Introduction
Type 2 diabetes mellitus (T2DM) accounts for ~90% of
all cases of diabetes mellitus (DM) and imparts a 1.5-2-fold
increase in morbidity compared with the general population
(1). Cardiovascular dysfunction is
a major morbidity and cause of mortality in T2DM patients. However,
the mechanisms underlying the high prevalence of cardiovascular
dysfunction in patients with T2DM have not been completely
elucidated (2,3) and therapeutic outcomes remain
unsatisfactory.
Autophagy is a highly conserved process that
preserves cellular homeostasis between normal and
pathophysiological conditions (4).
Although autophagy is primarily involved in cell survival,
continuous activation may lead to autophagic or apoptotic cell
death (5). Cardiomyocyte autophagy
is a crucial adaptive response of the myocardium to preserve the
cellular energy balance, particularly during stress (6,7).
T2DM patients exhibit increased cardiac autophagy and cleavage of
caspase-3, which was investigated by analysis of the right atrial
appendages from subjects receiving coronary artery bypass graft
surgery (8). Impaired cardiac
autophagy has been reported in metabolic syndrome and T2DM animals
(9,10). Since insulin is able to modulate
myocardial autophagy via phosphatidylinositol 3-kinase/RAC-α
serine/threonine protein kinase (Akt) signaling to inhibit the
serine/threonine protein kinase mTOR (mTOR) signaling pathway,
cardiac autophagy is impaired in insulin resistant T2DM (11). In addition, under conditions of
elevated nutrients, mTOR is activated in hyperglycemia, thereby
inhibiting autophagy. A decrease in autophagy results in an
aggregation of cytotoxic proteins and defective organelles that may
provoke apoptosis and damage cardiomyocytes (12). Therefore, modulation of autophagy
may decrease DM cardiomyopathy.
Histone deacetylases (HDACs) serve an essential role
in regulating cell proliferation, migration and death. Previously,
HDACs and inhibitors have been identified to be therapeutic targets
for type 1 DM and T2DM (13,14).
A pan HDAC inhibitor MPT0E014 has been demonstrated to decrease
cardiac fibrosis and profibrotic signaling protein expression in a
heart failure animal model (15).
In addition, MPT0E014 has been demonstrated to regulate cardiac
metabolism through its effects on peroxisome proliferator-activated
receptors (PPARs) and inflammatory cytokines to reduce the
accumulation of fatty acids in DM hearts (16). However, it is not clear whether
HDAC inhibition is able to regulate cardiac autophagy in T2DM
cardiomyopathy. Additionally, advanced glycation end products
(AGEs) are involved in the pathogenesis of vascular damage
resulting from hyperglycemia (17). AGEs cause detrimental effects by
directly altering the structure and function of AGE-modified
macromolecules, or by binding with the advanced glycosylation end
product-specific receptor (RAGE) (18). However, the effect of HDAC
inhibition on the expression of RAGE in DM cardiomyopathy remains
unknown.
Therefore, the purpose of the present study was to
investigate the effect of the HDAC inhibitor MPT0E014 on the
regulation of myocardial autophagy in rats with high-fat diet (HFD)
and low-dose streptozotocin (STZ)-induced T2DM.
Materials and methods
Animals, blood sampling and tissue
preparation
The present study was approved by the Institutional
Animal Care and Use Committee of Taipei Medical University
(approval no. LAC-2013-0085). Rats were housed under standard
environmental conditions (21±2°C; humidity 50–60%; 12-h light/dark
cycle) and maintained on commercial rat food and tap water ad
libitum. A total of 24 male 8-week old Wistar rats, weighing
260±4.0 g were purchased from BioLasco, Taiwan, Co., Ltd., (Taipei,
Taiwan) were used in the present study. To induce T2DM, 16 rats
were fed an HFD (60% fat, 18% protein and 21% carbohydrates;
Research Diet Inc., New Brunswick, NJ, USA) ad libitum
starting at 8 weeks old and received a low dose of STZ (35 mg/kg;
Sigma-Aldrich, Merck KGaA, Darmstadt, Germany) intraperitoneally at
10 weeks old (19). T2DM was
defined as high fasting plasma glucose (≥15 mmol/l) and was
measured with a glucometer (Ascensia Elite; Bayer, Pittsburgh, PA,
USA) (20,21). At 12 weeks of age, the rats were
grouped into three groups of eight rats each, including control,
HFD + low-dose STZ-induced T2DM and MPT0E014-treated HFD + low-dose
STZ-induced T2DM groups. MPT0E014 [a pan-HDAC inhibitor (15); 50 mg/kg in 50% polyethylene glycol
400 and 0.25% carboxymethyl cellulose (22)], or a vehicle (1 ml/kg of 50%
polyethylene glycol 400 and 0.25% carboxymethyl cellulose) was
given once daily for 7 days by oral gavage in the studied rats. The
rats were sacrificed with an intraperitoneal injection of sodium
pentobarbital (100 mg/kg) at 13 weeks of age. Body weight was
measured prior to sacrifice. Fasting plasma blood urea nitrogen,
creatinine, cholesterol, triglyceride and high-density
lipoprotein-cholesterol (HDL-C) were obtained with a SPOTCHEM
analyzer (Arkray, Inc., Kyoto, Japan) using SPOTCHEM II Inorganic
Phosphorous reagent strips. Plasma free fatty acid was measured
using a Free Fatty Acid Quantitation kit (Sigma-Aldrich; Merck
KGaA) and plasma fasting insulin was measured with a Mercodia
Ultrasensitive Rat Insulin ELISA (Mercodia AB, Uppsala, Sweden).
Transverse tissue pieces from the left ventricle (LV) weighing
0.55–0.65 g were snap-frozen in liquid nitrogen and stored at −80°C
for protein isolation.
Echocardiographic measurements
At 10 and 13 weeks of age, transthoracic
echocardiography was performed using a Vivid I ultrasound
cardiovascular system (GE Healthcare, Chicago, IL, USA) was
performed under isoflurane anesthesia (5% for induction and 2% for
maintenance) in the control and HFD + STZ T2DM rats with or without
treatment with MPT0E014. M-mode tracing of the LV was used to
measure the following cardiac structures: The LV end-diastolic
diameter (LVEDd), LV end-systolic diameter (LVESd),
interventricular septal thickness in diastole (IVSd), end diastolic
volume (EDV), end systolic volume (ESV), fractional shortening (FS)
and the ejection fraction (EF) (15).
Western blot analysis
Tissues were homogenized and lysized in M-PER™
Mammalian Protein Extraction Reagent (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and the protein concentration were
determination by Bradford assay. Equal amounts of proteins (40 µg)
were resolved by SDS-PAGE on a 8–15% gel followed by
electrophoretic transfer of proteins onto polyvinylidene difluoride
membranes. Blots were blocked with 5% skimmed milk for 1 h at room
temperature, then probed with antibodies against Light Chain (LC)
3-I (cat. no. APG8B; 1:1,000; Abgent, Inc., San Diego, CA, USA) and
LC3-II (cat. no. APG8B; 1:1,000; Abgent, Inc.), Beclin-1 (cat. no.
ADI-905-721; 1:1,000; Enzo Life Sciences, Inc., Farmingdale, NY,
USA), poly ADP-ribose polymerase 1 (PARP1; cat. no. sc-1561;
1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), 5′
adenosine monophosphate-activated protein kinase (AMPK) α2 (cat.
no. 07–363; 1:500; Upstate Biotechnology, Inc., Lake Placid, NY,
USA), phosphorylated (p)-AMPKα2 Thr172 (cat. no. 07-681; 1:1,000;
EMD Millipore, Billerica, MA, USA), anti-histone H3 (cat. no.
ab1791; 1:1,000; Abcam, Cambridge, MA, USA), anti-acetyl-histone H3
at Lys9 (cat. no. 06-942; 1:5,000; EMD Millipore), mTOR (cat. no.
2972; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
p-mTOR-Ser-2448 (cat. no. 2971; 1:1,000; Cell Signaling Technology,
Inc.), P70S6 kinase (cat. no. 9202; 1:1,000; Cell Signaling
Technology, Inc.) and p-P70S6K-Thr 389 (cat. no. 9205; 1:1,000;
Cell Signaling Technology, Inc.), tumor necrosis factor (TNF)-α
(cat. no. AB1837P; 1:500; EMD Millipore), interleukin (IL)-6 (cat.
no. ab6672; 1:1,000; Thermo Fisher Scientific, Inc.), advanced
glycosylation end product-specific receptor (RAGE; cat. no.
PA1-84173; 1:3,000; Thermo Fisher Scientific, Inc.), glucose
transporter (GLUT) 4 (cat. no. ab654; 1:2,000; Abcam), insulin
substrate receptor (IRS; cat. no. 2382; 1:1,000; Cell Signaling
Technology, Inc.), p-IRS-1 at Ser307 (cat. no. 2381; 1:1,000; Cell
Signaling Technology, Inc.), Akt (cat. no. 4685; 1:1,000; Cell
Signaling Technology, Inc.) and p-Akt (cat. no. 4060; 1:3,000; Cell
Signaling Technology, Inc.) for overnight at 4°C and secondary
antibodies conjugated with horseradish peroxidase (Leinco
Technologies, Inc., Fenton, MO, USA) for 1 h at room temperature.
Bound antibodies were detected with an enhanced chemiluminescence
detection system (EMD Millipore) and analyzed with AlphaEaseFC
software (version 6.0; ProteinSimple, San Jose, CA, USA). Targeted
bands were normalized to cardiac GAPDH (Sigma-Aldrich; Merck KGaA)
to confirm equal protein loading.
Statistical analysis
All quantitative data are expressed as the mean ±
standard error of the mean. Statistically significant differences
between different groups was determined using one-way analysis of
variance with Tukey's test for multiple comparisons as appropriate
using SigmaStat (version 3.5; Systat Software, Inc., San Jose, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Effect of MPT0E014 on biochemistry and
cardiac function
The body weights were similar among rats in the
control, HFD + low-dose STZ-induced T2DM and T2DM rats treated with
MPT0E014 groups (Table I).
However, HFD + low-dose STZ-induced T2DM rats had larger absolute
heart weights compared with control rats and T2DM rats treated with
MPT0E014. In addition, the heart-to-body weight ratios were greater
in the HFD + low-dose STZ-induced T2DM rats compared with the
control rats and T2DM rats treated with MPT0E014 (Table I). Compared with the control rats,
HFD + low-dose STZ-induced T2DM rats and MPT0E014-treated T2DM rats
had higher levels of blood glucose, triglycerides and free fatty
acids (Table I). However, the
MPT0E014-treated T2DM rats had lower levels of blood glucose,
triglycerides and free fatty acids compared with HFD + low-dose
STZ-induced T2DM rats. The level of HDL-C was lower in the HFD +
low-dose STZ-induced T2DM rats compared with the control and
MPT0E014-treated T2DM rats (Table
I). Total cholesterol, blood urea nitrogen, creatinine and
insulin levels were not significantly different between the three
groups (P>0.05).
 | Table I.Physical and biochemical
characteristics of the control, T2DM and T2DM rats treated with
MPT0E014 at 13 weeks. |
Table I.
Physical and biochemical
characteristics of the control, T2DM and T2DM rats treated with
MPT0E014 at 13 weeks.
| Physical
characteristics | Control | T2DM | MPT0E104-treated
T2DM |
|---|
| BW, g |
369.2±10.4 |
370.7±17.1 |
366.2±11.0 |
| HW, g |
1.3±0.1 |
1.6±0.1a |
1.3±0.1b |
| HW/BW ratio,
g/kg |
3.5±0.1 |
4.2±0.2a |
3.6±0.1b |
| Biochemical
characteristics |
| Fasting
blood glucose, mmol/l |
5.9±0.3 |
19.1±0.9a |
12.9±1.8a,b |
| BUN,
mmol/l |
5.5±0.4 |
5.5±0.4 |
5.1±0.2 |
|
Creatinine, mmol/l |
34.2±6.8 |
42.7±2.7 |
39.8±4.4 |
|
Cholesterol, mmol/l |
1.8±0.2 |
1.9±0.1 |
1.7±0.1 |
|
Triglyceride, mmol/l |
0.7±0.1 |
2.5±0.4a |
1.4±0.2b |
| HDL-C,
mmol/l |
0.5±0.1 |
0.3±0.1a |
0.5±0.1b |
| Free
fatty acid µmol/l |
26.3±3.3 |
56.5±6.6a |
38.7±3.2b |
| Fasting
insulin, pmol/l |
48.8±10.5 |
28.2±6.6 |
29.2±2.7 |
Table II
illustrates echocardiograms of control, HFD + STZ-induced T2DM and
MPT0E014-treated T2DM rats. The HFD + STZ-induced T2DM rats had
higher LVEDd, LVESd, EDV and ESV values, and lower EF and FS values
compared with control and MPT0E014-treated T2DM rats.
| Table II.Echocardiogram of control, T2DM and
T2DM rats treated with MPT0E104 at 13 weeks. |
Table II.
Echocardiogram of control, T2DM and
T2DM rats treated with MPT0E104 at 13 weeks.
| Group | LVEDd (mm) | LVEDs (mm) | EDV (mm) | ESV (mm) | EF (%) | FS (%) |
|---|
| Control |
7.1±0.1 |
3.0±0.2 |
0.8±0.1 |
0.08±0.1 |
90.8±1.3 |
57.9±2.6 |
| T2DM |
8.0±0.2a |
4.1±0.2a |
1.1±0.1a |
0.2±0.1a |
80.9±1.2a |
44.8±1.3a |
| MPT0E104-treated
T2DM |
7.1±0.2b |
3.2±0.2b |
0.8±0.1b |
0.09±0.1b |
88.9±0.9b |
53.1±0.5b |
Effects of MPT0E014 on cardiac
autophagy and cell death
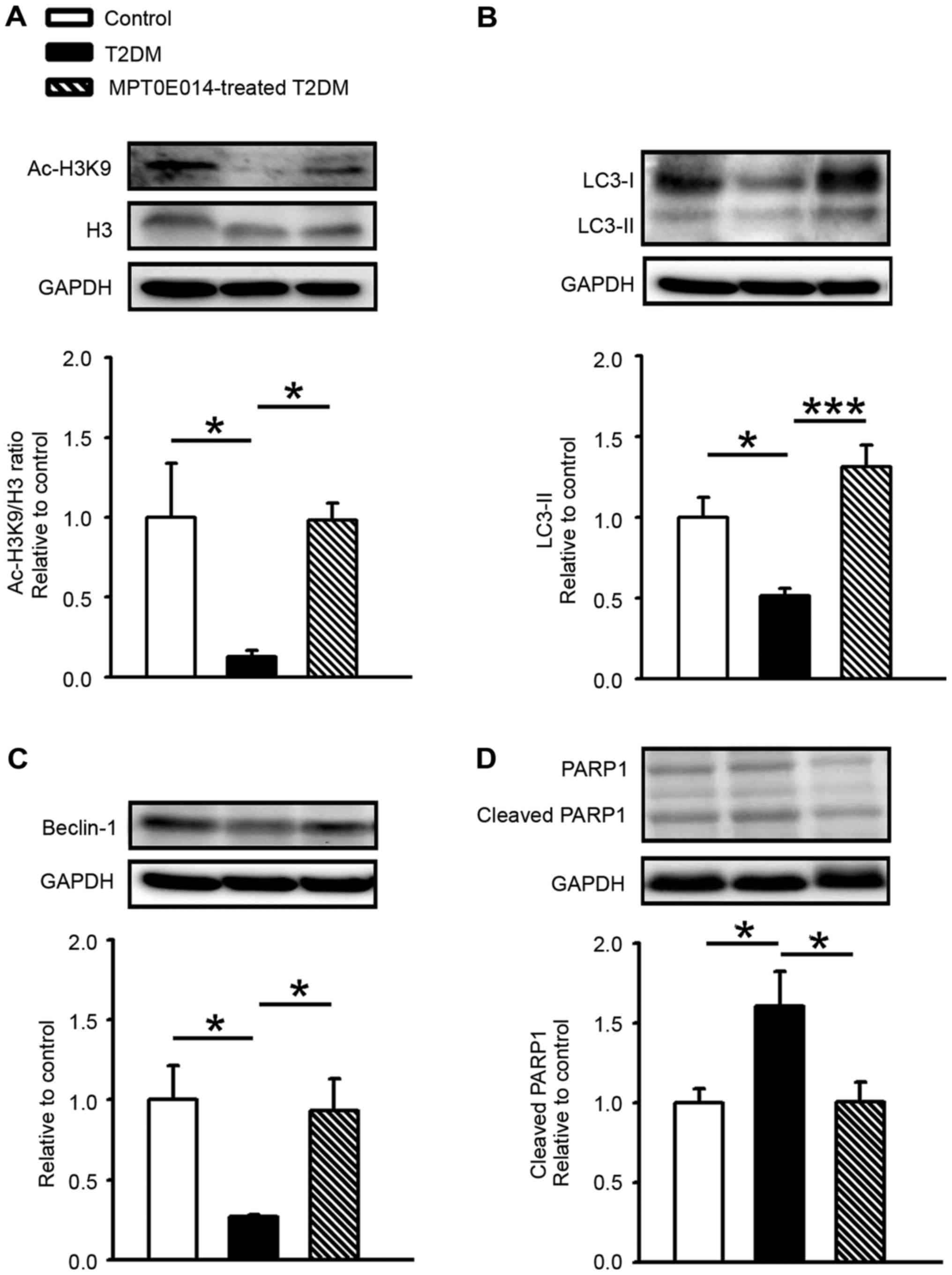
Compared with the control group, the acetyl histone
H3 (Ac-H3K9)/histone 3 (H3) ratio was decreased in the HFD +
STZ-induced T2DM hearts by 0.87. Ac-H3K9/H3 ratio was increased in
the T2DM rats treated with MPT0E104 compared with the T2DM group
(Fig. 1A). In order to assess the
role of autophagy in T2DM cardiomyopathy, cardiac LC3-II protein
expression was measured and it was demonstrated that LC3-II levels
significantly decreased in HFD + STZ-induced T2DM hearts compared
with control hearts by 0.49 (P<0.05). Similar to control hearts,
MPT0E014-treated T2DM hearts had higher LC3-II levels compared with
HFD + STZ-induced T2DM hearts (P<0.001; Fig. 1B).
The role of Beclin-1 in DM cardiomyocytes was
examined and it was demonstrated that the level of Beclin-1 protein
was significantly lower in the hearts of HFD + STZ-induced T2DM
hearts compared with the control or T2DM hearts treated with
MPT0E014 (P<0.05; Fig. 1C). In
addition, the level of cleaved PARP1 in the T2DM cardiomyocytes was
demonstrated to be increased by 0.6 compared with the control
hearts; however, in the DM hearts treated with MPT0E014 the cleaved
PARP protein was significantly decreased compared with the
untreated DM hearts. (P<0.05; Fig.
1D).
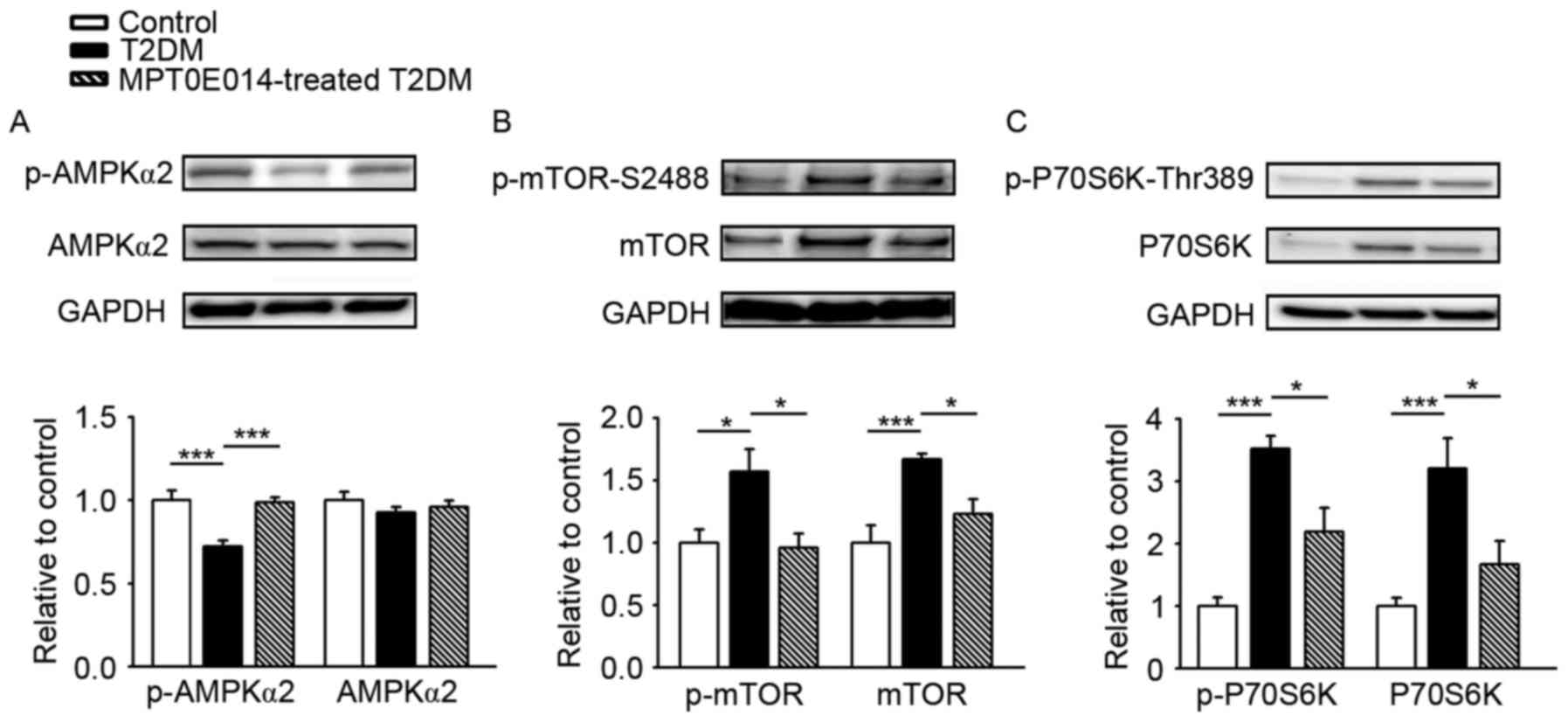
The regulatory role of AMPK in autophagy of the
heart was examined and it was demonstrated that the expression of
AMPKα2 was similar in the three groups. However, the p-AMPKα2
significantly decreased in the hearts of HFD + STZ-induced T2DM
hearts compared with the control hearts by 0.28, and this was
ameliorated in the hearts of T2DM rats treated with MPT0E014
(P<0.001; Fig. 2A). The effects
of MPT0E014 on the tuberin (TSC)-mTOR signaling pathway were
investigated. T2DM hearts exhibited activation of the TSC-mTOR
signaling pathway, as reflected by increased protein expression of
p-mTOR-S2448 (Fig. 2B) and its
downstream effector p-P70S6K-Thr389 (Fig. 2C), by 0.8 and 2.5, respectively,
compared with the control hearts; this effect was reversed in the
MPT0E014-treated HFD + STZ T2DM hearts.
Effects of MPT0E014 on the myocardial
insulin signaling pathway, RAGE and proinflammatory cytokines
As demonstrated in Fig.
3A, the protein expression of GLUT 4 was decreased in the HFD +
STZ-induced T2DM hearts compared with control hearts by 0.76.
Similar to the control hearts, hearts treated with MPT0E014 T2DM
had a higher level of GLUT 4 compared with the HFD + STZ-induced
T2DM hearts. In addition, the hearts of the HFD + STZ-induced T2DM
rats had decreased protein expression of p-Akt (Fig. 3B) and p-IRS-1 at Ser307 (Fig. 3C), compared with the control and
MPT0E014-treated T2DM hearts.
As demonstrated in Fig.
4, the hearts of the HFD + STZ-induced T2DM exhibited greater
protein expression of RAGE, TNF-α, and IL-6 compared with the
control and MPT0E014-treated T2DM hearts, while the control and
MPT0E014-treated T2DM hearts had similar expression of RAGE, TNF-α,
and IL-6.
Discussion
In the present study, it was demonstrated that HDAC
inhibition may restore myocardial autophagy and improve insulin
resistance in a T2DM rat model. T2DM is a progressive disorder that
is associated with insulin resistance and is correlated with
elevated, normal or low insulin levels depending on the stage when
pancreatic function is measured (23). The association between T2DM and
derangement in lipid metabolism, including elevated levels of
triglycerides and small low-density lipoprotein-cholesterol with
reduced HDL-C levels, have been reported (24,25).
An increase in plasma free fatty acids is common in patients with
T2DM (26) and this may contribute
to insulin resistance (27).
Similar to previous studies (19,28),
the rats in the present study fed with a HFD in a low-dose STZ T2DM
model exhibited hyperglycemia with increased levels of
triglycerides and free fatty acids, and decreased HDL-C levels
mimicking T2DM patients, except for a nonsignificant elevation of
cholesterol. In addition, a modest reduction in blood sugar and
reversed alterations in plasma triglyceride and HDL-C were
demonstrated following treatment with MPT0E104, suggesting
antihyperglycemic and hypolipidemic action.
Autophagy is a beneficial mechanism for preserving
homeostasis, and the growth and development of cells (29,30).
Previous studies have demonstrated that cardiac autophagy is
suppressed during metabolic derangement (31,32).
Beclin-1 and sequesteosome-1 (p62) are involved in the formation of
autophagosomes (29) and a
reduction in LC3-II levels with p62 accumulation in rodents with
HFD-induced obesity was identified to indicate a decrease in
autophagosome formation (33,34).
Similarly, the present study displayed a reduction in cardiac
autophagy in T2DM rats, as indicated by decreased protein
expression of LC3-II and Beclin-1. The reduction in cardiac
autophagy in the present study was accompanied by suppressed
myocardial phosphorylation of AMPKα2 and exacerbated cardiac
dysfunction. The inhibition of cardiac autophagy may have been
caused by AMPK dysregulation, which is involved in the pathogenesis
of DM cardiomyopathy. AMPK negatively regulates mTOR activity
(35) and serves an important role
in mediating starvation-induced autophagy (36). Likewise, it was demonstrated that
hyperglycemia increased the phosphorylation of mTOR, and activated
protein expression of the mTOR downstream effector P70S6K,
suggesting activation of the TSC-mTOR signaling pathway. In
addition, PARP1 cleavage, a hallmark of apoptosis, was increased in
the T2DM rat heart. In the present study, administration of the
HDAC inhibitor MPT0E014 activated AMPKα2, and enhanced cardiac
autophagic activity by modulating Beclin-1, LC3-II, TSC-mTOR and
decreasing cleaved PARP1 expression. The results of the present
study suggested that the restoration of autophagy following HDAC
inhibition may be a novel therapeutic mechanism of MPT0E014 against
DM cardiomyopathy.
RAGE accelerates vascular inflammation and cellular
stress to cause atherosclerosis (37). In the present study, upregulation
of the myocardial protein levels of RAGE was demonstrated, in
addition to TNF-α and IL-6 in T2DM rats. Additionally, it was
demonstrated that treatment with MPT0E104 attenuated the
upregulation of RAGE and pro-inflammatory cytokines in T2DM hearts.
To the best of our knowledge, this is the first study to
demonstrate the effect of HDAC inhibition on the expression of
cardiac RAGE in T2DM hearts.
Considering the important role of insulin resistance
in the development of T2DM in rats fed an HFD with low-dose STZ, a
reduction in the GLUT 4 protein has been implicated during insulin
resistance and impaired glucose metabolism (38). Although the results of the present
study demonstrated that MPT0E104 did not induce an increase in
plasma insulin concentrations, the total GLUT 4 protein expression
was significantly increased compared with the untreated hearts.
This demonstrated that MPT0E104 was able to restore the expression
of GLUT 4 protein through an insulin-independent pathway. Elevated
phosphorylation of Akt and IRS-1 has been reported to be essential
for the membrane translocation of GLUT 4 (39,40).
In the present study, MPT0E104-treated T2DM hearts were observed to
exhibit increased GLUT 4 protein in cardiomyocytes and restoration
of p-Akt and p-IRS-1 (Ser 307) protein levels. The results of the
present study suggested that enhanced insulin signaling
transduction may be responsible for improving insulin sensitivity
by HDAC inhibition. Although MPT0E014 was able to alter the
expression of cardiac GLUT 4 and the insulin signaling pathway, it
may be useful to investigate the direct effects of MPT0E014 by
measuring the utilization of carbohydrates.
In conclusion, HDAC inhibition improved myocardial
insulin sensitivity and attenuated diabetes-induced dysregulation
of cardiac autophagy. HDAC inhibition provides a novel scenario in
which autophagy reactivation may represent a potential therapeutic
target to reduce cardiac dysfunction in patients with DM
cardiomyopathy.
Acknowledgements
The present study was supported by grants from
Taipei Medical University, Wan Fang Hospital (grant nos.
104CGH-TMU-03, 104-wf-eva-03, 104swf07, 103TMU-SHH-23, 104swf02,
104-wf-eva-01 and 105-wf-eva-06) and the Ministry of Science and
Technology of Taiwan (grant nos. MOST103-2314-B-038-041-MY2,
MOST103-2314-B-281-005-MY2, MOST103-2314-B-281-006,
MOST103-2314-B-038-055, MOST104-2314-B-038-071-MY3,
MOST104-2314-B-038-073, MOST104-2314-B-038-032 and MOST
105-2314-B-038-026).
References
|
1
|
Simonson DC: Etiology and prevalence of
hypertension in diabetic patients. Diabetes Care. 11:821–827. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garcia MJ, McNamara PM, Gordon T and
Kannel WB: Morbidity and mortality in diabetics in the Framingham
population. Sixteen year follow-up study. Diabetes. 23:105–111.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haffner SM, Lehto S, Rönnemaa T, Pyörälä K
and Laakso M: Mortality from coronary heart disease in subjects
with type 2 diabetes and in nondiabetic subjects with and without
prior myocardial infarction. N Engl J Med. 339:229–234. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nishida K, Kyoi S, Yamaguchi O, Sadoshima
J and Otsu K: The role of autophagy in the heart. Cell Death
Differ. 16:31–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gustafsson AB and Gottlieb RA: Autophagy
in ischemic heart disease. Circ Res. 104:150–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Munasinghe PE, Riu F, Dixit P, Edamatsu M,
Saxena P, Hamer NS, Galvin IF, Bunton RW, Lequeux S, Jones G, et
al: Type-2 diabetes increases autophagy in the human heart through
promotion of Beclin-1 mediated pathway. Int J Cardiol. 202:13–20.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li ZL, Woollard JR, Ebrahimi B, Crane JA,
Jordan KL, Lerman A, Wang SM and Lerman LO: Transition from obesity
to metabolic syndrome is associated with altered myocardial
autophagy and apoptosis. Arterioscler Thromb Vasc Biol.
32:1132–1141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mellor KM, Bell JR, Young MJ, Ritchie RH
and Delbridge LM: Myocardial autophagy activation and suppressed
survival signaling is associated with insulin resistance in
fructose-fed mice. J Mol Cell Cardiol. 50:1035–1043. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sciarretta S, Volpe M and Sadoshima J:
Mammalian target of rapamycin signaling in cardiac physiology and
disease. Circ Res. 114:549–564. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kubli DA and Gustafsson AB: Cardiomyocyte
health: Adapting to metabolic changes through autophagy. Trends
Endocrinol Metab. 25:156–164. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Christensen DP, Dahllöf M, Lundh M,
Rasmussen DN, Nielsen MD, Billestrup N, Grunnet LG and
Mandrup-Poulsen T: Histone deacetylase (HDAC) inhibition as a novel
treatment for diabetes mellitus. Mol Med. 17:378–390. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sharma S and Taliyan R: Histone
deacetylase inhibitors: Future therapeutics for insulin resistance
and type 2 diabetes. Pharmacol Res. 113:320–326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kao YH, Liou JP, Chung CC, Lien GS, Kuo
CC, Chen SA and Chen YJ: Histone deacetylase inhibition improved
cardiac functions with direct antifibrotic activity in heart
failure. Int J Cardiol. 168:4178–4183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee TI, Kao YH, Tsai WC, Chung CC, Chen YC
and Chen YJ: HDAC inhibition modulates cardiac PPARs and fatty acid
metabolism in diabetic cardiomyopathy. PPAR Res. 2016:59387402016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barlovic DP, Soro-Paavonen A and
Jandeleit-Dahm KA: RAGE biology, atherosclerosis and diabetes. Clin
Sci (Lond). 121:43–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mansor LS, Gonzalez ER, Cole MA, Tyler DJ,
Beeson JH, Clarke K, Carr CA and Heather LC: Cardiac metabolism in
a new rat model of type 2 diabetes using high-fat diet with low
dose streptozotocin. Cardiovasc Diabetol. 12:1362013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee TI, Kao YH, Chen YC, Pan NH, Lin YK
and Chen YJ: Cardiac peroxisome-proliferator-activated receptor
expression in hypertension co-existing with diabetes. Clin Sci
(Lond). 121:305–312. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee TI, Chen YC, Kao YH, Hsiao FC, Lin YK
and Chen YJ: Rosiglitazone induces arrhythmogenesis in diabetic
hypertensive rats with calcium handling alteration. Int J Cardiol.
165:299–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lkhagva B, Lin YK, Kao YH, Chazo TF, Chung
CC, Chen SA and Chen YJ: Novel histone deacetylase inhibitor
modulates cardiac peroxisome proliferator-activated receptors and
inflammatory cytokines in heart failure. Pharmacology. 96:184–191.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cefalu WT: Animal models of type 2
diabetes: Clinical presentation and pathophysiological relevance to
the human condition. ILAR J. 47:186–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boden G, Lebed B, Schatz M, Homko C and
Lemieux S: Effects of acute changes of plasma free fatty acids on
intramyocellular fat content and insulin resistance in healthy
subjects. Diabetes. 50:1612–1617. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haffner SM: American Diabetes Association:
Management of dyslipidemia in adults with diabetes. Diabetes care.
26 Suppl 1:S83–S86. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reaven GM, Hollenbeck C, Jeng CY, Wu MS
and Chen YD: Measurement of plasma glucose, free fatty acid,
lactate, and insulin for 24 h in patients with NIDDM. Diabetes.
37:1020–1024. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shulman GI: Cellular mechanisms of insulin
resistance. J Clin Invest. 106:171–176. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Srinivasan K, Viswanad B, Asrat L, Kaul CL
and Ramarao P: Combination of high-fat diet-fed and low-dose
streptozotocin-treated rat: A model for type 2 diabetes and
pharmacological screening. Pharmacol Res. 52:313–320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh R and Cuervo AM: Autophagy in the
cellular energetic balance. Cell Metab. 13:495–504. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sciarretta S, Zhai P, Shao D, Maejima Y,
Robbins J, Volpe M, Condorelli G and Sadoshima J: Rheb is a
critical regulator of autophagy during myocardial ischemia:
Pathophysiological implications in obesity and metabolic syndrome.
Circulation. 125:1134–1146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He C, Zhu H, Li H, Zou MH and Xie Z:
Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances
cardiac autophagy and protects against cardiomyocyte apoptosis in
diabetes. Diabetes. 62:1270–1281. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo R, Zhang Y, Turdi S and Ren J:
Adiponectin knockout accentuates high fat diet-induced obesity and
cardiac dysfunction: Role of autophagy. Biochim Biophys Acta.
1832:1136–1148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu X and Ren J: Macrophage migration
inhibitory factor (MIF) knockout preserves cardiac homeostasis
through alleviating Akt-mediated myocardial autophagy suppression
in high-fat diet-induced obesity. Int J Obes (Lond). 39:387–396.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Inoki K, Zhu T and Guan KL: TSC2 mediates
cellular energy response to control cell growth and survival. Cell.
115:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lum JJ, DeBerardinis RJ and Thompson CB:
Autophagy in metazoans: Cell survival in the land of plenty. Nat
Rev Mol Cell Biol. 6:439–448. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun L, Ishida T, Yasuda T, Kojima Y, Honjo
T, Yamamoto Y, Yamamoto H, Ishibashi S, Hirata K and Hayashi Y:
RAGE mediates oxidized LDL-induced pro-inflammatory effects and
atherosclerosis in non-diabetic LDL receptor-deficient mice.
Cardiovasc Res. 82:371–381. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zisman A, Peroni OD, Abel ED, Michael MD,
Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF,
Virkamaki A, Goodyear LJ, et al: Targeted disruption of the glucose
transporter 4 selectively in muscle causes insulin resistance and
glucose intolerance. Nat Med. 6:924–928. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin HV, Ren H, Samuel VT, Lee HY, Lu TY,
Shulman GI and Accili D: Diabetes in mice with selective impairment
of insulin action in Glut4-expressing tissues. Diabetes.
60:700–709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chao KC, Chao KF, Fu YS and Liu SH:
Islet-like clusters derived from mesenchymal stem cells in
Wharton's Jelly of the human umbilical cord for transplantation to
control type 1 diabetes. PLoS One. 3:e14512008. View Article : Google Scholar : PubMed/NCBI
|