Introduction
The vascular endothelium forms a semi-permeable
barrier between blood and tissue compartments, and serves an
important role in regulating vascular physiological functions.
However, the disruption of the endothelial barrier, marked by an
increased vascular permeability, contributes to the pathogenesis of
various inflammatory disease processes (1).
Sphingosine-1-phosphate (S1P), a bioactive
sphingolipid abundant in plasma, is emerging as a potent modulator
of a variety of biological functions in endothelial cells,
including cell cytoskeleton regulation, cellular locomotion,
angiogenesis and vascular maturation (2). Thrombocyte was originally considered
as the primary source of S1P (3).
However, previous work has suggested that the main source of plasma
S1P is erythrocytes, which mediates the release of S1P independent
of stimuli and may serve as a ‘buffer system’ against S1P depletion
(4). The next candidate for
non-hematopoietic derived S1P is endothelial cells, which
contribute to 40% of plasma S1P in mice (5,6).
It was previously demonstrated that S1P may be
protective in burn injury by enhancing the endothelial barrier
function (7). Mounting evidence
has demonstrated that S1P mediates different cellular responses by
interacting with S1P receptors (S1PRs). The primary receptors
expressed in endothelial cells are S1PR1, S1PR2 and S1PR3 (8). Among these, S1PR1 activates the Rac
signaling pathway, which promotes vascular integrity (9). By contrast, S1PR2 activates the Rho
pathway and leads to disruption of endothelial barrier integrity.
Previous studies have revealed that S1PR2 is an attractive target
for the treatment of disorders of both macro- and micro-vasculature
(2,10,11).
Kim et al (12)
demonstrated that S1PR2 serves a critical role in the disruption of
cerebrovascular integrity in experimental stroke models. Evidence
has demonstrated that S1PR2 serves a key role in the permeability
and inflammatory responses of the vascular endothelium during
endotoxemia (13). It was
previously reported that low-dose S1P enhanced endothelial barrier
integrity through S1PR1, whereas high-dose S1P induced endothelial
hyperpermeability via S1PR2 (14).
Consistently, Sammani et al (15) revealed the therapeutic effects of
S1PR1 in ameliorating inflammatory lung injury, whereas by
contrast, the activation of S1PR2 or S1PR3 induced significant
alveolar and vascular barrier disruption.
Ezrin, radixin and moesin (ERM) proteins organize
the cortical cytoskeleton by linking filamentous actin to the
plasma membrane. S1P promotes ERM translocation to the cell
periphery and its subsequent phosphorylation on a threonine residue
(16). These actin-binding
proteins are engaged differently in endothelial barrier responses.
A previous study demonstrated that moesin promoted thrombin-induced
endothelial barrier dysfunction, whereas radixin exerted the
opposite effect (17).
Moesin is the primary ERM protein expressed in
endothelial cells. It was previously demonstrated that moesin
phosphorylation was required in advanced glycation end products
(AGEs)-induced F-actin rearrangement and hyperpermeability
responses in endothelial cells (18). In the present study, it was
questioned whether moesin phosphorylation was also involved in
excessive S1P-induced endothelial barrier disruption. Therefore,
the role of moesin phosphorylation in modulating high-dose
S1P-induced endothelial hyperpermeability responses was
investigated. The present study may contribute to the
identification of novel drug targets for the treatment of vascular
disorders.
Materials and methods
Reagents
Human umbilical vein endothelial cells (HUVECs) were
purchased from Scien Cell Research Laboratories (Carlsbad, CA,
USA). Dulbecco's modified Eagle's medium (DMEM)/F12 medium, fetal
bovine serum (FBS), trypsin, glutamine, penicillin and streptomycin
were all from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). S1P was obtained from Sigma-Aldrich; Merck KGaA (Darmstadt,
Germany). The S1PR1 antagonist W146 was from Avanti Polar Lipids,
Inc. (Alabaster, AL, USA). The S1PR2 antagonist, JTE013, and the
S1PR3 antagonist, CAY10444, were purchased from Cayman Chemical
Company (Ann Arbor, MI, USA). An antibody against phosphorylated
(p)-moesin (Thr558) was from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA; cat. no. sc-12895); a moesin antibody was from
Cell Signaling Technology, Inc. (Danvers, MA, USA; cat. no. 3150).
An anti-β-actin antibody was from OriGene Technologies, Inc.
(Beijing, China; cat. no. TA310155). The secondary antibodies,
rabbit anti-mouse IgG horseradish peroxidase-conjugated (HRP)
antibody (cat. no. bs-0296R-HRP) and goat anti-rabbit IgG
HRP-conjugated antibody (cat. no. bs-0295G-HRP) were from Beijing
Biosynthesis Biotechnology Co., Ltd. (Beijing, China). Human moesin
small interfering (si)RNA and control nonsense siRNA were purchased
from Shanghai GenePharma Co., Ltd. (Shanghai, China).
Rhodamine-phalloidin recognizing F-actin was obtained from
Molecular Probe; Thermo Fisher Scientific, Inc. (cat. no. R415).
Biochemical reagents were obtained from Sigma-Aldrich unless
otherwise indicated.
Cell culture
HUVECs were cultured in DMEM/F12 containing 10% FBS
at 37°C in a humidified atmosphere with 5% CO2 and 95%
air. In all experiments, HUVECs were grown to 90% confluence and
starved of serum for 12 h. HUVECs were then stimulated with 10
µmol/l S1P for 0, 5, 10, 20, 40, 60 and 90 min, respectively. In
order to study the effect dose effect, HUVECs were also stimulated
with 0, 0.5, 1, 5 and 10 µmol/l S1P for 20 min. The following
investigations were performed under S1P (10 µmol/l) treatment at
37°C for 20 min; HUVECs treated with DMEM onlly were used as
control. For detection of the effects on S1P receptors in HUVEC
responses, specific antagonists of S1PR1 (10 µmol/l), S1PR2 (10
µmol/l) and S1PR3 (10 µmol/l) were each added to HUVECs at 37°C for
30 min before the relevant S1P application.
Transfection of siRNA
HUVECs were seeded into 6-well plates at 70–80%
confluence. After 24 h, HUVECs were incubated at 37°C with siRNA
Mate Transfection reagent (Shanghai Gene Pharma Co., Ltd.) and
siRNA targeting moesin (sense 5′-GGGAUGUCAACUGACCUAAdTdT-3′ and
antisense 5′-UUAGGUCAGUUGACAUCCCdTdG-3′), or control nonsense siRNA
(sense 5′-UUCUCCGAACGUGUCACGUUTdTdG-3′ and antisense
5′-ACGUGACACGUUCGGAGAATTdTdG-3′), according to the manufacturer's
protocol. The transfected cells were harvested 48–72 h later.
Western blotting
Total cellular extracts were prepared using lysis
buffer (20 mmol/l Tris at pH 7.4, 2.5 mmol/l EDTA, 1% Triton X-100,
1% deoxycholic acid, 0.1% SDS, 100 mmol/l NaCl, 10 mmol/l NaF and 1
mmol/l Na3VO4) supplemented with protease and
phosphatase inhibitors. Total proteins concentrations were measured
by Bicinchoninic Acid assay, and then 30 µg of protein samples were
subjected to 12% SDS-PAGE separation, and transferred onto
polyvinylidene difluoride membranes. After blocking with 5% bovine
serum albumin (BSA; GBCBIO Technologies, Inc., Guangdong, China),
the membranes were incubated with primary antibodies against
p-moesin, moesin and β-actin at 1:1,000 dilution at 4°C overnight,
with agitation. Following washing three times with TBST containing
0.1% Tween-20 (each time for 10 min), membranes were probed with an
HRP-conjugated rabbit anti-mouse or goat anti-rabbit IgG antibody
according to the source of the primary antibody at 1:8,000 dilution
at room temperature for 1 h. After washing the membrane three times
with TBST for 10 min each time, the protein bands were visualized
using a chemiluminescence reagent (EMD Millipore, Billerica, MA,
USA). Images were acquired using a Kodak IS4000R Imaging instrument
(Kodak, Rochester, NY, USA), and then densitometric analysis was
performed using ImageJ version 1.48i (National Institutes of
Health, Bethesda, MD, USA).
Fluorescence staining
The distribution of the cytoskeletal F-actin in
HUVECs was determined. HUVECs were plated on gelatin-coated
glass-bottom microwell plates (MatTek Corporation, MA, USA) and
cultured until 70–80% confluent. Cells were transfected with moesin
siRNA or control nonsense siRNA, which was followed by further
treatment with or without S1P. The untransfected cells treated with
DMEM alone were used as control. Then, the medium was removed, and
the cells were washed with PBS and permeabilized for 15 min at room
temperature in PBS containing 3.7% formaldehyde and 0.5% Triton
X-100. Subsequently, cells were washed with PBS twice and blocked
in 5% BSA at room temperature for 1 h. After a thorough wash in
PBS, the cells were stained with a conjugated rhodamine-phalloidin
antibody (2 U ml−1) at room temperature for 1 h. Cells
were further incubated with diamidino-2-phenylindole (DAPI;
1:1,000) at room temperature for 15 min and then washed with PBS
again. DAPI was used to stain nuclear DNA. The staining results
were imaged using a Zeiss LSM780 laser confocal scanning microscope
(Zeiss GmbH, Jena, Germany), and analyzed using ImageJ version
1.48i (National Institutes of Health).
Transendothelial electrical resistance
(TER)
TER of the HUVEC monolayer was determined using a
STX2 electrode and EVOM2 meter according to the
manufacturer's protocol (World Precision Instruments, Sarasota, FL,
USA) (19). Briefly, HUVECs were
seeded at 0.5×105/well in gelatin-coated, 6.5 mm
Transwell filters (0.4-mm pore size) until confluent. Resistance
values of multiple Transwell inserts within an experimental group
were measured sequentially and the mean was expressed in the common
unit (Ωcm2) after subtraction of the value of a blank
cell-free filter.
Statistical analysis
All data are expressed as the mean ± standard
deviation from at least three independent experiments and analyzed
using SPSS version 16.0 software (SPSS, Inc., Chicago, IL, USA).
Statistical comparisons were performed using a one-way analysis of
variance with Bonferroni post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
S1P induces moesin phosphorylation in
HUVECs
Previously published results suggested that moesin
is phosphorylated in AGE-induced endothelial barrier dysfunction
(18,20). To determine whether moesin
phosphorylation serves a role in S1P-mediated endothelial
hyperpermeability, moesin phosphorylation levels were measured
after S1P treatment. HUVECs were incubated with a high dose of S1P
(10 µmol/l) for different durations, and then p-moesin expression
levels were determined by western blotting. The results
demonstrated that phosphorylation of moesin was significantly
enhanced after a 5-min S1P treatment compared with untreated cells,
and gradually reached a peak at 20 min, before the expression
decreased to a relatively stable level until 90 min S1P stimulation
(Fig. 1A). The influence of S1P
treatment at different doses on moesin phosphorylation were also
investigated in endothelial cells after S1P incubation for 20 min.
It was demonstrated that the phosphorylation levels of moesin were
significantly increased by S1P in a concentration-dependent manner
compared with untreated cells, ranging from 0.5 to 10 µmol/l
(Fig. 1B). These results suggested
that S1P induces moesin phosphorylation in a time- and
dose-dependent manner.
High-dose S1P mediates moesin
phosphorylation via S1PR2
It was previously demonstrated that low-dose S1P
(0.5–1 µmol/l) enhances endothelial barrier integrity via S1PR1,
whereas high-dose S1P (5–10 µmol/l) induced endothelial monolayer
hyperpermeability responses via S1PR2 (14). Given that moesin phosphorylation is
involved in endothelial barrier disruption, it was questioned
whether S1PR2 is required for S1P-induced moesin phosphorylation.
Thus, HUVECs were subjected to S1P treatment (10 µmol/l) for 20
min. In order to confirm the effects of different S1PRs, specific
S1PR antagonists were used. Pretreatment of the S1PR2 antagonist
(JTE013) significantly attenuated moesin phosphorylation compared
with the S1P group. However, the S1PR1 antagonist (W146) and S1PR3
antagonist (CAY10444) did not affect high-dose S1P-induced moesin
phosphorylation (Fig. 2). This
suggested that S1PR2 is required to enhance moesin phosphorylation
induced by high-doses of S1P.
 | Figure 2.Involvement of S1PR2 in high-dose
S1P-induced moesin phosphorylation. HUVECs were pretreated with
either each of the S1PR1, S1PR2 or S1PR3 antagonists for 30 min, or
without antagonist treatment, prior to the addition of 10 µmol/l
S1P for 20 min. p-moesin or total moesin was examined using western
blot analysis. The results are expressed as the mean ± standard
deviation from three independent experiments. **P<0.01 vs.
untreated control; #P<0.05 vs. S1P. p-,
phosphorylated; S1P, sphingosine-1-phosphate; W+S1P, S1PR1
antagonist W146 and sphingosine-1-phosphate; J+S1P, S1PR2
antagonist JTE013 and sphingosine-1-phosphate; C+S1P, S1PR3
antagonist CAY10444 and sphingosine-1-phosphate; S1PR1,
sphingosine-1-phosphate receptor 1; S1PR2, sphingosine-1-phosphate
receptor 2; S1PR3, sphingosine-1-phosphate receptor 3. |
Moesin phosphorylation is involved in
high-dose S1P-induced endothelial barrier dysfunction
To determine whether moesin phosphorylation is
involved in high-dose S1P-induced responses in endothelial cells,
siRNA targeting moesin was used. HUVECs transfected with moesin
siRNA exhibited significantly reduced moesin expression levels
compared with untransfected control cells, whereas negative control
siRNA had no effect on moesin expression (Fig. 3A). Consistent with the above
findings, high-dose S1P increased moesin phosphorylation, however,
knockdown of moesin reduced the levels of phosphorylated moesin in
HUVECs treated with high-dose S1P (Fig. 3B).
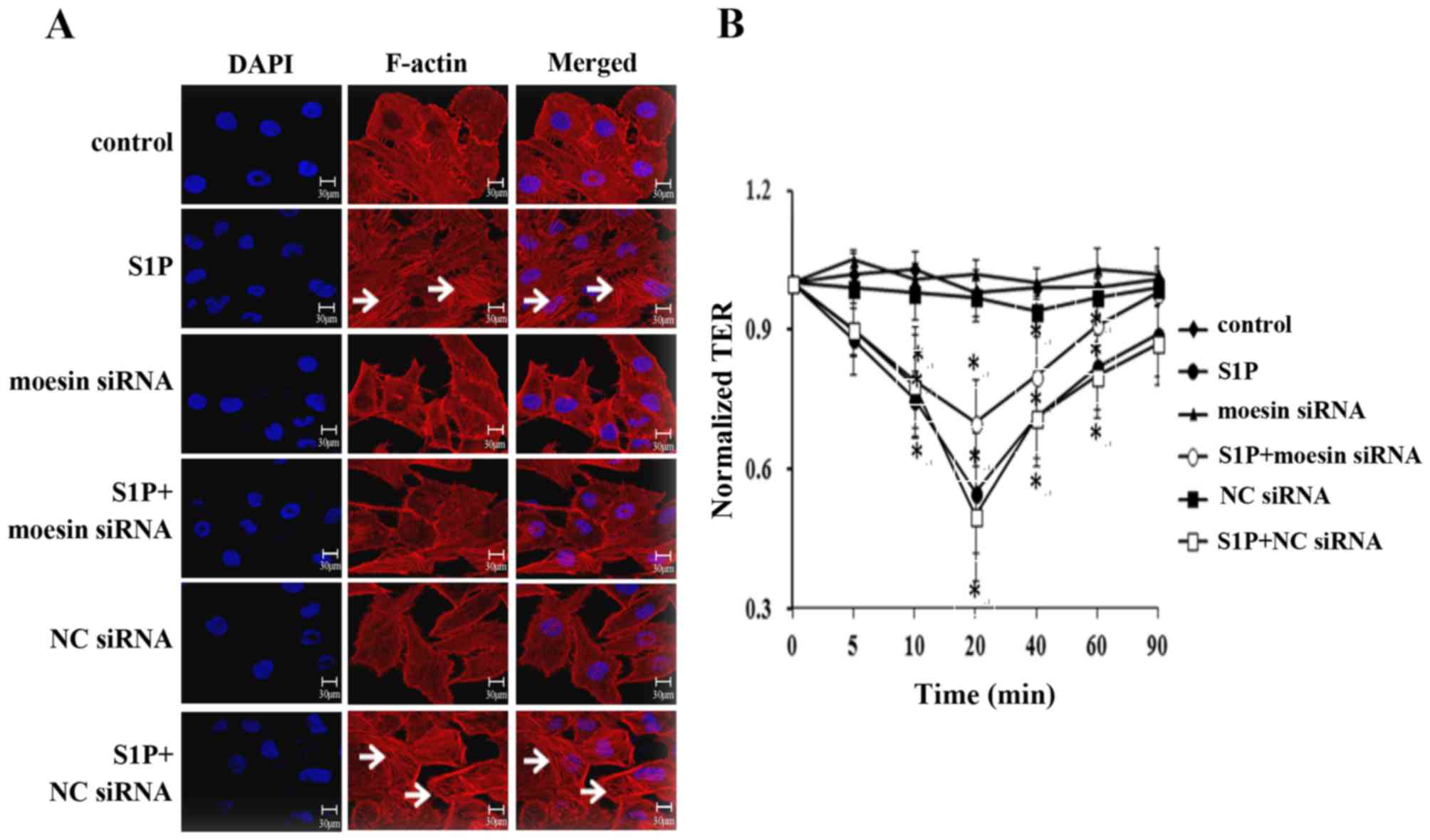
Next, it was determined whether moesin
phosphorylation was involved in the structural and functional
changes in high-dose S1P-treated endothelial cells. The role of
moesin in modulating endothelial cell structural responses induced
by S1P was examined by visualizing the distribution of F-actin
(Fig. 4A). When compared with the
control, the application of high-dose S1P resulted in the
disorganization of F-actin and the formation of stress fibers, as
well as the opening of intercellular gaps (panel 2 in Fig. 4A). This effect was markedly
attenuated by moesin siRNA (panel 4 in Fig. 4A), while the application of 10
µmol/l S1P in control siRNA-treated cells caused the same
polymerization of F-actin and formation of stress fibers (panel 6
in Fig. 4A) as S1P-treated alone.
No significant alterations were observed in the moesin siRNA or the
control siRNA groups. These results suggested that moesin may have
a critical role in mediating F-actin rearrangement induced by
high-dose S1P.
To further confirm the effect of moesin on
S1P-induced endothelial barrier responses, a TER test was performed
(Fig. 4B). The monolayer TER was
measured intermittently within 90 min. Accordingly, compared with
control group, the S1P-treated and S1P+NC siRNA groups exhibited a
significant decrease in the TER value and reached a plateau at 20
min, and then restored to a level lower than 0 min in the later
stage. Pretreatment with moesin siRNA attenuated the S1P-induced
decline in TER and the value at 90 min was similar to the value to
0 min. No significant differences were observed between the TER
value in the moesin siRNA group or the NC siRNA group. Taken
together, it was concluded that moesin phosphorylation serves an
important role in S1P-induced F-actin rearrangement and endothelial
barrier disruption.
Discussion
Physiological plasma levels of circulating S1P are
vasculoprotective, whereas abnormal activation of S1P signaling is
associated with a diverse range of diseases, including diabetes,
fibrosis and cancer (21,22). In certain pathological conditions,
the serum S1P concentration may be increased and contribute to
vascular hyperpermeability responses. It was previously
demonstrated that physiological concentrations of S1P, ranging from
0.1 to 1 µmol/l, contribute to endothelial barrier-protective
responses via S1PR1. By contrast, high concentrations of S1P (>5
µmol/l) hampers endothelial barrier integrity via S1PR2 (14). The underlying molecular mechanisms
of the protective role of physiological-dose S1P in endothelial
integrity enhancement have been widely reported (23–26).
However, signaling pathways underlying high-dose S1P-induced
endothelial barrier dysfunction remains to be fully clarified.
Moesin is the primary ERM protein expressed in
endothelium. Phosphorylation of moesin is critically implicated in
endothelial barrier dysfunction. Previously, the role of moesin
phosphorylation in AGE-induced endothelial hyperpermeability was
described (18). The present study
investigated the effect of moesin phosphorylation on excessive
S1P-induced endothelial barrier responses. S1P induced significant
moesin phosphorylation in a time- and dose-dependent manner.
High-dose S1P induced sustained threonine phosphorylation of
moesin, which reached maximum levels at 20 min and remained
elevated for ≥90 min. These results were consistent with the prior
observation that physiological doses of S1P induced a significant
elevation in moesin phosphorylation (27). However, an S1PR1 specific agonist,
SEW2871, had no effect on moesin phosphorylation (27). In addition, siRNA depletion of
S1PR1 failed to attenuate S1P-induced ERM phosphorylation, whereas
antagonists for S1PR2 (JTE013) or S1PR3 (CAY10444) exhibited
dramatic decrease of ERM phosphorylation (27). These results suggested that S1PR2
and/or S1PR3, but not S1PR1, are likely to participate in
S1P-induced moesin phosphorylation. In the present study, it was
demonstrated that the S1PR2 antagonist, but not S1PR1 or S1PR3
antagonists, abolished the effect of S1P on moesin phosphorylation.
These results suggested that high-dose S1P induced moesin
phosphorylation via S1PR2 rather than S1PR1 or S1PR3. Furthermore,
moesin depletion ameliorated S1P-induced F-actin rearrangement and
stress fiber formation, as well as endothelial barrier disruption.
Collectively, the data suggested that high-dose S1P induced moesin
phosphorylation via S1PR2, and thus mediated endothelial barrier
disruption responses. These findings were consistent with a
previous report that suggested that S1P-induced cytoskeletal
rearrangement was dependent on ERM phosphorylation, with the
involvement of S1PR2 (28).
The discrepant effect between low- and high-dose S1P
may result from the different roles of S1PRs and ERM proteins. The
basic expression level of S1PR2 in endothelial cells was much lower
than S1PR1 and S1PR3 (29). A
previous study demonstrated that physiological doses of S1P
enhanced the TER of the endothelial monolayer, whereas an S1PR1
antagonist significantly attenuated this effect (14). By contrast, high-dose S1P triggered
a decrease in the TER value, whereas an S1PR2 inhibitor partially
ameliorated this effect (14).
Therefore, the present study hypothesized that when low-dose S1P
encountered those receptors, most of which may bind to S1PR1,
prominently mediating a barrier-protective effect. However, when
high-dose S1P overwhelmed these receptors, more S1PR2 and S1PR3
were activated and exerted the opposite effect. It was previously
unveiled that Rho-associated protein kinase (ROCK) activation
served an important role in high-dose S1P-mediated endothelial
hyperpermeability through S1PR2 (27). In addition, ROCK activation was
indispensable for AGE-induced moesin phosphorylation (18). Thus, it was speculated that moesin
may act as a downstream molecule of the S1PR2-ROCK axis and mediate
high-dose S1P-induced endothelial barrier disruption.
How individual ERM proteins differentially regulate
endothelial barrier requires further investigation. Adyshev et
al (27) reported that
transfection with either radixin or pan-ERM markedly ameliorated
physiological-dose S1P-mediated endothelial barrier enhancement.
Ezrin depletion partially attenuated S1P-induced endothelial
barrier enhancement and cytoskeletal alterations (27). Moesin was phosphorylated under
physiological doses of S1P stimulation; however, the depletion of
moesin contributed to the elevated TER value (27). In the present study, it was
demonstrated that moesin phosphorylation resulted in high-dose
S1P-induced F-actin polymerization, stress fiber formation and
endothelial barrier dysfunction, which were dependent on S1PR2.
Taken together, it is possible that under low-dose
S1P treatment, radixin serves a key barrier-enhancing role and this
process may be associated with S1PR1. Whereas under excessive S1P
stimulation, S1PR2 was dominantly activated and mediated
moesin-exerted endothelial barrier disruption, and this process may
be associated with ROCK activation.
It should be noted that the present study only
examined the effect of excessive S1P on endothelial integrity in
vitro, and it has not yet been investigated in vivo. In
an LPS-induced murine model of acute lung injury, intravenous
injection of S1P (85 µg/kg) significantly rescued vascular leak
(30). However, the specific high
dose of S1P for in vivo study remains to be investigated. As
reported previously, the effectiveness of high-dose S1P may be more
complex in vivo, as S1PR2 expression was upregulated in
certain inflammatory conditions (29). The possibility exists that
excessive S1P combines with inducible S1PR2 and exacerbates
inflammatory responses. The authors of the present study intend to
investigate whether excessive S1P functions synergistically with
inflammatory mediators such as lipopolysaccharide-binding protein
or tumor necrosis factor-α.
In conclusion, the present study suggested that
induction of moesin phosphorylation by excessive S1P occurs through
S1PR2 and further induces endothelial barrier damage responses. The
findings further highlighted the potential utility of a
pharmacological target of the S1PR2-moesin axis in vascular barrier
dysfunction.
Acknowledgements
The present study was supported by the General
Program from Natural Science Foundation of China (grant nos.
81370226 and 81170297) and The Team-Project of Natural Science
Foundation of Guangdong, China (grant no. S2013030013217).
References
|
1
|
Yoo H, Ku SK, Baek YD and Bae JS:
Anti-inflammatory effects of rutin on HMGB1-induced inflammatory
responses in vitro and in vivo. Inflamm Res. 63:197–206. 2014.
View Article : Google Scholar
|
|
2
|
Chen S, Yang J, Xiang H, Chen W, Zhong H,
Yang G, Fang T, Deng H, Yuan H, Chen AF and Lu H: Role of
sphingosine-1-phosphate receptor 1 and sphingosine-1-phosphate
receptor 2 in hyperglycemia-induced endothelial cell dysfunction.
Int J Mol Med. 35:1103–1108. 2015. View Article : Google Scholar
|
|
3
|
Yatomi Y, Ruan F, Hakomori S and Igarashi
Y: Sphingosine-1-phosphate: A platelet-activating sphingolipid
released from agonist-stimulated human platelets. Blood.
86:193–202. 1995.
|
|
4
|
Hänel P, Andréani P and Gräler MH:
Erythrocytes store and release sphingosine 1-phosphate in blood.
FASEB J. 21:1202–1209. 2007. View Article : Google Scholar
|
|
5
|
Hisano Y, Kobayashi N, Yamaguchi A and
Nishi T: Mouse SPNS2 functions as a sphingosine-1-phosphate
transporter in vascular endothelial cells. PloS One. 7:e389412012.
View Article : Google Scholar :
|
|
6
|
Nijnik A, Clare S, Hale C, Chen J, Raisen
C, Mottram L, Lucas M, Estabel J, Ryder E, Adissu H, et al: The
role of sphingosine-1-phosphate transporter Spns2 in immune system
function. J immunol. 189:102–111. 2012. View Article : Google Scholar :
|
|
7
|
Liu X, Wu W, Li Q, Huang X, Chen B, Du J,
Zhao K and Huang Q: Effect of sphingosine 1-phosphate on
morphological and functional responses in endothelia and venules
after scalding injury. Burns. 35:1171–1179. 2009. View Article : Google Scholar
|
|
8
|
Sanchez T and Hla T: Structural and
functional characteristics of S1P receptors. J Cell Biochem.
92:913–922. 2004. View Article : Google Scholar
|
|
9
|
Singleton PA, Dudek SM, Chiang ET and
Garcia JG: Regulation of sphingosine 1-phosphate-induced
endothelial cytoskeletal rearrangement and barrier enhancement by
S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J.
19:1646–1656. 2005. View Article : Google Scholar
|
|
10
|
Sanchez T: Sphingosine-1-Phosphate
signaling in endothelial disorders. Curr Atheroscler Rep.
18:312016. View Article : Google Scholar
|
|
11
|
Liu W, Liu B, Liu S, Zhang J and Lin S:
Sphingosine-1-phosphate receptor 2 mediates endothelial cells
dysfunction by PI3K-Akt pathway under high glucose condition. Eur J
Pharmacol. 776:19–25. 2016. View Article : Google Scholar
|
|
12
|
Kim GS, Yang L, Zhang G, Zhao H, Selim M,
McCullough LD, Kluk MJ and Sanchez T: Critical role of
sphingosine-1-phosphate receptor-2 in the disruption of
cerebrovascular integrity in experimental stroke. Nat Commun.
6:78932015. View Article : Google Scholar :
|
|
13
|
Zhang G, Yang L, Kim GS, Ryan K, Lu S,
O'Donnell RK, Spokes K, Shapiro N, Aird WC, Kluk MJ, et al:
Critical role of sphingosine-1-phosphate receptor 2 (S1PR2) in
acute vascular inflammation. Blood. 122:443–455. 2013. View Article : Google Scholar :
|
|
14
|
Li Q, Chen B, Zeng C, Fan A, Yuan Y, Guo
X, Huang X and Huang Q: Differential activation of receptors and
signal pathways upon stimulation by different doses of
sphingosine-1-phosphate in endothelial cells. Exp Physiol.
100:95–107. 2015. View Article : Google Scholar
|
|
15
|
Sammani S, Moreno-Vinasco L, Mirzapoiazova
T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain
A, Moitra J, et al: Differential effects of sphingosine 1-phosphate
receptors on airway and vascular barrier function in the murine
lung. Am J Respir Cell Mol Biol. 43:394–402. 2010. View Article : Google Scholar
|
|
16
|
Canals D, Jenkins RW, Roddy P,
Hernández-Corbacho MJ, Obeid LM and Hannun YA: Differential effects
of ceramide and sphingosine 1-phosphate on ERM phosphorylation:
Probing sphingolipid signaling at the outer plasma membrane. J Biol
Chem. 285:32476–32485. 2010. View Article : Google Scholar :
|
|
17
|
Adyshev DM, Dudek SM, Moldobaeva N, Kim
KM, Ma SF, Kasa A, Garcia JG and Verin AD: Ezrin/radixin/moesin
proteins differentially regulate endothelial hyperpermeability
after thrombin. Am J Physiol Lung Cell Mol Physiol. 305:L240–L255.
2013. View Article : Google Scholar :
|
|
18
|
Guo X, Wang L, Chen B, Li Q, Wang J, Zhao
M, Wu W, Zhu P, Huang X and Huang Q: ERM protein moesin is
phosphorylated by advanced glycation end products and modulates
endothelial permeability. Am J Physiol Heart Circ Physiol.
297:H238–H246. 2009. View Article : Google Scholar
|
|
19
|
Patabendige A, Skinner RA and Abbott NJ:
Establishment of a simplified in vitro porcine blood-brain barrier
model with high transendothelial electrical resistance. Brain Res.
1521:1–15. 2013. View Article : Google Scholar :
|
|
20
|
Zhang W, Xu Q, Wu J, Zhou X, Weng J, Xu J,
Wang W, Huang Q and Guo X: Role of Src in Vascular
Hyperpermeability induced by advanced glycation end products. Sci
Rep. 5:140902015. View Article : Google Scholar :
|
|
21
|
Maceyka M, Milstien S and Spiegel S:
Sphingosine-1-phosphate: The Swiss army knife of sphingolipid
signaling. J Lipid Res. 50 Suppl:S272–S276. 2009. View Article : Google Scholar :
|
|
22
|
Russo SB, Ross JS and Cowart LA:
Sphingolipids in obesity, type 2 diabetes, and metabolic disease.
Handb Exp Pharmacol. 373–401. 2013. View Article : Google Scholar :
|
|
23
|
Finigan JH, Dudek SM, Singleton PA, Chiang
ET, Jacobson JR, Camp SM, Ye SQ and Garcia JG: Activated protein C
mediates novel lung endothelial barrier enhancement: Role of
sphingosine 1-phosphate receptor transactivation. J Biol Chem.
280:17286–17293. 2005. View Article : Google Scholar
|
|
24
|
Feistritzer C and Riewald M: Endothelial
barrier protection by activated protein C through PAR1-dependent
sphingosine 1-phosphate receptor-1 crossactivation. Blood.
105:3178–3184. 2005. View Article : Google Scholar
|
|
25
|
Garcia JG, Liu F, Verin AD, Birukova A,
Dechert MA, Gerthoffer WT, Bamberg JR and English D: Sphingosine
1-phosphate promotes endothelial cell barrier integrity by
Edg-dependent cytoskeletal rearrangement. J Clin Invest.
108:689–701. 2001. View
Article : Google Scholar :
|
|
26
|
Schaphorst KL, Chiang E, Jacobs KN, Zaiman
A, Natarajan V, Wigley F and Garcia JG: Role of sphingosine-1
phosphate in the enhancement of endothelial barrier integrity by
platelet-released products. Am J Physiol Lung Cell Mol Physiol.
285:L258–L267. 2003. View Article : Google Scholar
|
|
27
|
Adyshev DM, Moldobaeva NK, Elangovan VR,
Garcia JG and Dudek SM: Differential involvement of
ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced
human pulmonary endothelial cell barrier enhancement. Cell Signal.
23:2086–2096. 2011. View Article : Google Scholar :
|
|
28
|
Gandy KA, Canals D, Adada M, Wada M, Roddy
P, Snider AJ, Hannun YA and Obeid LM: Sphingosine 1-phosphate
induces filopodia formation through S1PR2 activation of ERM
proteins. Biochem J. 449:661–672. 2013. View Article : Google Scholar :
|
|
29
|
Du J, Zeng C, Li Q, Chen B, Liu H, Huang X
and Huang Q: LPS and TNF-alpha induce expression of
sphingosine-1-phosphate receptor-2 in human microvascular
endothelial cells. Pathol Res Pract. 208:82–88. 2012. View Article : Google Scholar
|
|
30
|
McVerry BJ, Peng X, Hassoun PM, Sammani S,
Simon BA and Garcia JG: Sphingosine 1-phosphate reduces vascular
leak in murine and canine models of acute lung injury. Am J Respir
Crit Care Med. 170:987–993. 2004. View Article : Google Scholar
|