cat no. ab97023) (both from Abcam) antibodies were used
as secondary antibodies for 1 h at room temperature. Protein bands
were detected using the 0.01% diaminobenzidine (cat no. PA110;
Tiangen Biotech Co., Ltd., Beijing, China) chromogenic reagent and
the signal intensity was semi-quantified using ImageJ v1.42q
software (NIH).
Statistical analysis
The results of each experiment are representative of
at least three independent experiments. Data were analyzed using a
one-way analysis of variance with Tukey's multiple comparison test
using GraphPad Prism 5.0 software (GraphPad Software, Inc., La
Jolla, CA, USA) and were expressed as the mean ± standard error of
the mean. P<0.05 was considered to indicate a statistically
significant difference.
Results
Purification of rtTGFβRII protein
As shown in Fig. 1,
the purified rtTGFβRII exhibited a single band following SDS-PAGE
with Coomassie brilliant blue staining. The molecular weight of
rtTGFβRII was ~13 kDa.
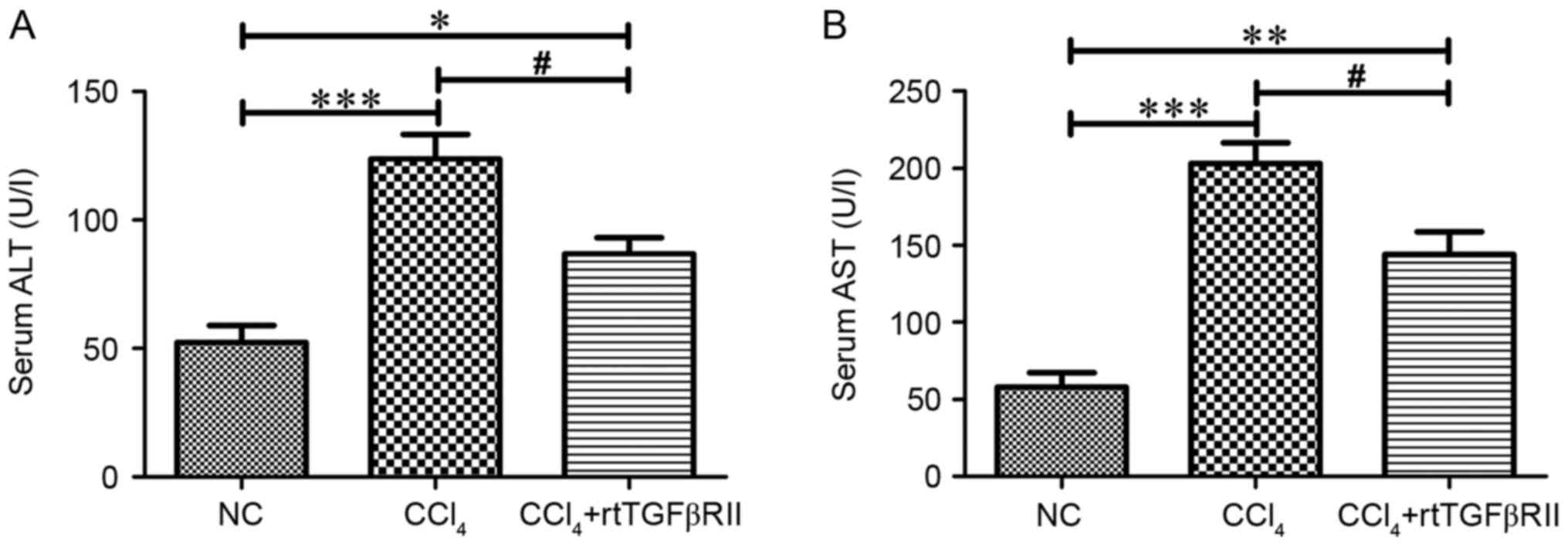
Activities of serum alanine
aminotransferase (ALT) and aspartate aminotransferase (AST)
activities
As shown in Fig. 2,
the serum activities of ALT and AST were markedly increased
(P<0.001) in the CCl4 model group compared with those
in the normal control group. Treatment with rtTGFβRII had a
beneficial effect on ALT and AST activities; decreased ALT and AST
activities (P<0.05) were detected in the CCl4 +
rtTGFβRII group compared with in the CCl4 model
group.
Histopathological alterations in the
liver
H&E staining detected pathological alterations
in the CCl4 model group, including prominent hepatocyte
degeneration, necrosis, inflammatory cell infiltration, periportal
expansion and formation of pseudolobules (Fig. 3A). MTS and Sirius red staining
detected a large amount of collagen fiber proliferation in the
CCl4 model group compared with in the NC group (Fig. 3B and C); however, and the
surrounding fibrous tissue was significantly reduced (P<0.05) in
the rtTGFβRII treatment group compared with the CCl4
model group (Fig. 3D and E).
 | Figure 3.rtTGFβRII attenuates
CCl4-induced liver injury, fibrosis and collagen
deposition. (A) H&E, (B) MTS and (C) Sirius red staining
(magnification, ×100) were performed on liver sections. Scale bars,
50 µm. (D and E) Semi-quantitative analysis for MTS and Sirius red
staining, respectively. Data are presented as the mean ± standard
error of the mean, n=8. *P<0.05, **P<0.01, ***P<0.001
compared with the NC group; #P<0.05 compared with the
CCl4 group. H&E, hematoxylin and eosin; MTS,
Masson's trichrome; NC, normal control; rtTGFβRII, recombinant
truncated transforming growth factor β receptor II. |
Effects of rtTGFβRII on ECM deposition
and EMT expression in liver fibrosis
To evaluate whether rtTGFβRII modulates the
accumulation of ECM and EMT expression in liver fibrosis, the mRNA
expression levels of EMT-associated markers were investigated by
RT-qPCR. A significant decrease in mRNA expression levels of the
epithelial marker E-cadherin, and an increase in the mRNA
expression levels of the mesenchymal cell markers fibronectin,
α-SMA, vimentin, FSP-1 and collagen I were detected in the
CCl4 model group. However, rtTGFβRII treatment
downregulated mesenchymal cell marker mRNA expression and slightly
upregulated E-cadherin expression compared with the CCl4
model group (Fig. 4).
Immunofluorescence staining analysis indicated that the rtTGFβRII
group exhibited reduced vimentin, α-SMA and fibronectin expression,
and increased E-cadherin expression compared with the
CCl4 model group (Fig.
5). The present study further evaluated the expression of
E-cadherin, collagen I, fibronectin and p-Smad2/3 via western
blotting. There results demonstrated that collagen I, fibronectin
and p-Smad2/3 protein levels were significantly higher in the
CCl4 model group compared with in the control group.
Treatment with rtTGFβRII significantly decreased collagen I,
fibronectin and p-Smad2/3 expression and increased the expression
of E-cadherin (Fig. 6). The
results indicated that rtTGFβRII could prevent the expression of
fibroblast and mesenchymal markers, thus slowing down the
progression of EMT in liver fibrosis.
 | Figure 4.Effects of rtTGFβRII treatment on the
mRNA expression levels of EMT markers. The mRNA expression levels
of (A) α-SMA, (B) vimentin, (C) FSP-1, (D) collagen I, (E)
fibronectin and (F) E-cadherin were detected by quantitative
polymerase chain reaction analysis. mRNA expression levels were
normalized to β-actin. Data are presented as the mean ± standard
error of the mean, n=8. *P<0.05, **P<0.01, ***P<0.001
compared with the NC group; #P<0.05 compared with the
CCl4 group. α-SMA, α-smooth muscle actin; EMT,
epithelial-mesenchymal transition; FSP-1, fibroblast specific
protein-1; rtTGFβRII, recombinant truncated transforming growth
factor β receptor II. |
 | Figure 5.rtTGFβRII decreased
CCl4-induced α-SMA, vimentin, and fibronectin
expression, and increased the expression of E-cadherin.
Immunofluorescence staining (magnification, ×100) was performed for
(A) α-SMA, (B) vimentin, (C) fibronectin and (D) E-cadherin. Scale
bars, 100 µm. (E-H) Semi-quantitative analysis for
immunofluorescence staining. Data are presented as the mean ±
standard error of the mean, n=8. **P<0.01, ***P<0.001
compared with the NC group; #P<0.05,
##P<0.01, ###P<0.001 compared with the
CCl4 group. α-SMA, α-smooth muscle actin; rtTGFβRII,
recombinant truncated transforming growth factor β receptor II. |
 | Figure 6.Effects of rtTGFβRII treatment on the
protein expression of fibronectin, collagen I and E-cadherin,
Smad2/3 and p-Smad2/3 in rat liver. (A) Western blot analysis of
fibronectin, collagen I and E-cadherin protein levels. (B-D)
Semi-quantitative data from densitometric analysis of fibronectin,
collagen I and E-cadherin presented as the relative ratio of each
protein to GAPDH. (E) Western blot analysis of p-Smad2/3 protein
levels. (F) Semi-quantitative data from densitometric analysis of
p-Smad2/3 presented as the relative ratio of protein to Smad2/3.
Data are presented as the mean ± standard error of the mean of the
optical density of each band, n=8. *P<0.05, **P<0.01,
***P<0.001 compared with the NC group; #P<0.05,
##P<0.01 compared with the CCl4 group.
p-Smad2/3, phosphorylated Smad2/3; rtTGFβRII, recombinant truncated
transforming growth factor β receptor II. |
Discussion
The present study is the first, to the best of our
knowledge, to demonstrate that rtTGFβRII may ameliorate
CCl4-induced liver fibrosis through the inhibition of
EMT. The anti-EMT effects of rtTGFβRII may be mediated by
inhibition of TGF-β1 activation.
Liver fibrosis is marked by the accumulation of
collagen and associated molecules in the liver. EMT is a process
accompanied by a loss of epithelial markers, including E-cadherin,
and a concurrent increase in mesenchymal and fibroblast markers,
including α-SMA, collagens and FSP-1 (21,22).
CCl4 is widely used to generate animal models of liver
fibrosis, and is a potent hepatotoxin that results in centrilobular
hepatic necrosis (23). Altering
the balance of ECM synthesis and degradation may be a key factor in
the development of liver fibrosis (24). The role of TGF-β1 as a fibrogenic
factor has been reported to trigger fibrogenesis. TGF-β1 promotes
fibronectin and fibrillar collagens, inhibits ECM degradation
through the expression of tissue inhibitors of matrix
metalloproteinases and regulates ECM deposition. It has been
demonstrated that myofibroblasts can be generated from parenchymal
epithelial cells in the liver during fibrogenesis (25). In particular, it is accepted that
hepatocyte EMT serves a key role in the perpetuation of liver
fibrosis.
TGF-β is a highly pleiotropic cytokine. Mammals have
three different forms of TGF-β: TGF-β1, TGF-β2 and TGF-β3 (26). These three TGF-βs bind to similar
receptors: TGFβRI; TGFβRII and TGFβRIII, respectively (27,28).
TGFβRI and TGFβRII are endowed with similar transmembrane
serine/threonine kinases activity (29,30).
In the TGF-β signaling pathway, signaling is initiated through
binding to TGFβRII and TGFβRI serine/threonine kinases by active
TGF-β1 ligands. This is a critical event in the TGF-β signaling
pathway, which serves as the initiation point for downstream
events.
TGF-β is considered a key mediator of fibrosis that
induces EMT. TGF-β1 is an important cytokine that induces fibrosis
and the profibrogenic pathway in the liver. Smad2/3 are important
molecules of the TGF-β/Smad signaling pathway. It has previously
been suggested that overexpression of TGF-β1 is associated with
chronic liver fibrosis in patients and in various animal disease
models, whereas inhibition of TGF-β1 has been revealed to reduce
the development of fibrosis in experimental animal disease models
(31). TGFβRII is a member of the
serine/threonine kinase receptor family that includes activin
receptors. The cytoplasmic region of these receptors contains the
apparent kinase domain. Active TGF-β binds to TGFβRII, triggering
the kinase activity of the cytoplasmic domain that in turn
activates TGFβRI. The truncated 27–123 residues of TGFβRII are a
partial fragment of the TGFβRII extracellular domain, which can
efficiently trap TGF-β1; however, a lack of a kinase domain
achieves the purpose of blocking TGF-β1-triggered signals.
Glutathione-S-transferase-pull down assays and a yeast two-hybrid
screening experiment were performed to verify the affinity of
tTGFβRII to TGF-β1. A previous study demonstrated that rtTGFβRII is
able to bind to ligands and associates with TGFβRI, but is unable
to activate TGFβRI (18), thus
blocking TGF-β1 activation and achieving the purpose of treating
liver fibrosis by inhibiting EMT.
The present study investigated the effects of
rtTGFβRII on liver fibrosis using an experimental
CCl4-induced rat model. The results demonstrated that
rtTGFβRII exerted its effects by reducing the serum levels of ALT,
AST and the degree of liver fibrosis in rats. Enzyme serum levels
are markers of liver damage and are key biomarkers reflecting the
degree of liver function and liver fibrosis. H&E, MTS and
Sirius red staining indicated that treatment with rtTGFβRII could
alleviate liver fibrosis. RT-qPCR, immunofluorescence staining and
western blotting results also demonstrated that rtTGFβRII induced
inhibition of ECM component deposition, which was associated with
the reversion of EMT.
In conclusion, the results of the present study
indicated that rtTGFβRII may alter the balance of EMT in
vivo via the inhibition of TGF-β1 activation. Inhibition of
TGF-β1 by rtTGFβRII may lead to the attenuation of liver fibrosis
and improvement of liver function. This study demonstrated that
rtTGFβRII may represent a novel therapeutic approach for the
treatment of EMT-associated diseases.
Acknowledgements
The present study was supported in part by the
National Natural Science Funding of China (grant no. 81573068 to
Y.H.C and grant no. 81200305 to C.Y.N) and the National Natural
Science Funding of Heilongjiang Province (grant no. H2015081 to
Y.H.C and grant no. LC2015037 to C.Y.N).
References
|
1
|
Zhong W, Shen WF, Ning BF, Hu PF, Lin Y,
Yue HY, Yin C, Hou JL, Chen YX, Zhang JP, et al: Inhibition of
extracellular signal-regulated kinase 1 by adenovirus mediated
small interfering RNA attenuates hepatic fibrosis in rats.
Hepatology. 50:1524–1536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhan YY, Wang JH, Tian X, Feng SX, Xue L
and Tian LP: Protective effects of seed melon extract on
CCl4-induced hepatic fibrosis in mice. J Ethnopharmacol.
193:531–537. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Y, Yang P, Chen N, Lin S and Liu M:
Effects of recombinant human adenovirus-p53 on the regression of
hepatic fibrosis. Int J Mol Med. 38:1093–1100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Ying X, Zhang W, Chen Y, Shi C,
Hou Y and Zhang Y: The hepatoprotective effect of fraxetin on
carbon tetrachloride induced hepatic fibrosis by antioxidative
activities in rats. Int Immunopharmacol. 17:543–547. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi SS and Diehl AM:
Epithelial-to-mesenchymal transitions in the liver. Hepatology.
50:2007–2013. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thenappan A, Li Y, Kitisin K, Rashid A,
Shetty K, Johnson L and Mishra L: Role of transforming growth
factor beta signaling and expansion of progenitor cells in
regenerating liver. Hepatology. 51:1373–1382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fabris L and Strazzabosco M:
Epithelial-mesenchymal interactions in biliary diseases. Semin
Liver Dis. 31:11–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee SJ, Kim KH and Park KK: Mechanisms of
fibrogenesis in liver cirrhosis: The molecular aspects of
epithelial-mesenchymal transition. World J Hepatol. 6:207–216.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pinzani M: Epithelial-mesenchymal
transition in chronic liver disease: Fibrogenesis or escape from
death? J Hepatol. 55:459–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taura K, Iwaisako K, Hatano E and Uemoto
S: Controversies over the epithelial-to-mesenchymal transition in
liver fibrosis. J Clinical Med. 5:pii: E9. 2016. View Article : Google Scholar
|
|
13
|
Wells RG: Fibrogenesis. V. TGF-beta
signaling pathways. Am J Physiol Gastrointest Liver Physiol.
279:G845–G850. 2000.PubMed/NCBI
|
|
14
|
Verrecchia F and Mauviel A: Transforming
growth factor-beta and fibrosis. World J Gastroenterol.
13:3056–3062. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zavadil J and Böttinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fabregat I, Moreno-Càceres J, Sánchez A,
Dooley S, Dewidar B, Giannelli G and Ten Dijke P:
IT-LIVERConsortium: TGF-β signalling and liver disease. FEBS
J. 283:2219–2232. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shrestha N, Chand L, Han MK, Lee SO, Kim
CY and Jeong YJ: Glutamine inhibits CCl4 induced liver fibrosis in
mice and TGF-β1 mediated epithelial-mesenchymal transition
in mouse hepatocytes. Food Chem Toxicol. 93:129–137. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chu Y, Guo F, Li Y, Li X, Zhou T and Guo
Y: A novel truncated TGF-beta receptor II downregulates collagen
synthesis and TGF-beta I secretion of keloid fibroblasts. Connect
Tissue Res. 49:92–98. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Institute for Laboratory Animal
ResearchGuide for the Care and Ue of Laboratory Animals. 8th
edition. National Academy Press; Washington, DC: 2011
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sicklick JK, Choi SS, Bustamante M, McCall
SJ, Pérez EH, Huang J, Li YX, Rojkind M and Diehl AM: Evidence for
epithelial-mesenchymal transitions in adult liver cells. Am J
Physiol Gastrointest Liver Physiol. 291:G575–G583. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rygiel KA, Robertson H, Marshall HL,
Pekalski M, Zhao L, Booth TA, Jones DE, Burt AD and Kirby JA:
Epithelial-mesenchymal transition contributes to portal tract
fibrogenesis during human chronic liver disease. Lab Invest.
88:112–123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen S, Zou L, Li L and Wu T: The
protective effect of glycyrrhetinic acid on carbon
tetrachloride-induced chronic liver fibrosis in mice via
upregulation of Nrf2. PLoS One. 8:e536622013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He X, Lv R, Wang K, Huang X, Wu W, Yin L
and Liu Y: Cytoglobin exhibits anti-fibrosis activity on liver in
vivo and in vitro. Protein J. 30:437–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gressner OA and Gao C: Monitoring
fibrogenic progression in the liver. Clin Chim Acta. 433:111–122.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Prud'homme GJ: Pathobiology of
transforming growth factor beta in cancer, fibrosis and immunologic
disease, and therapeutic considerations. Lab Invest. 87:1077–1091.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li MO, Wan YY, Sanjabi S, Robertson AK and
Flavell RA: Transforming growth factor-beta regulation of immune
responses. Ann Rev Immunol. 24:99–146. 2006. View Article : Google Scholar
|
|
28
|
Rubtsov YP and Rudensky AY: TGFbeta
signalling in control of T-cell-mediated self-reactivity. Nat Rev
Immunol. 7:443–453. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Massague J: TGFβ signalling in
context. Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
ten Dijke P and Hill CS: New insights into
TGF-β-Smad signalling. Trends Biochem Sci. 29:265–273. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wynn TA and Ramalingam TR: Mechanisms of
fibrosis: Therapeutic translation for fibrotic disease. Nat Med.
18:1028–1040. 2012. View
Article : Google Scholar : PubMed/NCBI
|