Introduction
The signal transducer and activator of transcription
3 (STAT3) has been demonstrated to be one of the regulators in
cardiac dysfunction (1). STAT3
possesses multiple functions, with its central role described as a
transcription factor. Moreover, STAT3 has been demonstrated to
function as a signaling molecule, as a factor involved in cellular
respiration, and as a protein interacting with the mitochondrial
pore (2–5). Therefore, in cardiomyocytes, STAT3
plays an important role in survival, growth, sarcomere
architecture, energetics, and metabolism (6–8).
Hyperglycemia is important in the pathogenesis of
diabetic disorders. Hyperglycemia was found to increase the STAT3
either through the gene expression or the phosphorylation (9). STAT3 is known as a cytoplasmic
transcription factor that transmits extracellular signals to the
nucleus (10). Activated STAT3 in
the nucleus binds to specific DNA promoter sequences to regulate
the gene expression (11). Recent
studies have indicated that hyperglycemia increases STAT3
activation, thereby contributing to the pathophysiology of tissue
injury (12). STAT3 activation,
increased phosphorylated STAT3 (p-STAT3) and p-STAT3 nuclear
translocation, are reportedly some of the underlining mechanisms of
STAT3 under high glucose condition. However, p-STAT3 was induced at
Y705 and S727 in cells for STAT3 activation by high glucose levels
(13). STAT3 has been demonstrated
to shuttle between the cytoplasm and nucleus independently of
tyrosine phosphorylation (14)
while unphosphorylated STAT3 in nucleus also can drive gene
expression (15).
Lipopolysaccharide (LPS) is mainly obtained from the
outer membrane of gram-negative bacteria, and the inflammatory
cytokines produced as a consequence of LPS exposure are implicated
in cardiac dysfunction (16,17).
The rapid activation of STAT3 by LPS through phosphorylation in
cardiomyocytes has been identified (18), and it is suggested as a direct
receptor-mediated activation (19). However, STAT3 activation by LPS in
hepatocytes is slower than in cardiomyocytes (20). Toll-like receptor 4 (TLR4) is known
as the binding site of LPS (21).
Activation of TLR4 by LPS has also been indicated to induce an
inflammatory response that decreases cardiomyocytes contractility
(22). Moreover, the
Janus-activated kinase 2 (JAK2) and the STAT3 pathway (JAK2/STAT3
pathway) is also coupled to the signaling of cytokine receptors
including TLR4 (23). Otherwise,
erythropoietin (EPO) is also produced effectiveness through
activation of the specific cell-surface receptor, erythropoietin
receptor (EPOR) (24). It has been
established that JAK2/STAT3 signaling pathway is also coupled to
EPOR (25). Interestingly, agent
improves left ventricular performance via activation of JAK2/STAT3
pathway in rats (26). Therefore,
we included the effects of EPO in this study, because EPO produced
actions also through an activation of receptors, EPOR, which is
similar to the action of LPS (27).
Additionally, STAT3 is introduced to involve in
cardiac fibrosis of diabetes (28), while high glucose increased STAT3
activated by angiotensin II has been demonstrated to be produced
mainly through a reactive oxygen species (ROS)-dependent mechanism
(29). Recently, the ROS-activated
STAT3 pathway has been characterized in early reperfusion of heart
(30). High glucose is known as
the main factor for inducing diabetes-associated cardiovascular
dysfunctions. It seems that activation of STAT3 through
phosphorylation by hyperglycemia differs from the promotion of
STAT3 via receptor-coupled signaling in the regulation of cardiac
function. However, variations in the phosphorylation of STAT3
between high glucose-induced change and promotion by
receptor-coupled signaling remained unclear in cardiomyocytes.
In the present study, we focused on STAT3
phosphorylation that is important in regulation of cardiac
function. Also, we are interested to know the difference whether
STAT3 phosphorylation induced by receptor signaling is varied with
that induced by pathologic disorders such as hyperglycemia.
Therefore, we used the embryonic rat cardiomyoblast cell line H9c2
which offers the advantage of being an animal-free alternative
(31). Moreover, H9c2 expressed
TLR4 (32) and EPOR (33). Therefore, it is suitable to apply
in the present study.
Additionally, we link it to signals-associated
fibrosis, including connective tissue growth factor (CTGF) and
metalloproteinase (MMP)-9, to determine its association in cardiac
disorders.
Materials and methods
Cell cultures
It has been confirmed that H9c2 cells possess the
advantage of being an animal-free alternative (31). The H9C2 cells (BCRC, no. 60096)
were cultured according to a previous method (34). In brief, H9c2 cells were maintained
in Dulbecco's modified Eagle's medium (pH 7.2) supplemented with
10% fetal bovine serum. The H9c2 cells were plated at a density of
6,000 cells/cm2 and allowed to proliferate in growth
medium. The medium was changed every 48 h.
Drug treatment
The cultured H9c2 cells were treated at indicated
times with Salmonella typhosa LPS (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany), as described previously (18). The stock solution of EPO containing
epoetin beta (Recormon, 5,000 IU/0.3 ml), purchased from Roche
Diagnostics (Mannheim, Germany), was diluted in culture medium. A
fresh solution diluted to the indicated dose was applied to treat
the H9c2 cells. Incubation of hyperglycemia with H9c2 cells was
also performed according to our previous report (35).
Western blot analysis
Protein was extracted and separated by SDS-PAGE,
following our previous method (27). Proteins were detected using
antibodies (1:1,000) against p-STAT3, STAT3, CTGF and MMP-9, while
antibody against β-actin serving as the internal control. After
comparing with the marker, the immunoblots of TLR4 (95 kDa), EPOR
(55 kDa), p-JAK2 (130 kDa), JAK2 (130 kDa), p-STAT3 (88 kDa), STAT3
(88 kDa), CTGF (38 kDa), MMP-9 (92 kDa) and β-actin (43 kDa) were
then quantified.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cell lysates with
TRIzol (Qiagen, Hilden, Germany). Two microgram of total RNA was
used for the reverse transcription reaction, along with
Superscriptase II (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), oligo-dT, and random primers. Web-based
assay-design software from the Universal Probe Library Assay Design
Center (http://www.roche-applied-science.com/sis/rtpcr/upl/adc.jsp)
was utilized to design TaqMan primer pairs and to select
appropriate hybridization probes (Table I). For quantification, real-time
PCR analysis was performed using Light Cycler 480 SYBR-Green I
Master on a Light Cycler 480 II (Roche Diagnostics). The relative
fold changes were quantified using the comparative threshold cycle
method, and β-actin was used as a control, according to our
previous reports (36,37).
 | Table I.Primers used for targets
amplification in this study. |
Table I.
Primers used for targets
amplification in this study.
| Target | Primer | Sequence
(5′-3′) |
|---|
| STAT3 | F |
5′-GGCTTCAGCCCCAGAGAC-3′ |
|
| R |
5′-CTCCAGGTAGCGCGTGTC-3′ |
| CTGF | F |
5′-ATGCTGTGAGGAGTGGGTGT-3′ |
|
| R |
5′-GGCCAAATGTGTCTTCCAGT-3′ |
| MMP-9 | F |
5′-TCGTGGCTCTAAACCTGACC-3′ |
|
| R |
5′-GAGCTGTCGGCTGTGGTT-3′ |
| β-actin | F |
5′-CTCTCTTCCAGCCTTCCTTC-3′ |
|
| R |
5′-GGTCTTTACGGATGTCAACG-3′ |
Statistical analysis
Data were indicated as the mean ± standard error of
the mean (SEM) from the sample number (n) of each group. The
differences between two groups were analyzed using a Student's
two-sided t-test. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of LPS on the phosphorylation
of STAT3 in H9c2 cells
Incubation of LPS dose-dependently induced a marked
elevation of STAT3 phosphorylation within 30 min in H9c2 cells, as
shown in Fig. 1A. The results were
obtained from TLR4 through phosphorylated JAK2 (p-JAK2) to STAT3
phosphorylation (Fig. 1A and B).
However, the expression of STAT3 was still not modified in terms of
both protein and mRNA levels (Fig.
1C). Consequently, the downstream signals both CTGF and MMP-9
were also not activated by LPS in this condition (Fig. 1A and B).
 | Figure 1.Effects of LPS on the changes in
expression of STAT3 and associated signals in H9c2 cells. (A) The
representative changes in signals in addition to the p-STAT3,
STAT3, CTGF and MMP-9 expressions by LPS at indicated concentration
in western blots. (B) The protein levels, using phosphorylated
signal over the original one or each signal over β-actin (Actin),
are indicated as mean ± SEM (n=6 per group) in each column.
*P<0.05 and **P<0.01 compared to the vehicle-treated control
shown at 0 concentration. (C) Related mRNA expression as detected
using RT-PCR and the quantified mRNA STAT3 level is represented as
mean ± SEM (n=6 per group). LPS, lipopolysaccharide; STAT3, signal
transducer and activator of transcription 3; JAK2, Janus-activated
kinase 2; p-, phosphorylated; CTGF, connective tissue growth
factor; MMP-9, matrix metalloproteinase-9; SEM, standard error of
the mean. |
Effects of EPO on the phosphorylation
of STAT3 in H9c2 cells
The same incubation of EPO with H9c2 cells also
produced a similar change in STAT3 phosphorylation, as shown in
Fig. 2A, except that the EPOR was
activated by EPO. Additionally, the expression of STAT3 was also
not changed by EPO in terms of both protein and mRNA levels
(Fig. 2B and C). Similarly, the
consequent downstream signals, both CTGF and MMP-9, were also not
activated by EPO in this condition.
 | Figure 2.Effects of EPO on the changes in
expression of STAT3 and associated signals in H9c2 cells. (A) The
representative changes in receptor signals in addition to the
p-STAT3, STAT3, CTGF and MMP-9 expressions by EPO at indicated
concentration in western blots. (B) The protein levels, using
phosphorylated signal over the original one or each signal over
β-actin (Actin), are indicated as mean ± SEM (n=6 per group) in
each column. *P<0.05 and **P<0.01 compared to the
vehicle-treated control shown at 0 concentration. (C) Related mRNA
expression as detected using RT-PCR and the quantified mRNA STAT3
level is represented as mean ± SEM (n=6 per group). EPO.
erythropoietin; STAT3, signal transducer and activator of
transcription 3; p-, phosphorylated; CTGF, connective tissue growth
factor; MMP-9, matrix metalloproteinase-9; SEM, standard error of
the mean; EPOR; erythropoietin receptor; JAK2, Janus-activated
kinase 2. |
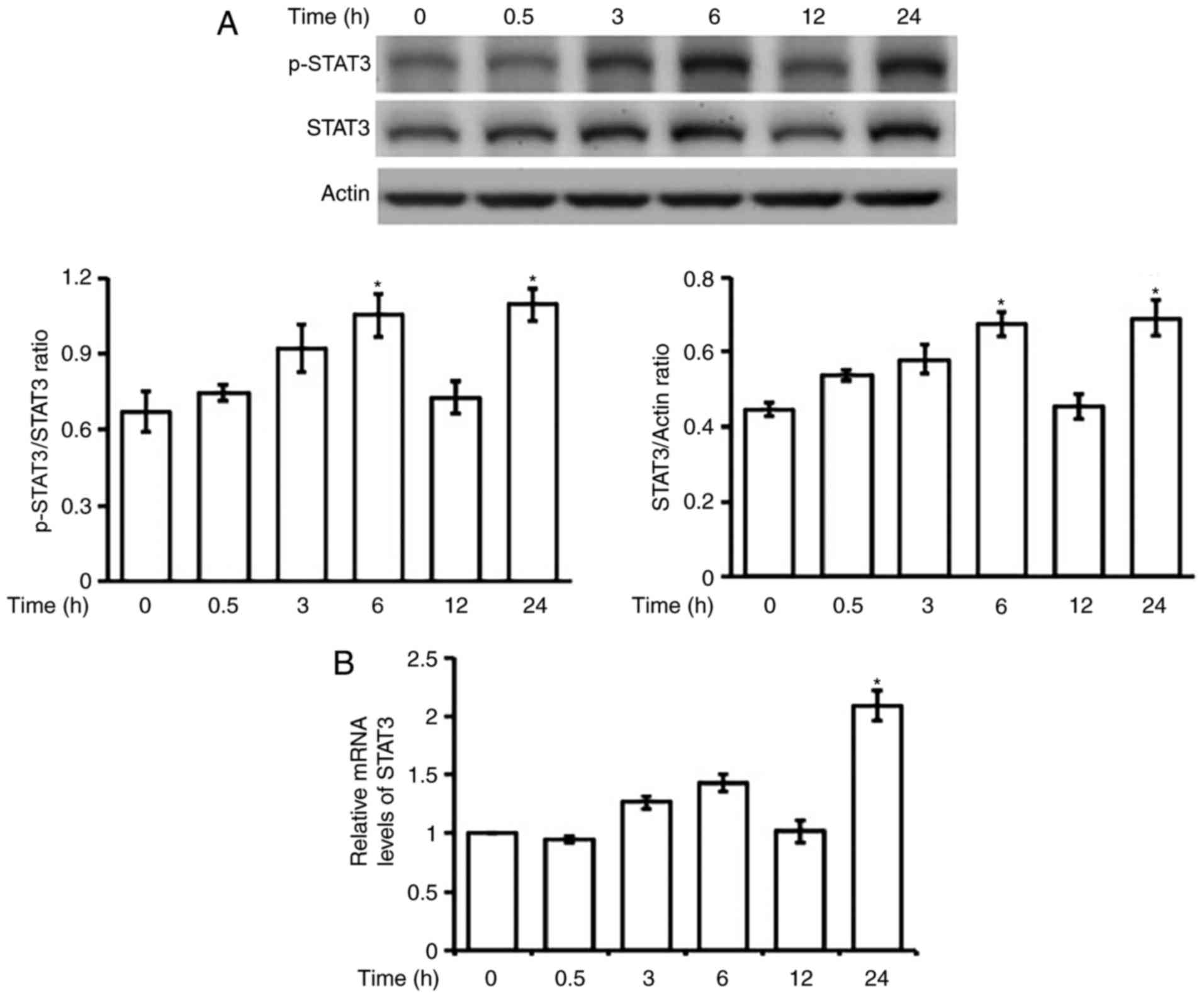
Effect of hyperglycemia on the
phosphorylation of STAT3 in H9c2 cells
Incubation of high glucose (30 mM) with H9c2 cells
at the time same as LPS failed to induce changes in STAT3
phosphorylation. Therefore, we incubated H9c2 cells for a longer
time with high glucose in the medium. As shown in Fig. 3A, changes in STAT3 phosphorylation
were not stable except at 24 h post-incubation. Both protein and
mRNA levels of STAT3 were also markedly elevated after 24-h
incubation with high glucose (Fig.
3B).
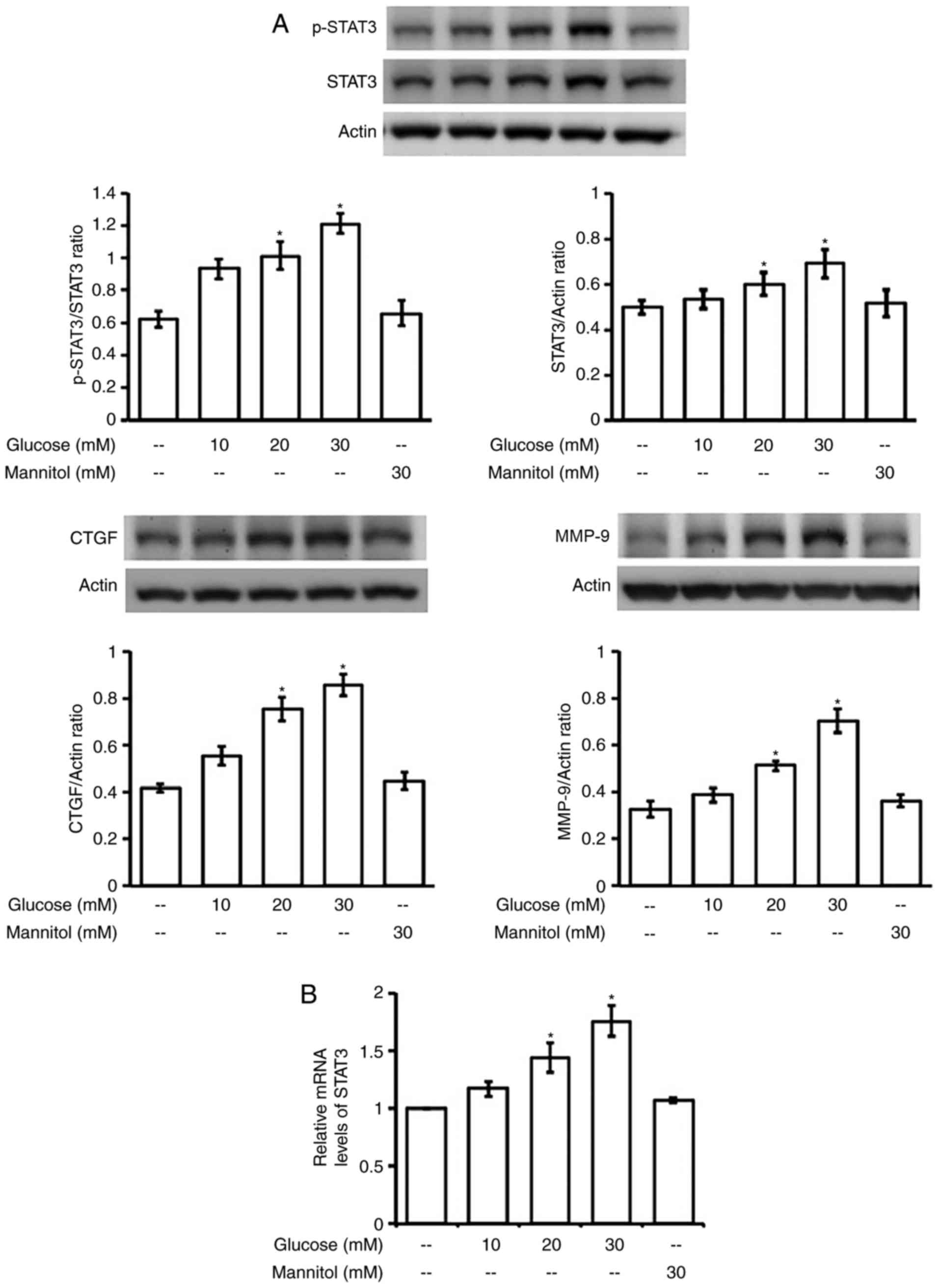
Additionally, increase of STAT3 expression, both in
terms of protein (Fig. 4A) and
mRNA (Fig. 4B) levels, was
produced in a dose-dependent manner by hyperglycemia after 24-h
incubation. However, changes were not observed in H9C2 cells that
received similar incubation with manitol (30 mM), which produced
the same osmolarity as high glucose (30 mM), as described in our
previous report (35). Thus, the
possible influence of osmolarity in the changes of STAT3 expression
can be excluded.
Moreover, the downstream signals for fibrosis,
including CTGF and MMP-9, were also enhanced by hyperglycemia in
the same dose-dependent fashion (Fig.
4A). This change was also not related to osmolarity as shown in
manitol-treated cells. However, fibrosis-related signals were not
modified in H9c2 cells treated with LPS or EPO at the effective
dose in above.
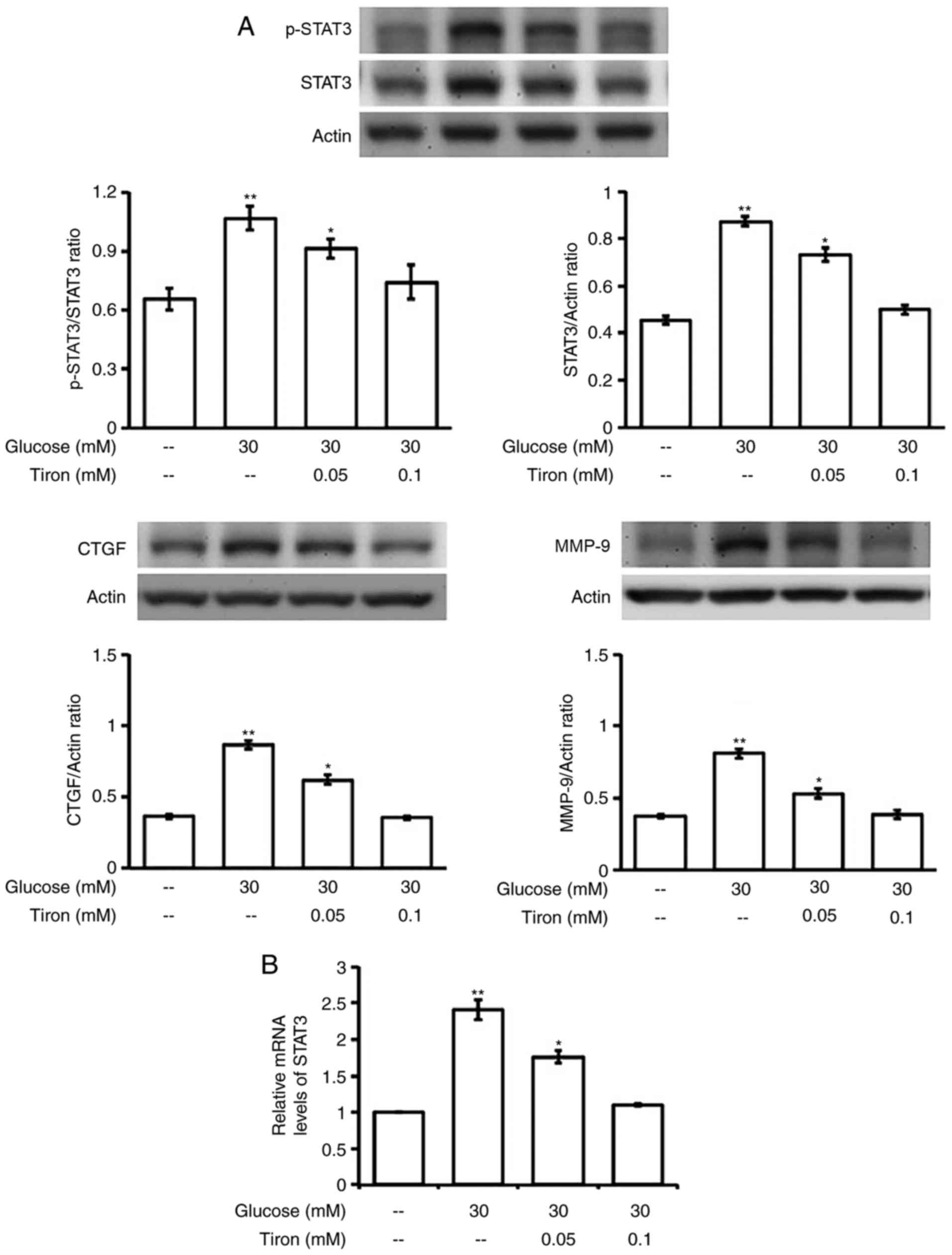
Mediation of oxidative stress in
hyperglycemia increased expressions of STAT3 in H9c2 cells
We applied the antioxidant, tiron, to examine the
role of oxidative stress in the changes of STAT3 expression by
hyperglycemia. As shown in Fig.
5A, tiron inhibits the elevation of STAT3 activation in a
dose-related manner. Moreover, the promotion in mRNA level of STAT3
by hyperglycemia was also reduced by tiron in the same fashion
(Fig. 5B). Consequently,
fibrosis-associated signals, including CTGF and MMP-9, elevated by
hyperglycemia were markedly reduced by tiron in the same manner
(Fig. 4A). Mediation of oxidative
stress can thus be confirmed.
Discussion
In the present study, we demonstrated that STAT3
phosphorylation occurred within 30 min after exposure to LPS in
H9c2 cells. Additionally, the dose-dependent effect of LPS was
produced through TLR4 to link the p-JAK2 for STAT3 phosphorylation;
it is fully consistent with the previous reports (22,26).
However, LPS did not influence the expressions of STAT3 and the
downstream signals, including CTGF and MMP-9. Similar results were
also obtained in EPO-treated H9c2 cells except that EPOR was
involved in the STAT3 phosphorylation by EPO (24). Otherwise, high glucose increased
STAT3 phosphorylation after a longer time, particularly 24 h
post-incubation. Additionally, expression of STAT3 was also
augmented by high glucose in a dose-dependent manner after 24-h
incubation. In parallel, fibrosis-related signals, including CTGF
and MMP-9, were both elevated. Therefore, STAT3 phosphorylation
induced by LPS or EPO is quite different from that by
hyperglycemia. The possible reason might be due to the treatment of
H9c2 with LPS or EPO may stimulatee cardiac mitochondrial function
through a highly regulated, receptor-mediated, eNOS/Akt1 and
JAK-STAT-dependent cascade that activates the transcriptional
program of mitochondrial biogenesis in a short time (38–40).
However, hyperglycemia-induced oxidative stress attenuated the
mitochondrial function and affected the proliferation and survival
of cells. Increased ROS in diabetes is implicated in the
development of diabetic cardiomyopathy (41). To the best of our knowledge, the
present study is the first to conduct this finding.
Inflammatory cytokines produced as a consequence of
LPS exposure are implicated in myocardial dysfunction (17,42).
Paradoxically, sub-lethal doses of LPS provided cardioprotective
effects against ischemia-reperfusion injury (43). In addition, STAT3 is demonstrated
to be a key modulator of an integrated signaling network in the
heart (1). Increase of STAT3
phosphorylation by LPS in a CD14-independent manner has been
indicated in cardiomyocytes (18).
Moreover, EPO is known to protect heart from chemical damage
(44) and to improve experimental
heart failure (45). In the
present study, we observed that STAT3 phosphorylation is raised by
EPO in a way that is similar to LPS in H9c2 cells. Interestingly,
both effects were induced by activation of each specific receptors,
LPS via TLR4 (21) and EPO through
specific receptor EPOR (25).
Moreover, both effects on STAT3 phosphorylation were produced
through p-JAK2, as described previously (23). Therefore, a rapid increase of
p-STAT3 seems helpful in protection against cardiac damage. This
view is consistent to previous reports that demonstrated the
cardio-protective effects of propofol (46) and morphine (47) through an increase in STAT3
phosphorylation.
In contrast, as shown in Fig. 3, STAT3 phosphorylation was not
immediately increased by high glucose, but rather changed in
unstable way. After a longer time of incubation with H9c2 cells,
approximately 24 h later, STAT3 phosphorylation was stable and
markedly increased in high glucose medium. Moreover, expression of
STAT3 was also promoted after the same incubation. Basically,
phosphorylation is known as the major way for the activation of
STAT. Moreover, p-STAT3 has been shown to enter the nucleus easily
(48). However, nuclear
accumulation of STAT3 without phosporylation has also been
demonstrated (49), and the
unphosphorylated STAT3 in nucleus may activate gene expression both
in cancer and in responses to cytokines (15). Therefore, STAT3 is effective to
promote transcription in cardiac cells that have been characterized
in this study. We demonstrated that fibrosis-related signals,
including CTGF and MMP-9, were both enhanced in high glucose medium
with the increased STAT3. This novel view is useful to explain the
role of cardiac fibrosis in diabetes (50,51).
Cardiac fibrosis is a prominent component of
diabetic cardiomyopathy (52,53).
High glucose has been demonstrated to promote fibrosis in
vitro, including changes in MMP activity (28). Hyperglycemia may sustain the
progression of heart failure through excessive interstitial
myocardial collagen accumulation, thus leading to impaired
diastolic and systolic function (54). STAT3 is also shown to mediate the
proliferation of cardiac fibroblasts and collagen synthesis induced
by high glucose (55). Although
STAT3 may participate in the transcription of target genes in
ischemia/reperfusion injury (54)
and pressure overload hypertrophy (55), the role of STAT3 in cardiac
fibrosis induced by hyperglycemia is critical.
The pathogenesis of cardiovascular diseases almost
invariably involves, the occurrence of oxidative stress (56), a major cause of progressive
cellular sufferance and death. Moreover, diabetic cardiomyopathy is
a condition in which oxidative stress seems to play a major
pathogenic role (57). Therefore,
we focused on the role of oxidative stress in the changes of STAT3
induced by hyperglycemia in H9c2 cells. In the presence of the
antioxidant, tiron (58), increase
of STAT3 by high glucose was markedly reduced in H9c2 cells.
Additionally, the augmented fibrosis-related signals, including
CTGF and MMP-9, were also attenuated in parallel. Different to the
effects induced by stimulation of receptors, such as LPS and EPO,
posphorylation of STAT3 by hyperglycemia needs a longer time in
incubation. It seems that enough oxidative stress induced by
hyperglycemia needs a time to accumulate and STAT3 phosphorylation
via oxidative stress is varied with that rapidly induced via the
p-JAK2. Taken together, we found that expression of STAT3 increased
by hyperglycemia is mainly through oxidative stress to promote the
expressions of fibrosis-related signals, including CTGF and MMP-9,
in H9c2 cells. This mechanism is quite different with that induced
by receptor activation, both LPS and EPO.
In conclusion, we identified that STAT3
phosphorylation is rapidly raised by LPS or EPO via
receptor-mediated signaling, but different from high glucose, in
H9c2 cells. Additionally, STAT3 increased by hyperglycemia needs a
longer time because it is mainly through an accumulation of
oxidative stress which is effective to promote the transcription of
downstream signals for fibrosis, including CTGF and MMP-9, in H9c2
cells. Therefore, we suggest that phosphorylation of STAT3 seems
suitable for rapidly identification of receptor-mediated signaling
while the nuclear STAT3 is more reliable in cardiac cells receiving
hyperglycemic stress. Furthermore, nuclear STAT3 may be a potential
clinical indicator of cardiac fibrosis and heart dysfunction. The
developments of new drugs that not only prevent myofibroblast
formation but also alleviate the hyperglycemia-induced STAT3
phosphorylation may be useful to prevent cardiac dysfunction.
Acknowledgements
We thank Y.C. Chen for assistance with the
experiments, and we acknowledge Jake Carpenter for editing. The
present study was partially supported by a grant from the Chi-Mei
Medical Center-Liouying (CLFHR10407), Tainan, Taiwan, R.O.C.
References
|
1
|
Haghikia A, Ricke-Hoch M, Stapel B, Gorst
I and Hilfiker-Kleiner D: STAT3, a key regulator of cell-to-cell
communication in the heart. Cardiovasc Res. 102:281–289. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boengler K, Hilfiker-Kleiner D, Heusch G
and Schulz R: Inhibition of permeability transition pore opening by
mitochondrial STAT3 and its role in myocardial
ischemia/reperfusion. Basic Res Cardiol. 105:771–785. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elschami M, Scherr M, Philippens B and
Gerardy-Schahn R: Reduction of STAT3 expression induces
mitochondrial dysfunction and autophagy in cardiac HL-1 cells. Eur
J Cell Biol. 92:21–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heusch G, Musiolik J, Gedik N and
Skyschally A: Mitochondrial STAT3 activation and cardioprotection
by ischemic postconditioning in pigs with regional myocardial
ischemia/reperfusion. Circ Res. 109:1302–1308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wegrzyn J, Potla R, Chwae YJ, Sepuri NB,
Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et
al: Function of mitochondrial Stat3 in cellular respiration.
Science. 323:793–797. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haghikia A, Stapel B, Hoch M and
Hilfiker-Kleiner D: STAT3 and cardiac remodeling. Heart Fail Rev.
16:35–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hilfiker-Kleiner D, Hilfiker A, Fuchs M,
Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z,
Podewski E, et al: Signal transducer and activator of transcription
3 is required for myocardial capillary growth, control of
interstitial matrix deposition and heart protection from ischemic
injury. Circ Res. 95:187–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zouein FA, Kurdi M and Booz GW: Dancing
rhinos in stilettos: The amazing saga of the genomic and nongenomic
actions of STAT3 in the heart. JAKSTAT. 2:e243522013.PubMed/NCBI
|
|
9
|
Wang S, Li B, Li C, Cui W and Miao L:
Potential renoprotective agents through inhibiting CTGF/CCN2 in
diabetic nephropathy. J Diabetes Res. 2015:9623832015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miller AM, Wang H, Bertola A, Park O,
Horiguchi N, Ki SH, Yin S, Lafdil F and Gao B:
Inflammation-associated interleukin-6/signal transducer and
activator of transcription 3 activation ameliorates alcoholic and
nonalcoholic fatty liver diseases in interleukin-10-deficient mice.
Hepatology. 54:846–856. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jung JE, Lee HG, Cho IH, Chung DH, Yoon
SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK and Chung MH: STAT3 is
a potential modulator of HIF-1-mediated VEGF expression in human
renal carcinoma cells. FASEB J. 19:1296–1298. 2005.PubMed/NCBI
|
|
12
|
Chen Y, Wang JJ, Li J, Hosoya KI, Ratan R,
Townes T and Zhang SX: Activating transcription factor 4 mediates
hyperglycaemia-induced endothelial inflammation and retinal
vascular leakage through activation of STAT3 in a mouse model of
type 1 diabetes. Diabetologia. 55:2533–2545. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saengboonmee C, Seubwai W, Pairojkul C and
Wongkham S: High glucose enhances progression of cholangiocarcinoma
cells via STAT3 activation. Sci Rep. 6:189952016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu L, McBride KM and Reich NC: STAT3
nuclear import is independent of tyrosine phosphorylation and
mediated by importin-alpha3. Proc Natl Acad Sci USA. 102:8150–8155.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang J, Chatterjee-Kishore M, Staugaitis
SM, Nguyen H, Schlessinger K, Levy DE and Stark GR: Novel roles of
unphosphorylated STAT3 in oncogenesis and transcriptional
regulation. Cancer Res. 65:939–947. 2005.PubMed/NCBI
|
|
16
|
Meldrum DR: Tumor necrosis factor in the
heart. Am J Physiol. 274:R577–R595. 1998.PubMed/NCBI
|
|
17
|
Wagner DR, McTiernan C, Sanders VJ and
Feldman AM: Adenosine inhibits lipopolysaccharide-induced secretion
of tumor necrosis factor-alpha in the failing human heart.
Circulation. 97:521–524. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cowan DB, Poutias DN, Del Nido PJ and
McGowan FX Jr: CD14-independent activation of cardiomyocyte signal
transduction by bacterial endotoxin. Am J Physiol Heart Circ
Physiol. 279:H619–H629. 2000.PubMed/NCBI
|
|
19
|
Darnell JE Jr: STATs and gene regulation.
Science. 277:1630–1635. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruff-Jamison S, Zhong Z, Wen Z, Chen K,
Darnell JE Jr and Cohen S: Epidermal growth factor and
lipopolysaccharide activate Stat3 transcription factor in mouse
liver. J Biol Chem. 269:21933–21935. 1994.PubMed/NCBI
|
|
21
|
Chow JC, Young DW, Golenbock DT, Christ WJ
and Gusovsky F: Toll-like receptor-4 mediates
lipopolysaccharide-induced signal transduction. J Biol Chem.
274:10689–10692. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Avlas O, Fallach R, Shainberg A, Porat E
and Hochhauser E: Toll-like receptor 4 stimulation initiates an
inflammatory response that decreases cardiomyocyte contractility.
Antioxid Redox Signal. 15:1895–1909. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Seavey MM and Dobrzanski P: The many faces
of Janus kinase. Biochem Pharmacol. 83:1136–1145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao L, Li Z, Xu P, Li Z, Xu J and Yang Z:
The expression of EPOR in renal cortex during postnatal
development. PloS one. 7:e419932012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu X, Shacka JJ, Eells JB, Suarez-Quian C,
Przygodzki RM, Beleslin-Cokic B, Lin CS, Nikodem VM, Hempstead B,
Flanders KC, et al: Erythropoietin receptor signalling is required
for normal brain development. Development. 129:505–516.
2002.PubMed/NCBI
|
|
26
|
Qiao S, Mao X, Wang Y, Lei S, Liu Y, Wang
T, Wong GT, Cheung CW, Xia Z, Irwin MG, et al: Remifentanil
preconditioning reduces postischemic myocardial infarction and
improves left ventricular performance via activation of the janus
activated kinase-2/signal transducers and activators of
transcription-3 signal pathway and subsequent inhibition of
glycogen synthase kinase-3beta in rats. Crit Care Med.
44:e131–e145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fenton MJ and Golenbock DT: LPS-binding
proteins and receptors. J Leukoc Biol. 64:25–32. 1998.PubMed/NCBI
|
|
28
|
Dai B, Cui M, Zhu M, Su WL, Qiu MC and
Zhang H: STAT1/3 and ERK1/2 synergistically regulate cardiac
fibrosis induced by high glucose. J Leukoc Biol. 32:960–971.
2013.
|
|
29
|
Fiaschi T, Magherini F, Gamberi T,
Lucchese G, Faggian G, Modesti A and Modesti PA: Hyperglycemia and
angiotensin II cooperate to enhance collagen I deposition by
cardiac fibroblasts through a ROS-STAT3-dependent mechanism.
Biochim Biophys Acta. 1843:2603–2610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu L, Tan JL, Wang ZH, Chen YX, Gao L, Liu
JL, Shi YH, Endoh M and Yang HT: ROS generated during early
reperfusion contribute to intermittent hypobaric hypoxia-afforded
cardioprotection against postischemia-induced Ca(2+) overload and
contractile dysfunction via the JAK2/STAT3 pathway. J Mol Cell
Cardiol. 81:150–161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Watkins SJ, Borthwick GM and Arthur HM:
The H9C2 cell line and primary neonatal cardiomyocyte cells show
similar hypertrophic responses in vitro. In Vitro Cell Dev Biol
Anim. 47:125–131. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fan MJ, Huang-Liu R, Shen CY, Ju DT, Lin
YM, Pai P, Huang PY, Ho TJ, Tsai FJ, Tsai CH and Huang CY:
Reduction of TLR4 mRNA stability and protein expressions through
inhibiting cytoplasmic translocation of HuR transcription factor by
E2 and/or ERα in LPS-treated H9c2 cardiomyoblast cells.
Chin J Physiol. 57:8–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Parvin A, Pranap R, Shalini U, Devendran
A, Baker JE and Dhanasekaran A: Erythropoietin protects
cardiomyocytes from cell death during hypoxia/reperfusion injury
through activation of survival signaling pathways. PLoS One.
9:e1074532014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang L, Luo C, Chen C, Wang X, Shi W and
Liu J: All-trans retinoic acid protects against doxorubicin-induced
cardiotoxicity by activating the ERK2 signalling pathway. Br J
Pharmacol. 173:357–371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mar GY, Ku PM, Chen LJ, Cheng KC, Li YX
and Cheng JT: Increase in cardiac M2-muscarinic receptor expression
is regulated by GATA binding protein 4 (GATA-4) in
streptozotocin-induced diabetic rats. Int J Cardiol. 167:436–441.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheng YZ, Chen LJ, Lee WJ, Chen MF, Jung
Lin H and Cheng JT: Increase of myocardial performance by
rhodiola-ethanol extract in diabetic rats. J Ethnopharmacol.
144:234–239. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cheng JT, Yu BC and Tong YC: Changes of
M3-muscarinic receptor protein and mRNA expressions in the bladder
urothelium and muscle layer of streptozotocin-induced diabetic
rats. Neurosci Lett. 423:1–5. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Carraway MS, Suliman HB, Jones WS, Chen
CW, Babiker A and Piantadosi CA: Erythropoietin activates
mitochondrial biogenesis and couples red cell mass to mitochondrial
mass in the heart. Circ Res. 106:1722–1730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mudalagiri NR, Mocanu MM, Di Salvo C,
Kolvekar S, Hayward M, Yap J, Keogh B and Yellon DM: Erythropoietin
protects the human myocardium against hypoxia/reoxygenation injury
via phosphatidylinositol-3 kinase and ERK1/2 activation. Br J
Pharmacol. 153:50–56. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cimolai MC, Alvarez S, Bode C and Bugger
H: Mitochondrial mechanisms in septic cardiomyopathy. Int J Mol
Sci. 16:17763–17778. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Desco MC, Asensi M, Márquez R,
Martínez-Valls J, Vento M, Pallardó FV, Sastre J and Viña J:
Xanthine oxidase is involved in free radical production in type 1
diabetes: Protection by allopurinol. Diabetes. 51:1118–1124. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
MacKichan ML and DeFranco AL: Role of
ceramide in lipopolysaccharide (LPS)-induced signaling. LPS
increases ceramide rather than acting as a structural homolog. J
Biol Chem. 274:1767–1775. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng X, Ao L, Brown JM, Meldrum DR,
Sheridan BC, Cain BS, Banerjee A and Harken AH: LPS induces late
cardiac functional protection against ischemia independent of
cardiac and circulating TNF-alpha. Am J Physiol. 273:H1894–H1902.
1997.PubMed/NCBI
|
|
44
|
Li L, Takemura G, Li Y, Miyata S, Esaki M,
Okada H, Kanamori H, Khai NC, Maruyama R, Ogino A, et al:
Preventive effect of erythropoietin on cardiac dysfunction in
doxorubicin-induced cardiomyopathy. Circulation. 113:535–543. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lipsic E, Westenbrink BD, van der Meer P,
van der Harst P, Voors AA, van Veldhuisen DJ, Schoemaker RG and van
Gilst WH: Low-dose erythropoietin improves cardiac function in
experimental heart failure without increasing haematocrit. Eur J
Heart Fail. 10:22–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shravah J, Wang B, Pavlovic M, Kumar U,
Chen DD, Luo H and Ansley DM: Propofol mediates signal transducer
and activator of transcription 3 activation and crosstalk with
phosphoinositide 3-kinase/AKT. JAKSTAT. 3:e295542014.PubMed/NCBI
|
|
47
|
Gross ER, Hsu AK and Gross GJ: The
JAK/STAT pathway is essential for opioid-induced cardioprotection:
JAK2 as a mediator of STAT3, Akt and GSK-3 beta. Am J Physiol Heart
Circ Physiol. 291:H827–H834. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gu JJ, Montealegre ZF, Robert D, Engel MS,
Qiao GX and Ren D: Wing stridulation in a Jurassic katydid
(Insecta, Orthoptera) produced low-pitched musical calls to attract
females. Proc Natl Acad Sci USA. 109:3868–3873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Asbun J, Manso AM and Villarreal FJ:
Profibrotic influence of high glucose concentration on cardiac
fibroblast functions: Effects of losartan and vitamin E. Am J
Physiol Heart Circ Physiol. 288:H227–H234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Suskin N, McKelvie RS, Burns RJ, Latini R,
Pericak D, Probstfield J, Rouleau JL, Sigouin C, Solymoss CB,
Tsuyuki R, et al: Glucose and insulin abnormalities relate to
functional capacity in patients with congestive heart failure. Eur
Heart J. 21:1368–1375. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ather S, Chan W, Bozkurt B, Aguilar D,
Ramasubbu K, Zachariah AA, Wehrens XH and Deswal A: Impact of
noncardiac comorbidities on morbidity and mortality in a
predominantly male population with heart failure and preserved
versus reduced ejection fraction. J Am Coll Cardiol. 59:998–1005.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Paulus WJ and Tschöpe C: A novel paradigm
for heart failure with preserved ejection fraction: Comorbidities
drive myocardial dysfunction and remodeling through coronary
microvascular endothelial inflammation. J Am Coll Cardiol.
62:263–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ma H, Gong H, Chen Z, Liang Y, Yuan J,
Zhang G, Wu J, Ye Y, Yang C, Nakai A, et al: Association of Stat3
with HSF1 plays a critical role in G-CSF-induced cardio-protection
against ischemia/reperfusion injury. J Mol Cell Cardiol.
52:1282–1290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Butler KL, Huffman LC, Koch SE, Hahn HS
and Gwathmey JK: STAT-3 activation is necessary for ischemic
preconditioning in hypertrophied myocardium. Am J Physiol Heart
Circ Physiol. 291:H797–H803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Roul D and Recchia FA: Metabolic
alterations induce oxidative stress in diabetic and failing hearts:
Different pathways, same outcome. Antioxid Redox Signal.
22:1502–1514. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Varga ZV, Giricz Z, Liaudet L, Hasko G,
Ferdinandy P and Pacher P: Interplay of oxidative,
nitrosative/nitrative stress, inflammation, cell death and
autophagy in diabetic cardiomyopathy. Biochim Biophys Acta.
1852:232–242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Monticone M, Taherian R, Stigliani S,
Carra E, Monteghirfo S, Longo L, Daga A, Dono M, Zupo S, Giaretti W
and Castagnola P: NAC, tiron and trolox impair survival of cell
cultures containing glioblastoma tumorigenic initiating cells by
inhibition of cell cycle progression. PLoS One. 9:e900852014.
View Article : Google Scholar : PubMed/NCBI
|