Introduction
Colonic cancer is a common type of gastrointestinal
cancer, with stage II patients exhibiting risk factors and stage
III/IV patients requiring systemic chemotherapy. At present, the
FOLFOX regimen, folinic acid + oxaliplatin + 5-fluorouracil (5-FU)
(1–3), remains the first-line chemotherapy
for colonic cancer. However, ~90% of patients with malignant tumors
succumb as a result of gradually decreasing sensitivity to
chemotherapeutic drugs or resistance resulting from continuous use;
leading to ineffective treatment (4,5).
Therefore, to prolong the survival time of patients with colonic
cancer, it is important to study the mechanism of drug resistance
and develop an effective treatment method to increase drug
sensitivity.
Golgi phosphoprotein 3 (GOLPH3), also known as
GPP34, is localized on human chromosome 5p13 and encodes an ~34 kDa
protein of the Golgi complex. Numerous studies, domestic and
foreign, have confirmed that GOLPH3 is a proto-oncogene (6–9). The
authors previously demonstrated that GOLPH3 gene is overexpressed
in colorectal cancer tissues and promotes the proliferation of
colonic cancer cells by activating the
phosphatidylinositol-3-kinase/protein kinase B/mammalian target of
rapamycin and Wnt/β-catenin signaling pathways. GOLPH3
overexpression can be used as an important indicator to aid
assessing the prognosis of patients with colorectal cancer
(10–14). However, if and how GOLPH3 gene is
involved in inducing resistance to chemotherapy in colonic cancer
has not been reported to date.
According to a previous study on the GOLPH3 gene and
colorectal cancer (10–14), it is hypothesized that the high
expression of GOLPH3 may affect resistance of colonic cancer cells
to 5-FU chemotherapy. In the present study, the effect of GOLPH3
gene on chemotherapeutic drug resistance of human colonic cancer
cells by small interfering (si)RNA and other techniques in
vitro was investigated.
Materials and methods
Cell lines and culture
The human colorectal cancer cell line HT29 was
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). Cells were maintained in RPMI-1640
medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and grown in a humidified incubator with
5% CO2 at 37°C.
Cell groupings
The cells were divided into five groups: A control
group (HT29 cells); a transfection group (HT29 cells transfected
with siRNA-GOLPH3 (GCU UGC UUA AUC ATG GTT AT, Shanghai GenePharma
Co., Ltd, Shanghai, China) using the liposome method, according to
the manufacturer's protocol, for 50 nM siRNA transfection of HT29
cells for 48 h (15); experimental
group 1 (HT29 cells treated with 5-FU); experimental group 2
(transfected HT29 cells treated with 5-FU); and experimental group
3 [HT29 cells treated with the 5-FU and 50 µM extracellular
signal-regulated kinase (ERK)1/2 inhibitor MPD98059 (Cell Signaling
Technology, Inc., Danvers, MA, USA)]. Each experimental group was
maintained in culture medium containing 30 µM 5-FU (Qilu
Pharmaceutical Co., Ltd., Shandong, China) for 24 h.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The expression of GOLPH3 mRNA in the control and
siRNA transfection groups was detected. Total RNA was extracted
from cultured cells using the TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). A total of 2 µM of
total RNA from each sample was reverse transcribed into cDNA using
the SuperScript™ III Reverse Transcriptase kit
(Invitrogen; Thermo Fisher Scientific, Inc.). qPCR was performed
using the SYBR Ex Taq kit (Takara Bio, Inc., Otsu Japan) and the
ABI 9700 qRT-PCR detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The qPCR conditions were an initial cycle
of 20 sec at 95°C; followed by 45 cycles of denaturation for 10 sec
at 95°C, annealing for 20 sec at 60°C and extension for 20 sec at
72°C. All PCR primers were synthesized by Zimmer AG (Shanghai,
China). The primers sequences were as follows: GOLPH3 forward,
5′-AGGGCGACTCCAAGGAAA-3′ and reverse, 5′-TGATGTGTAACCCTCGCG-3′; and
GAPDH forward, 5′-GGTCATAAGCTTGCGTTGATTAAG-3′ and reverse,
5′-CTACGGAAACCTTGTTACGACTTT-3′. Taking GAPDH as an internal
reference, the relative expression level was calculated using the
2−ΔΔCq equation (16).
Cell proliferation (MTT) assay
A cell suspension was seeded into a 96-well plate,
with 103 cells/well and maintained for 24 h by the
anchorage-dependent method. A total of four double-wells and four
control wells, which contained cells without any other treatment,
were set up in each group. Following 48 h culture, 10 µl 5 mg/ml
MTT reagent (Sigma-Aldrich; Merck KGaA) was added to each well,
followed by further culture for 4 h and then the culture medium was
discarded. A total of 150 µl dimethylsulphoxide (Sigma-Aldrich;
Merck KGaA) was added to each well and the plate was agitated in
the dark. Optical density at 490 nm (OD490) was measured
with a microplate reader (Nanjing Huadong Electronics Group Co.,
Ltd, Nanjing, China). The above procedure was repeated three times.
The formula used: Rate of growth inhibition in cancer
cells=(1-average OD490 in treatment groups/average
OD490 in control groups) ×100.
Colony formation assay
Cells in the logarithmic growth phase were seeded
into six-well plates (500 cells/well) containing complete culture
medium. The medium was changed every 3 days over the next 3 weeks.
Once the clones became visible, their culture was terminated and
cells were washed with PBS and fixed in 100% methanol at 37°C for
20 min. Following staining with crystal violet dye for at 37°C 30
min, cells were washed three times with PBS and clones (≥50 cells)
were counted under an inverted microscope. The above steps were
repeated three times and the average cell count was calculated.
Apoptotic assay
The culture medium was centrifuged at 1,000 × g for
5 min at room temperature, the supernatant was discarded and the
cells were resuspended in PBS. A total of 5–10 million resuspended
cells were centrifuged at 1,000 × g for 5 min at room temperature
and the supernatant was discarded. Cells were resuspended in a
mixture containing 195 µl Annexin binding buffer, 5 µl Pacific Blue
Annexin V (both, Beckman Coulter, Inc., Brea, CA, USA) and 10 µl
propidium iodide (Sigma-Aldrich; Merck KGaA) solution for 10–20 min
at room temperature, and then placed in an ice bath and detected by
flow cytometry (CXP software, version 2.2; FC500 flow cytometer;
Beckman Coulter, Inc.).
Western blotting
Each cell line in the logarithmic growth phase was
washed with PBS. The cells were lyzed in radioimmunoprecipitation
assay lysis buffer [containing 50 mM Tris-HCl, (pH 7.4), 150 Mm
NaCl, 0.1% SDS, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF and 1 mM
protease inhibitor cocktail] for 20 min on ice with occasional
vortex mixing. Protein concentrations were determined using the
bicinchoninic acid assay kit (Shanghai Via-gene pro bio
Technologies Co., Ltd, Shanghai, China). Protein from cell lysates
(30 µg/lane) were separated by 10% SDS-PAGE and transferred
electrophoretically to nitrocellulose membranes (Beijing Solarbio
Science and Technology Co., Ltd., Beijing, China). Membranes were
blocked with 5% skimmed milk in TBS-Tween-20 for 1 h at room
temperature. The blots were incubated with rabbit antibodies
against GOLPH3 (1:1,000; cat. no. ab 98023; Abcam, Cambridge, UK),
rabbit antibodies against phosphorylated-glycoprotein (p-gp; 1:500;
cat. no. ab 103477; Abcam), mouse antibodies against β-catenin
(1:500; cat. no. sc-59737; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), rabbit antibodies against GAPDH (1:1,000; cat. no.
sc-365062; Santa), mouse antibodies against β-actin (1:1,000; cat.
no. sc-47778; Santa), rabbit antibodies against ERK1/2 (1:500; cat.
no. Ab 17942; Abcam) and rabbit antibodies against pERK1/2 (1:500;
cat. no. ab 65142; Abcam) at 4°C overnight. Following washing three
times with PBST for 10 min, the membranes were incubated with
horseradish peroxidase-conjugated anti-mouse and anti-rabbit
immunoglobulin G (both 1:5,000; cat. nos. sc-516102 and sc-2357,
respectively; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. The bands were visualized with Pierce enhanced
chemiluminescence detection kit (cat. no. 32109, Thermo Fisher
Scientific, Inc.) and the results were analyzed with Image J
Software (version 1.48; National Institutes of Health, Bethesda,
MD, USA). Relative intensity target protein = target band of gray
value/GAPDH (or β-actin) band gradation value. GAPDH (or β-actin)
was used as a loading control.
Statistical analysis
SPSS software (version 19.0; SPSS Inc., Chicago, IL,
USA) was used for data analysis and data were expressed as the mean
± standard deviation. Dunnett's t test was used to compare each
experimental group with the control group. Differences among the
experimental groups were compared by one-way analysis of variance.
Differences between paired experimental groups were compared by
Least-Significant Difference t test. P<0.05 was considered to
indicate a statistically significant difference.
Results
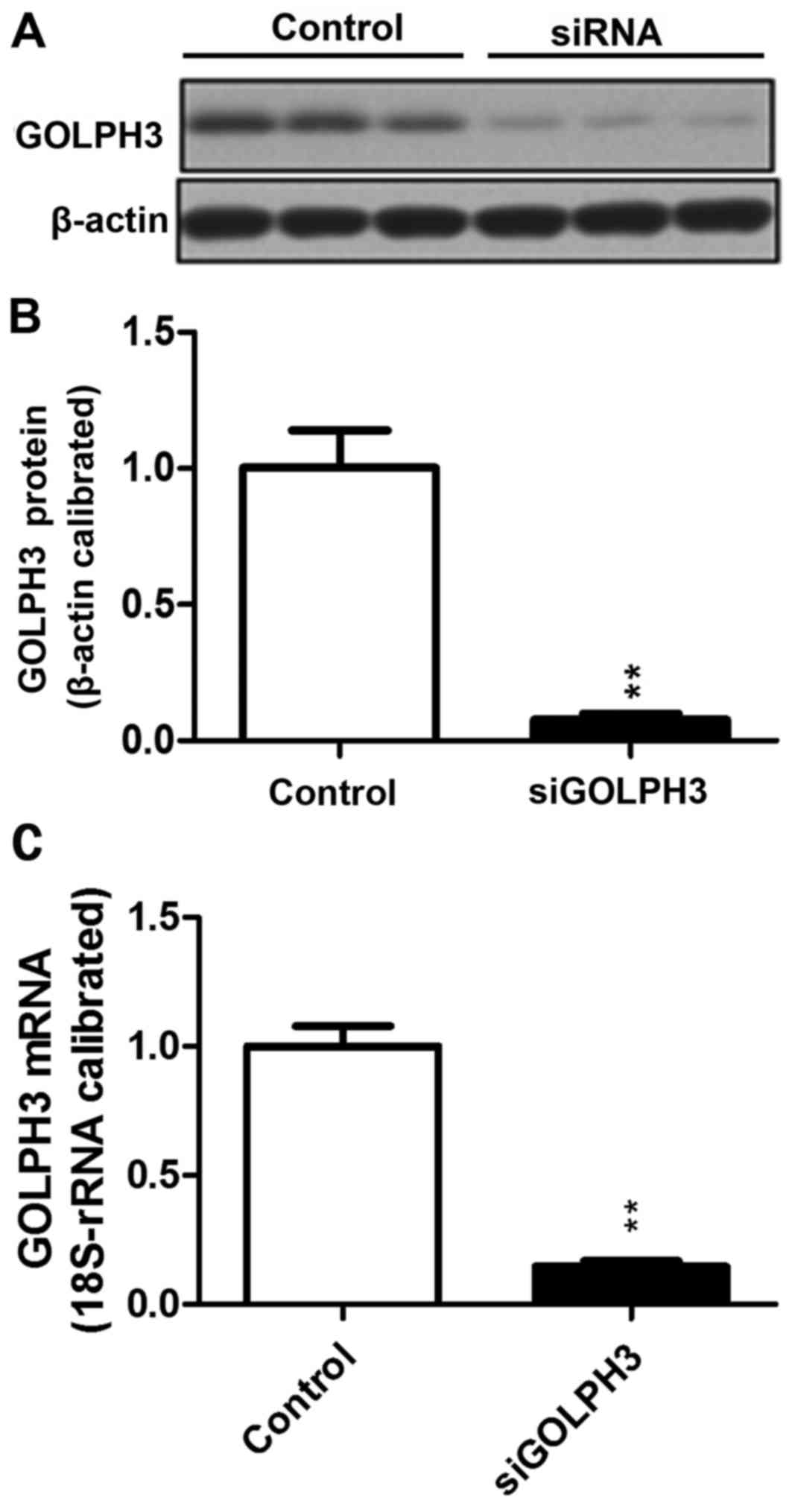
Detection of transfection efficiency
of HT29 colonic cancer cells
Expression of GOLPH3 in the HT29 human colonic
cancer cell line following transfection
Compared with the control group (1.0±0.07813), the
relative expression of GOLPH3 mRNA in the siRNA-transfected group
(0.1468±0.0213) decreased significantly (P<0.01). A similar
result was demonstrated using western blotting, which demonstrated
that the GOLPH3 protein expression in HT29 cells transfected with
siRNA-GOLPH3 (0.0768±0.3992) was decreased compared with the
control HT29 cells (1.0033±0.2353; P<0.01). siRNA transfection
silenced expression of GOLPH3 (Fig.
1).
Proliferation of cancer cells in the transfection
group
An MTT assay demonstrated that OD490 of
the transfected group was decreased compared with the control group
(P<0.01), which indicated that the proliferative ability of
GOLPH3 was significantly decreased as a result of silencing
(Fig. 2). Similar results were
demonstrated in the colony-formation assay, where the colony number
of tumor cells in the transfection group was significantly
decreased compared with the control group (P<0.01; Fig. 3), which demonstrated that the
tumorigenicity was significantly decreased following silencing
GOLPH3.
Investigation of the effect exerted by GOLPH3 on
the resistance of colonic cancer cells to treatment with 5-FU
Effect of silencing GOLPH3 on the proliferation
of HT29 cells treated with 5-FU
The MTT assay demonstrated that, in experimental
groups 1 (HT29 cells treated with 5-FU; P<0.05) and 2
(transfected HT29 cells treated with 5-FU; P<0.001),
OD490 was significantly decreased compared with the
control group. As a result of treatment with 5-FU, the
OD490 of experimental group 2 was significantly
decreased compared with experimental group 1 (P<0.001; Fig. 4). This demonstrates that knockout
of GOLPH3 could increase the sensitivity of HT29 colon cancer cells
to 5-FU. Similar results were demonstrated in the colony-formation
assay, where the colony number of the tumor cells in the
experimental group 1 and 2 was significantly decreased compared
with the control group. Treatment with 5-FU resulted in the colony
number in experimental group 2 being significantly decreased
compared with experimental group 1 (P<0.001; Fig. 5). These results demonstrated that
knockout of GOLPH3 decreased the colony-forming ability of HT29
colon cancer cells following treatment with 5-FU.
Effect of silencing GOLPH3 on apoptosis in HT29
cells following treatment with 5-FU
The percentage of apoptotic cancer cells in the
experimental groups was significantly increased in group 1
(P<0.01) and group 2 (P<0.001) compared with that in the
control group (Fig. 6). The
percentage of apoptotic cells in experimental group 2 was
significantly increased compared with experimental group 1
(P<0.001; Fig. 6). These
results demonstrated that knockdown of GOLPH3 increased the
percentage of apoptotic HT29 colon cancer cells following treatment
with 5-FU.
Investigation of the mechanism by which GOLPH3
affects the resistance of colonic cancer cells to 5-FU chemotherapy
by western blotting
Difference in the expression of GOLPH3, p-gp,
ERK1/2, β-catenin and pERK1/2 protein in HT29 colonic cancer cells
in each group were detected using western blotting and the
mechanism by which HT29 cells develop GOLPH3-induced resistance to
5-FU chemotherapy was investigated.
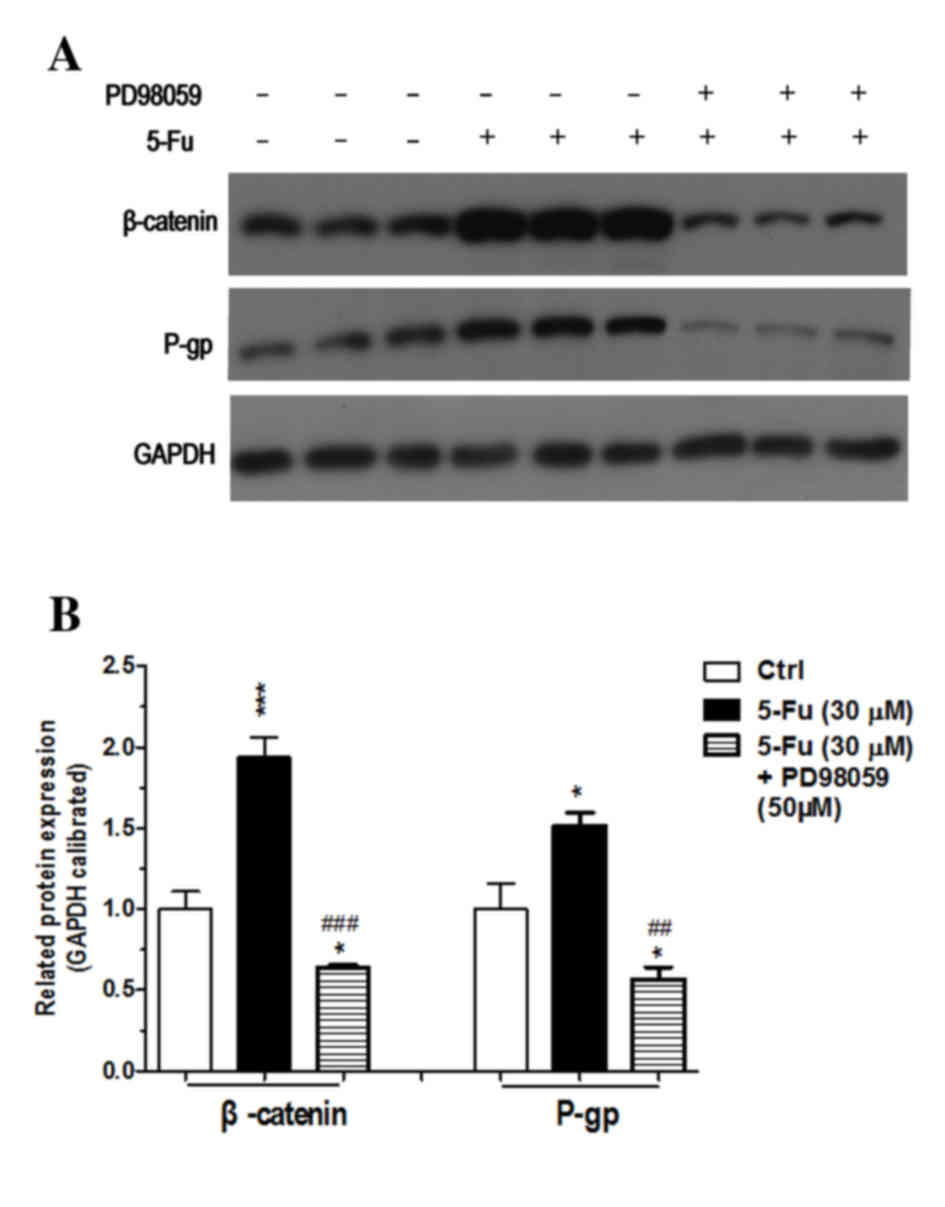
Compared with the control group, there was
significant increase in expression of p-gp in the cells treated
with 5-FU in group 1 (P<0.01; Fig.
7; Table I), which implied
that resistance was induced in colonic cancer cells treated with
5-FU, and expression of GOLPH3, pERK1/2 and β-catenin proteins also
significantly increased (all P<0.01 vs. control; Fig. 7). The results of the present study
suggested that the expression of GOLPH3 and the signaling pathways
of mitogen-activated protein kinase (MAPK)/ERK and Wnt/β-catenin
were involved in the mechanism of resistance to 5-FU.
 | Figure 7.Expression of proteins in experimental
groups 1 (5-FU) and 2 (5-FU+small interferring-GOLPH3).
Representative images of three individual experiments, GAPDH was
the loading control. (A) Western blotting protein band images. (B)
Quantitative analysis of western blotting. Values are expressed as
percentage of untreated Ctrl. *P<0.05, **P<0.01,
***P<0.001 vs. Ctrl; ##P<0.01,
###P<0.001 vs. 5-FU (30 µM). NS, not significant;
5-FU, 5 fluorouracil; GOLPH3, Golgi phosphoprotein 3; pERK1/2,
phosphorylated extracellular signal-regulated kinase; p-gp,
phosphorylated glycoprotein; Ctrl, control. |
 | Table I.Relative expression of proteins in
experimental groups 1 and 2. |
Table I.
Relative expression of proteins in
experimental groups 1 and 2.
| Protein | Control | Experimental group
1 | Experimental group
2 |
|---|
| GOLPH3 |
0.9667±0.09356 |
3.6335±0.5739a |
0.5201±0.17598b,c |
| p-ERK1/2 |
1.0002±0.33347 |
2.6614±0.43782d |
0.2391±0.08942b,c |
| β-catenin |
1.0003±0.10137 |
2.3235±0.41784d |
0.9330±0.04897e |
| p-gp |
1.0005±0.19883 |
2.4239±0.26066d |
0.4662±0.15896b,c |
Compared with the control group and experimental
group 1, when the HT29 cells with GOLPH3 stably silenced were
treated with 5-FU, the expression of GOLPH3 and p-gp was
significantly decreased in experimental group 2 (P<0.05;
Fig. 7; Table I), which implied that silencing
GOLPH3 would decrease the resistance of HT29 colonic cancer cells
to 5-FU to a certain extent and that the GOLPH3 gene is involved in
the resistance of colonic cancer cells to 5-FU.
The results also demonstrated that there was no
marked difference in the expression of ERK1/2 between all the
groups; however, the expression of pERK1/2 in experimental group 2
was significantly decreased compared with experimental group 1,
which demonstrated that silencing the GOLPH3 gene could reduce the
activation of the MAPK/ERK signaling pathway by reducing the
production of pERK1/2. The level of β-catenin protein expression in
group 2 was significantly decreased compared with group 1, which
implied that silencing the GOLPH3 gene by treatment with 5-FU could
decrease its expression and inhibit activation of the Wnt/β-catenin
signaling pathway. Therefore, GOLPH3 could affect the resistance of
HT29 cells to 5-FU by activating the MAPK/ERK and Wnt/β-catenin
signaling pathway.
Further experiments demonstrated that the expression
of β-catenin and p-gp in experiment group 3, which was treated with
an MAPK/ERK signaling pathway inhibitor, was significantly
decreased compared with the control group and experimental group 1
(Fig. 8; Table II). This result demonstrated that
inhibiting the MAPK/ERK signaling pathway would partly reverse
resistance of HT29 colonic cancer cells to 5-FU, which confirmed
the hypothesis suggested by the results of experimental group
2.
| Table II.Relative expression of proteins in
experimental groups 1 and 3. |
Table II.
Relative expression of proteins in
experimental groups 1 and 3.
|
| Control | Experimental group
1 | Experimental group
3 |
|---|
| β-catenin |
1.0000±0.1870 |
1.9372±0.2133b |
0.6375±0.02795a,d |
| p-gp |
1.0000±0.2796 |
1.5134±0.1428a |
0.5678±0.1275a,c |
Discussion
5-FU is a pyrimidine that inhibits thymidylate
synthetase (17). Metabolic
activation results in 5-FU being incorporated into RNA, which
interferes with RNA function and prevents DNA synthesis. This
restricts the proliferation of cancer cells and ultimately leads to
their death. There are several mechanisms for 5-FU resistance,
including drug metabolic pathway abnormalities, abnormal gene
expression and signaling pathway abnormalities (18,19).
The mechanisms of abnormal gene expression and signaling pathway
activation has become a popular area in current study on the
resistance of colonic cancer to chemotherapy. The novel
proto-oncogene GOLPH3 is overexpressed in a variety of tumor
tissues and is associated with poor prognosis, which has attracted
much attention.
Expression of p-gp and GOLPH3 was increased in HT29
cells following treatment with 5-FU, which caused the development
of drug resistance. Silencing GOLPH3 expression increased the
sensitivity of HT29 cells to 5-FU, reduced their tumorigenicity and
partly reversed their resistance to 5-FU. The expression of pERK1/2
and β-catenin was decreased, which indicated that the mechanism of
resistance was associated with the activation of the MAPK/ERK and
Wnt/β-catenin signaling pathways.
It has been demonstrated that the MAPK/ERK and
Wnt/β-catenin signaling pathways are involved in drug resistance in
a number of types of cancer including breast cancer (20,21).
The authors previously demonstrated that GOLPH3 could activate the
Wnt/β-catenin signaling pathway to promote the proliferation of
colonic cancer cells (14), which
was confirmed in the present study. In addition, it was
demonstrated that GOLPH3 may activate the MAPK/ERK signaling
pathway in colonic cancer.
It was also demonstrated that β-catenin expression
was significantly decreased following inhibition of the MAPK/ERK
signaling pathway, which confirmed that there is an association
between the Wnt/β-catenin and MAPK/ERK signaling pathways in the
mechanism of 5-FU resistance in colonic cancer. Jeon et al
(22) demonstrated that in hepatic
progenitor cells the Wnt/β-catenin and MAPK/ERK signaling pathways
worked together to promote or suppress tumors developing with Wnt3
alpha, adenomatous polyposis coli protein and Ras (23). Kim et al (24) hypothesize that there is a hidden
cancer-promoting, positive feedback cycle between the two pathways,
which is important for cancer formation. This suggests that
inhibiting certain molecules in the feedback cycle could be used as
a target for cancer therapy.
An important mechanism in 5-FU cytotoxicity in
colonic cancer cells is DNA damage (17,25).
Novel studies have demonstrated that DNA damage can act on the
DNA-pyruvate kinase (PK)-GOLPH3-unconventional myosin-XVIIIa
(MYO18a) signaling pathway, which increases the cell survival rate.
This pathway links the DNA damage response directly to the Golgi
apparatus. If any components of the pathway are removed the ability
of drugs that induce DNA damage to kill cancer cells could be
enhanced, by inhibiting cell proliferation and increasing apoptosis
(26,27). It is hypothesized that the
involvement of GOLPH3 in the resistance of HT29 cells to 5-FU may
be a result of DNA damage and activation of the
DNA-PK-GOLPH3-MYO18A pathway, which prevents HT29 colonic cancer
cells from being killed by 5-FU.
In conclusion, GOLPH3 expression could be involved
in the resistance of HT29 colonic cancer cells to 5-FU. The
mechanism of resistance may be that activation of the Wnt/β-catenin
and MAPK/ERK signaling pathways promote resistance to 5-FU.
Silencing expression of GOLPH3 partly reverses drug resistance.
GOLPH3 may be a potential novel target for reversing drug
resistance in colonic cancer.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Fujian Province, China (grant no.
2015J01438).
References
|
1
|
Cunningham D, Atkin W, Lenz HJ, Lynch HT,
Minsky B, Nordlinger B and Starling N: Colorectal cancer. Lancet.
375:1030–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loree JM and Cheung WY: Optimizing
adjuvant therapy and survivorship care of stage III colon cancer.
Future Oncol. 12:2021–2035. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rogowski W and Sulżyc-Bielicka V: Optimal
duration of a first-line palliative chemotherapy in disseminated
colorectal cancer-a review of the literature from a developing
country perspective. Contemp Oncol (Pozn). 20:210–214.
2016.PubMed/NCBI
|
|
4
|
Hu T, Li Z, Gao CY and Cho CH: Mechanisms
of drug resistance in colon cancer and its therapeutic strategies.
World J Gastroenterol. 22:6876–6889. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan S, Shi H, Ba M, Lin S, Tang H, Zeng X
and Zhang X: miR-409-3p sensitizes colon cancer cells to
oxaliplatin by inhibiting Beclin-1-mediated autophagy. Int J Mol
Med. 37:1030–1038. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scott KL, Kabbarah O, Liang MC, Ivanova E,
Anagnostou V, Wu J, Dhakal S, Wu M, Chen S, Feinberg T, et al:
GOLPH3 modulates mTOR signalling and rapamycin sensitivity in
cancer. Nature. 459:1085–1090. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scott KL and Chin L: Signaling from the
Golgi: Mechanisms and models for Golgi phosphoprotein 3-mediated
oncogenesis. Clin Cancer Res. 16:2229–2234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zeng Z, Lin H, Zhao X, Liu G, Wang X, Xu
R, Chen K, Li J and Song L: Overexpression of GOLPH3 promotes
proliferation and tumorigenicity in breast cancer via suppression
of the FOXO1 transcription factor. Clin Cancer Res. 18:4059–4069.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kunigou O, Nagao H, Kawabata N, Ishidou Y,
Nagano S, Maeda S, Komiya S and Setoguchi T: Role of Golph3 and
Golph3L in the proliferation of human rhabdomyosarcoma. Oncol Rep.
26:1337–1342. 2011.PubMed/NCBI
|
|
10
|
Qiu CZ, Yu WS and Wang CX: Expression and
clinical significance of Golgi phosphorylation protein 3 in
colorectal cancer tissues. Chin J Exp Surg. 30:461–463. 2013.(In
Chinese).
|
|
11
|
Yu WS, Qiu CZ and Wang CX: Golph3
expression and apoptosis in colorectal cancer cells. Clin Oncol.
40:1094–1463. 2013.(In Chinese).
|
|
12
|
Yang XF, Qiu CZ and Wang CX: Relationship
between golgi phosphorylation protein 3 expression and prognosis in
colorectal cancer. Chin J General Surg. 23:1362–1366. 2014.(In
Chinese).
|
|
13
|
Guo YT, Qiu CZ, Huang ZX, Yu WS, Yang XF
and Wang MZ: Correlational research of Golgi phosphorylation
protein 3 expression in colorectal cancer. World J Gastroenterol.
21:13473–13479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qiu CZ, Wang MZ, Yu WS, Guo YT, Wang CX
and Yang XF: Correlation of GOLPH3 gene with wnt signaling pathway
in human colon cancer cells. J Cancer. 7:928–934. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ah KK, Young KJ, Ah LY, Min A, Yil BY and
Heon SM: Entamoeba histolytica induces cell death of HT29 colonic
epithelial cells via NOX1-Derived ROS. Korean J Parasitol.
51:61–68. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wigmore PM, Mustafa S, El-Beltagy M, Lyons
L, Umka J and Bennett G: Effects of 5-fu. Adv Exp Med Biol.
678:157–164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang N, Yin Y, Xu SJ and Chen WS:
5-fluorouracil: Mechanisms of resistance and reversal strategies.
Molecules. 13:1551–1569. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Shi H, Tang H, Fang Z, Wang J and
Cui S: miR-218 inhibits the invasion and migration of colon cancer
cells by targeting the PI3K/Akt/mTOR signaling pathway. Int J Mol
Med. 35:1301–1308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Y, Moerkens M, Ramaiahgari S, de
Bont H, Price L, Meerman J and van de Water B: Elevated
insulin-like growth factor 1 receptor signaling induces
antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling
routes. Breast Cancer Res. 13:R522011. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Zhang X, Wu X, Li W, Su P, Cheng
H, Xiang L, Gao P and Zhou G: linterference of frizzled 1 (FZD1)
reverses multidrug resistance in breast cancer cells through the
Wnt/β-catenin pathway. Cancer Lett. 323:106–113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jeon SH, Yoon JY, Park YN, Jeong WJ, Kim
S, Jho EH, Surh YJ and Choi KY: Axin inhibits extracellular
signal-regulated kinase pathway by ras degradation via
beta-catenin. J Biol Chem. 28:14482–14492. 2007. View Article : Google Scholar
|
|
23
|
Jin C, Samuelson L, Cui CB, Sun Y and
Gerber DA: MAPK/ERK and Wnt/β-Catenin pathways are synergistically
involved in proliferation of Sca-1 positive hepatic progenitor
cells. Biochem Biophys Res Commun. 409:803–807. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim D, Rath O, Kolch W and Cho KH: A
hidden oncogenic positive feedback loop caused by crosstalk between
Wnt and ERK pathways. Oncogene. 26:4571–4579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matuo R, Sousa FG, Escargueil AE,
Grivicich I, Garcia-Santos D, Chies JA, Saffi J, Larsen AK and
Henriques JA: 5-Fluorouracil and its active metabolite FdUMP cause
Dna damage in human SW620 colon adenocarcinoma cell line. J Appl
Toxicol. 29:308–316. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Farber-Katz SE, Dippold HC, Buschman MD,
Peterman MC, Xing M, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM,
et al: DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3.
Cell. 156:413–427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Buschman MD, Rahajeng J and Field SJ:
GOLPH3 links the Golgi, DNA damage, and cancer. Cancer Res.
75:624–627. 2015. View Article : Google Scholar : PubMed/NCBI
|