Introduction
Atherosclerosis is a chronic inflammatory disease
involving numerous cytokines (1).
Tumor necrosis factor-like weak inducer of apoptosis (TWEAK), an
inflammatory cytokine of tumor necrosis factor (TNF) superfamily,
participates in regulation of multiple cellular responses,
including proinflammatory activity, angiogenesis and cell
proliferation (2). When binding to
its receptor fibroblast growth factor inducible molecule 14 (Fn14),
TWEAK exerts adverse biological functions in atherosclerosis,
resulting in dysfunction of endothelial cells (2,3) and
smooth muscle cells (4) and
inducing inflammatory response of monocytes/macrophages (5–7).
Endothelial dysfunction is an early hallmark of the
onset of atherosclerosis (8).
Excessive production of reactive oxygen species (ROS) and the
subsequent decrease in vascular bioavailability of nitric oxide
(NO) have long been proposed to be the common pathogenetic
mechanism of the endothelial dysfunction (9). The mitochondrial respiratory chain is
a major intracellular source of ROS (10) and an abnormal production of ROS in
the mitochondria plays a critical role in the development of
atherosclerosis, including oxidation of LDL and damage of
mitochondria DNA (mtDNA) (11).
Peroxisome proliferator-activated receptor-γ
coactivator-1α (PGC-1α), a transcriptional coactivator, recruits
transcription factors to regulate mitochondria numbers and
functions (12). PGC-1α plays a
crucial protective role in the regulation of mitochondrial
oxidative stress in endothelial cells (13,14).
Its underlying mechanism is to upregulate the mitochondrial
antioxidant defense system such as manganese superoxide dismutase
(MnSOD) (13). Furthermore, PGC-1α
is activated by AMP-activated protein kinase (AMPK) (14), which is an important metabolic
sensor.
In the present study, we demonstrated the role of
TWEAK/Fn14 on oxidative stress especially that derived from
mitochondrial and NO generation in human umbilical vein endothelial
cells (HUVECs). In addition, the underlying mechanism is implicated
in the AMPK/PGC-1α/MnSOD signaling pathway.
Materials and methods
Reagents
Recombinant human TWEAK was from Alexis
(Läufelfingen, Switzerland). GSK621 (S7898) was purchased from
Selleck Chemicals (Houston, TX, USA). Rabbit polyclonal antibody
against AMPK (ab131512) was purchased from Abcam (Cambridge, MA,
USA), and rabbit monoclonal antibody against GAPDH (2118S),
pho-AMPK (Thr172) (2535S), PGC-1α (2178S) and MnSOD
(13141S) were purchased from Cell Signaling (Beverly, MA, USA). The
Fn14-siRNA duplexes were designed and synthesized by Ribo-Bio
(Guangzhou, China).
Cell culture
HUVECs were obtained from American Type Culture
Collection (Manassas, VA, USA), and grown in Dulbecco's modified
Eagle's medium (Gibco, Carlsbad, CA, USA) supplemented with 10%
fetal bovine serum (Sciencell, Carlsbad, CA, USA). Cells were
incubated at 37°C in a humidified atmosphere of 5% CO2
and 95% air and grown to 70 to 80% confluence. For the experiments,
the HUVECs were treated with 50, 100 and 200 ng/ml TWEAK
respectively based on previous studies (15–17).
As for Fn14 siRNA, for each well of a 6-well plate, cells were
transfected with 5 µl siRNA (20 µM) or negative control (Ncontrol
group) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for
48 h. To confirm that AMPK activation is required for the
expression of PGC-1α and MnSOD, HUVECs were treated by 10 µmol/l
GSK621 as previous described (18,19).
Assessment of ROS production
To assess ROS and mitochondrial ROS production,
HUVECs were incubated with 2,7-dichlorofluorescein diacetate
(DCFH-DA, Beyotime, Shanghai, China) or MitoSOX Red (Thermofisher
Scientific, Waltham, MA, USA) in Hank's Buffered Salt Solution
(HBSS) at 37°C for 30 min. After washing two times in HBSS,
fluorescent images were captured using an Olympus fluorescent
microscope. The fluorescence intensity was quantified using Image J
software (National Institutes of Health, Bethesda, MD, USA). For
flow cytometry analysis, after 30 min loading of DCFH-DA or
MitoSOX, the cells were collected and resuspended in 200 µl of PBS
buffer. Then intracellular ROS and mitochondrial ROS levels were
performed by flow cytometry (BD Biosciences, San Jose, CA,
USA).
Measurement of NO production
Total NO production in culture medium was determined
by measuring the concentration of nitrate and nitrite, a stable
metabolite of NO, by modified Griess reaction method. The procedure
involed use of the Nitric Oxide Assay kit (Nanjing Jiancheng
Bioengineering, Nanjing, China).
MtDNA damage quantification. MtDNA damage was
determined by quantitative PCR in HUVECs as previously described
(20). Total DNA was extracted
using the Genomic DNA kit (TransGen Biotech, Beijing, China).
Quantitative PCR was performed using the Eppendorf Mastercycler ep
realplex PCR System and the sequences of the primers were as
follows: mtDNA primers: 5′-CCCCACAAACCCCATTACTAAACCCA-3′;
5′-TTTCATCATGCGGAGATGTTGGATGG-3′; β-globin primers:
5′-CGAGTAAGAGACCATTGTGGCAG-3′; 5′-GCTGTTCTGTCAATAAATTTCCTTC-3′. PCR
was performed under the following conditions: denaturation at 95°C
for 1 min, followed by 40 cycles of 95°C for 15 sec and 58°C for 20
sec. The values were determined relative to the control sample
after normalizing to β-globin gene control values and
calculated by the comparative cycle threshold (ΔΔCt) method.
Western blot
HUVECs after treatment were lysed with RIPA lysis
buffer (Beyotime, Shanghai, China) containing 10 mM
phenylmethylsulfonyl fluoride (PMSF, Beyotime, Shanghai, China).
Then the lysates were isolated by centrifugation and the protein
concentration was determined using the BCA Protein Assay kit
(Beyotime). Western blotting was performed as previously described
(21). After quantification, the
proteins were separated by 10% SDS-PAGE and proteins transferred to
polyvinylidene difluoride (PVDF) membrane (Millipore Corp.,
Billerica, MA, USA). Membranes were blocked with 5% nonfat dried
milk, and they were immunoblotted with anti-GAPDH (1:2,000),
anti-AMPK (1:1,000), anti-pho-AMPK (Thr172) (1:1,000),
anti-PGC-1α (1:1,000), and anti-MnSOD (1:1,000) antibodies at 4°C
overnight. Subsequently, the membranes were incubated with goat
anti-rabbit IR-Dye 800cw labeled secondary antisera in 0.1% Tween,
0.01% SDS LiCor blocking buffer for 1 h at room temperature.
Membranes were imaged using a LiCor Odyssey scanner.
Statistical analysis
All experiments were performed at least three times.
All statistical analysis was conducted with SPSS 18.0 software
(SPSS Inc., Chicago, IL, USA). Data were represented as means ±
standard deviation. Statistical significance of the data was
performed by unpaired Student test (2-tailed) between two groups or
one-way ANOVA followed by the post-hoc Tukey's test, as
appropriate. A value of P<0.05 was considered significant.
Results
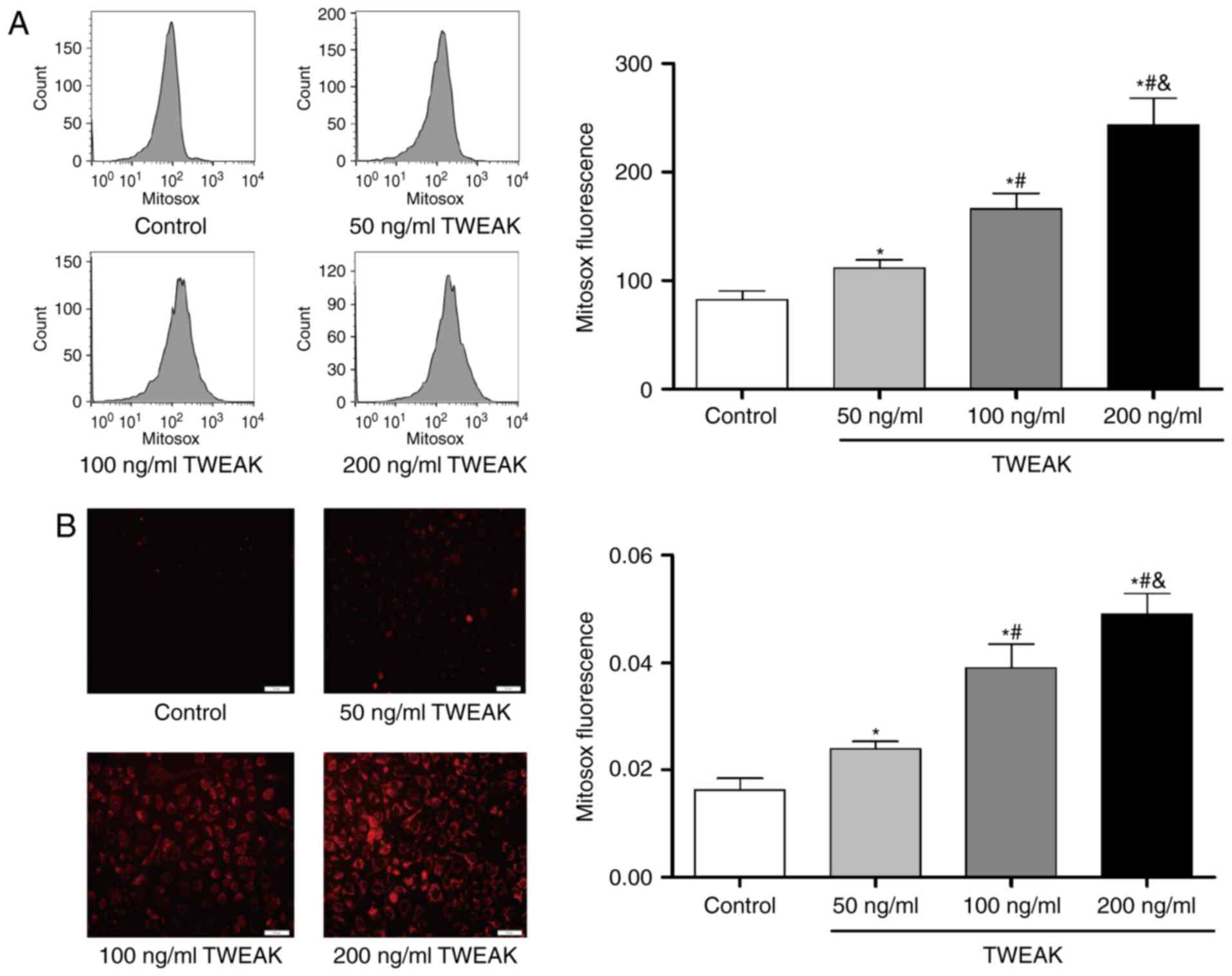
TWEAK induces production of ROS and mtROS and
decreases NO generation in HUVECs. After treated with TWEAK for 24
h, HUVECs were incubated with DCF-DA or MitoSOX probe for 30 min.
And then cell images were captured by a fluorescence microscope or
fluorescence was detected by flow cytometry. At the same time, NO
production in culture medium was determined by the Nitric Oxide
Assay kit. Compared to control group, TWEAK significantly increased
the production of ROS (Fig. 1) and
mtROS (Fig. 2), while it
significantly decreased the NO generation (Fig. 1). Furthermore, the effects were
dose-dependent within 50–200 ng/ml.
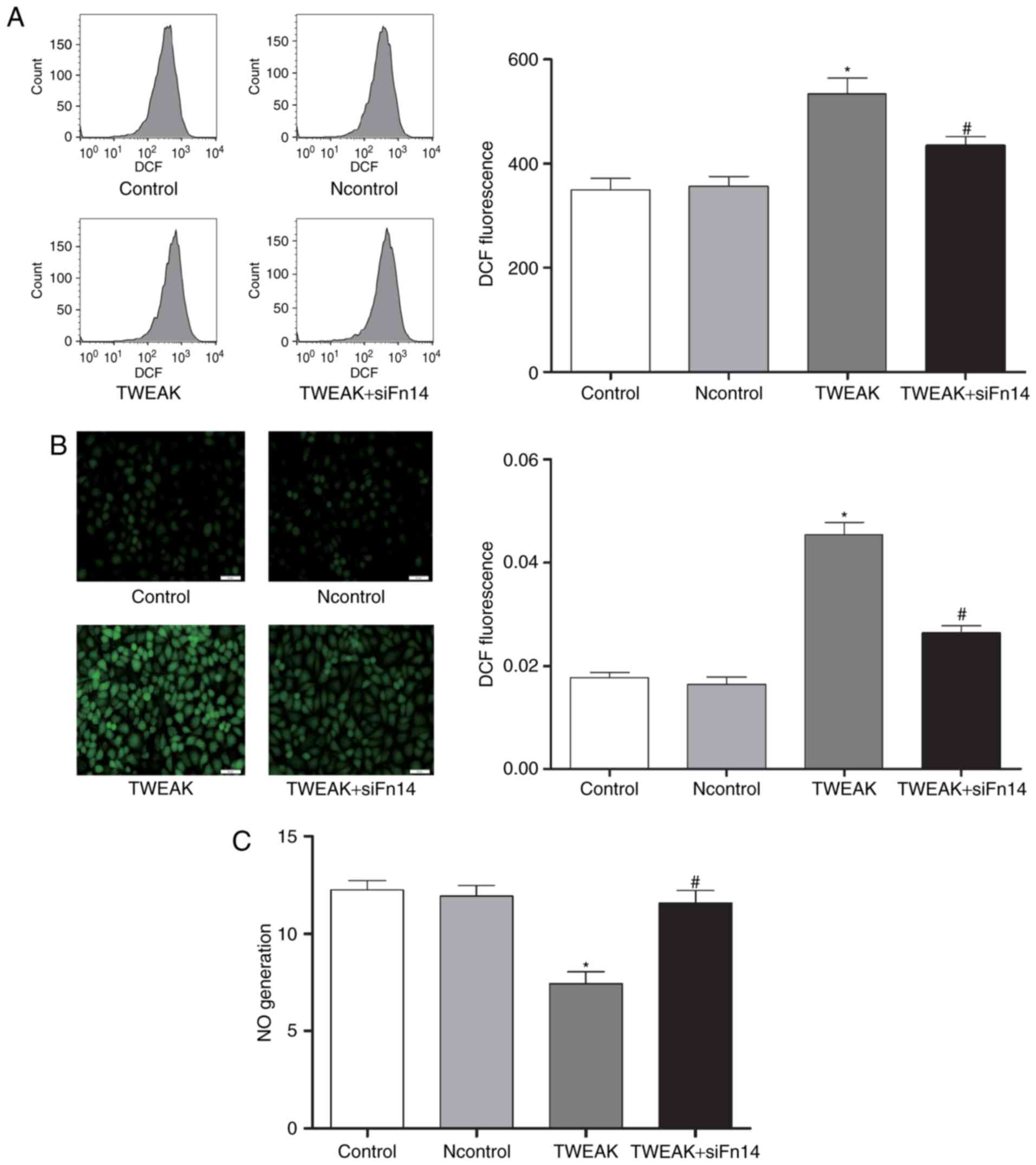
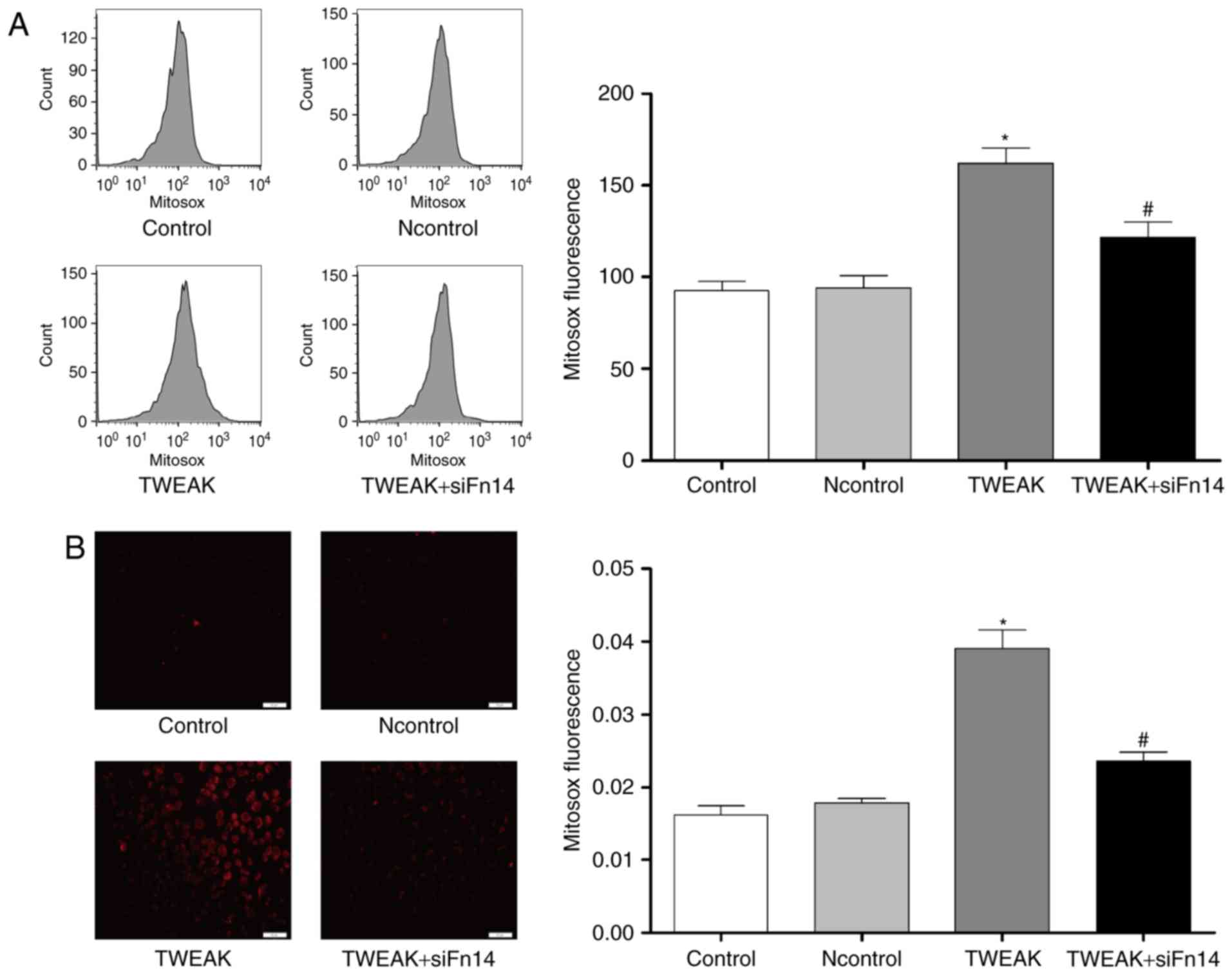
Fn14 mediates TWEAK-induced production of ROS and
mtROS and reduction of NO in HUVECs. To determine whether Fn14
mediate the effect of TWEAK on production of ROS and mtROS and
reduction of NO, HUVECs were treated with 100 ng/ml TWEAK for 24 h
after Fn14 siRNA or negative control siRNA pretreatment. Compared
to control group, the production of ROS and mtROS was increased
significantly while NO generation decreased markedly in TWEAK
treatment group. Furthermore, after Fn14 siRNA pretreatment, the
generation of ROS and mtROS decreased significantly while NO
generation increased markedly compared to TWEAK treatment group
(Figs. 3 and 4).
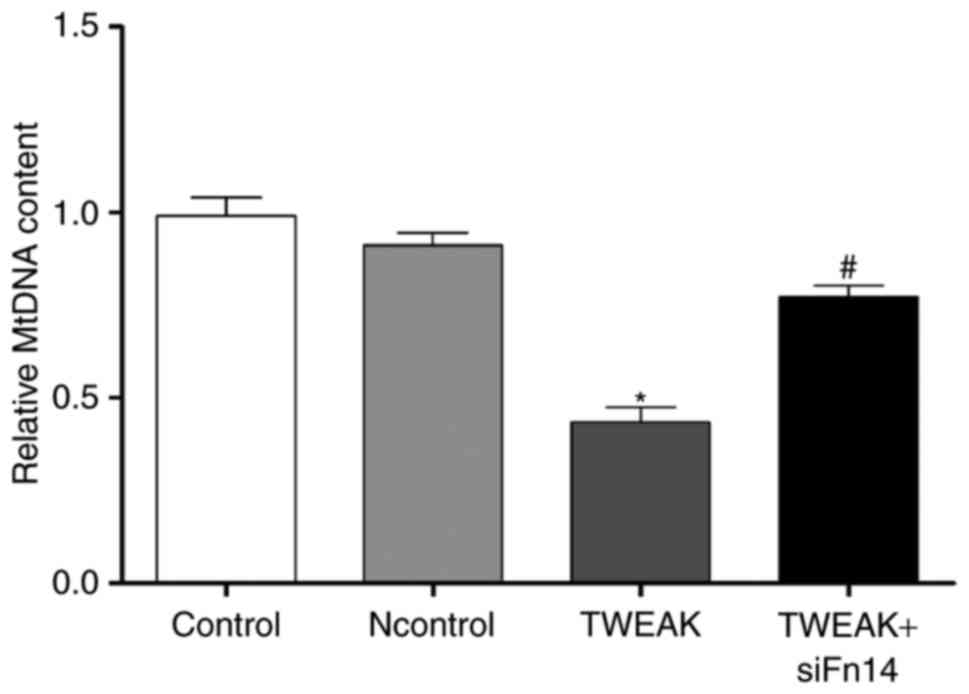
TWEAK/Fn14 promotes mtDNA damage in HUVECs. Given
mitochondrial oxidative stress leading to mitochondrial DNA damage,
we tested the impact of TWEAK/Fn14 on the damage of mtDNA in
HUVECs. MtDNA damage was assessed by the relative expression
quantity of DNA amplification. The lower relative expression
quantity of DNA would suggest more serious DNA damage. We observed
that mtDNA relative amplification was about 56.4% lower in the
group of TWEAK treatment, suggesting mtDNA damage increased
significantly compared to control group. After Fn14 siRNA
pretreatment, mtDNA damage was improved compared to TWEAK treatment
group (Fig. 5).
Essential role of TWEAK/Fn14 in the
expression of AMPK/PGC-1α/MnSOD in HUVECs
To understand the mechanism of TWEAK/Fn14 inducing
mtROS to increase the generation of ROS, we tested the expression
of PGC-1α and its downstream protein MnSOD. After 100 ng/ml TWEAK
treatment of HUVECs for 24 h, PGC-1α and MnSOD expressions were
significantly lower, suggesting PGC-1α/MnSOD may participate in the
process above. It also stated that the activation of PGC-1α
depended on AMPK activation (21).
Therefore, we further tested the pho-AMPK (Thr172) and the
expression of AMPK, and TWEAK treatment decreased the expression of
pho-AMPK in HUVECs. Furthermore, we also found that, compared with
the TWEAK treatment group, the expression of pho-AMPK, PGC-1α and
MnSOD were significantly increased in Fn14 siRNA pretreatment group
(Fig. 6). In addition, we further
tested whether AMPK activation was required for the expression of
PGC-1α and MnSOD. As shown in Fig.
7, the pho-AMPK (Thr172) increased significantly in HUVECs when
treated with the AMPK activator GSK621 (10 µmol/l), which resulted
in increased expression of PGC-1α and MnSOD.
Discussion
In present study, we first determined the effect of
one inflammatory factor, TWEAK, and its receptor Fn14 on inducing
ROS especially mtROS production and decreasing NO generation in
HUVECs. Furthermore, the underlying mechanism associated with the
AMPK/PGC-1α/MnSOD pathway was tried to demonstrate clearly.
Endothelial dysfunction resulting in disturbance in
endothelial homeostasis is considered as a characteristic feature
of atherosclerosis (22). The
hallmark of endothelial dysfunction is impaired
endothelium-dependent vasodilation, which is mediated by NO
(23). As an inflammatory factor
in the TNF family, TWEAK has been demonstrated to participate in
dysfunction of endothelial cells by inducing generation of adhesion
molecules (16), monocyte
chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8)
(3). In present study, we first
demonstrated that TWEAK reduced the generation of NO obviously.
Besides, a growing data suggests that increased production of ROS
have a pivotal role in reduction of NO (9). Excessive production of ROS induced by
TWEAK was measured in our study, and unsurprisingly, TWEAK
increased production of ROS in HUVECs and similar results were
found in monocytes/macrophages (24). In that article, the authors
demonstrated that TWEAK induces ROS through NADPH oxidase in
monocytes. As another major intracellular source of ROS (25), however, mitochondrial ROS was also
examined in cells for the first time in our study, and it is not
unexpected that TWEAK promoted production of mitochondrial ROS
significantly in HUVECs. Moreover, the increase of ROS especially
mitochondrial ROS and decrease of NO were curbed when treated with
Fn14 siRNA, suggesting that Fn14 may mediate these effects.
Given that human mtDNA lacks protective histones
(26) and is located proximal to
ROS generation, it is vulnerable to damage by ROS, which also
induced oxidative damage (26).
MtDNA damage was assessed by the relative expression quantity of
DNA amplification as previous described (20,27).
As for our study, TWEAK/Fn14 axis was capable of inducing mtDNA
damage, resulting from excessive production of ROS in the cells,
which was not reported before. Meanwhile, as a vicious cycle, mtDNA
damage further leads to an increase in oxidative stress, and both
of two effects promote atherosclerosis by contributing to
endothelial dysfunction (27–29).
However, previous studies shown that TWEAK/Fn14-repressed
mitochondrial biogenesis might lead to decrease of mtDNA content
(30,31). Therefore, it is needed to determine
that this is due to the inhibition of DNA synthesis or induction of
DNA damage, as well as whether there is interaction between
them.
As a positive regulator of oxidative metabolism,
PGC-1α upregulates the induction of a set of antioxidant proteins
response to mitochondrial oxidative stress, which increases the
cellular capacity to detoxify mitochondrial ROS in turn, preventing
endothelial dysfunction in response to oxidative stress conditions
(13). Until recently, the only
well-known and primary antioxidant mitochondrial protein is MnSOD
(32), which has been strongly
implicated in endothelial function via regulation of ROS within
mitochondria (33). In our study,
we found that the decreased expression of PGC-1α and subsequently
that of MnSOD were induced by TWEAK, which was mediated by binding
to Fn14. Besides, the induction of PGC-1α has been reported to
depend on the activation of AMPK via phosphorylating the enzyme at
Thr172 (21). In this
study, we also demonstrated that TWEAK/Fn14 axis decreased the
relative expression of phosphorylation levels of AMPK. Furthermore,
we reconfirmed that AMPK activation increased the expression of
PGC-1α and its downstream protein MnSOD. All these results suggest
that TWEAK/Fn14 induces mitochondrial oxidative stress through
regulation of the AMPK/PGC-1α/MnSOD pathway. However, previous
studies reported that TWEAK/Fn14 repressed PGC-1αand mitochondrial
biogenesis (30,31). It is not clear that whether
TWEAK/Fn14-repressed mitochondrial biogenesis results in the
production of ROS and mtROS. Therefore, further research is needed
to confirm.
In conclusion, this study first described the role
of TWEAK/Fn14 in upregulation of ROS and mtROS generation in
HUVECs, which provide novel evidence that TWEAK/Fn14 may become a
key target of interference in atherosclerosis development.
Furthermore, the AMPK/PGC-1α/MnSOD pathway may be involved in the
potential mechanism, providing a new treatment strategy.
Acknowledgements
The present study was supported by funding from the
National Natural Science Foundation of China (81470593), the
National Basic Research Program of China (2014CB542400), the key
research and development project of Hunan Province (2017SK2020) and
the Research Innovation Program for Graduate Students of Central
South University (grant no. 2016zzts152).
References
|
1
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar
|
|
2
|
Burkly LC, Michaelson JS, Hahm K,
Jakubowski A and Zheng TS: TWEAKing tissue remodeling by a
multifunctional cytokine: Role of TWEAK/Fn14 pathway in health and
disease. Cytokine. 40:1–16. 2007. View Article : Google Scholar
|
|
3
|
Blanco-Colio LM, Martin-Ventura JL,
Munoz-Garcia B, Moreno JA, Meilhac O, Ortiz A and Egido J: TWEAK
and Fn14. New players in the pathogenesis of atherosclerosis. Front
Biosci. 12:3648–3655. 2007. View
Article : Google Scholar
|
|
4
|
Muñoz-García B, Madrigal-Matute J, Moreno
JA, Martin-Ventura JL, López-Franco O, Sastre C, Ortega L, Burkly
LC, Egido J and Blanco-Colio LM: TWEAK-Fn14 interaction enhances
plasminogen activator inhibitor 1 and tissue factor expression in
atherosclerotic plaques and in cultured vascular smooth muscle
cells. Cardiovasc Res. 89:225–233. 2011. View Article : Google Scholar
|
|
5
|
Kim SH, Kang YJ, Kim WJ, Woo DK, Lee Y,
Kim DI, Park YB, Kwon BS, Park JE and Lee WH: TWEAK can induce
pro-inflammatory cytokines and matrix metalloproteinase-9 in
macrophages. Circ J. 68:396–399. 2004. View Article : Google Scholar
|
|
6
|
Schapira K, Burkly LC, Zheng TS, Wu P,
Groeneweg M, Rousch M, Kockx MM, Daemen MJ and Heeneman S: Fn14-Fc
fusion protein regulates atherosclerosis in ApoE−/− mice and
inhibits macrophage lipid uptake in vitro. Arterioscler Thromb Vasc
Biol. 29:2021–2027. 2009. View Article : Google Scholar
|
|
7
|
Sastre C, Fernández-Laso V,
Madrigal-Matute J, Muñoz-García B, Moreno JA, Pastor-Vargas C,
Llamas-Granda P, Burkly LC, Egido J, Martín-Ventura JL and
Blanco-Colio LM: Genetic deletion or TWEAK blocking antibody
administration reduce atherosclerosis and enhance plaque stability
in mice. J Cell Mol Med. 18:721–734. 2014. View Article : Google Scholar :
|
|
8
|
Lüscher TF and Barton M: Biology of the
endothelium. Clin Cardiol. 20 11 Suppl 2:(II): 3–10. 1997.
|
|
9
|
Münzel T, Gori T, Bruno RM and Taddei S:
Is oxidative stress a therapeutic target in cardiovascular disease?
Eur Heart J. 31:2741–2748. 2010. View Article : Google Scholar
|
|
10
|
Victor VM, Apostolova N, Herance R,
Hernandez-Mijares A and Rocha M: Oxidative stress and mitochondrial
dysfunction in atherosclerosis: Mitochondria-targeted antioxidants
as potential therapy. Curr Med Chem. 16:4654–4667. 2009. View Article : Google Scholar
|
|
11
|
Lane RK, Hilsabeck T and Rea SL: The role
of mitochondrial dysfunction in age-related diseases. Biochim
Biophys Acta. 1847:1387–1400. 2015. View Article : Google Scholar
|
|
12
|
Kadlec AO, Chabowski DS, Ait-Aissa K and
Gutterman DD: Role of PGC-1α in vascular regulation: Implications
for atherosclerosis. Arterioscler Thromb Vasc Biol. 36:1467–1474.
2016. View Article : Google Scholar :
|
|
13
|
Valle I, Alvarez-Barrientos A, Arza E,
Lamas S and Monsalve M: PGC-1alpha regulates the mitochondrial
antioxidant defense system in vascular endothelial cells.
Cardiovasc Res. 66:562–573. 2005. View Article : Google Scholar
|
|
14
|
Kahn BB, Alquier T, Carling D and Hardie
DG: AMP-activated protein kinase: Ancient energy gauge provides
clues to modern understanding of metabolism. Cell Metab. 1:15–25.
2005. View Article : Google Scholar
|
|
15
|
Ruiz-Andres O, Suarez-Alvarez B,
Sánchez-Ramos C, Monsalve M, Sanchez-Niño MD, Ruiz-Ortega M, Egido
J, Ortiz A and Sanz AB: The inflammatory cytokine TWEAK decreases
PGC-1α expression and mitochondrial function in acute kidney
injury. Kidney Int. 89:399–410. 2016. View Article : Google Scholar
|
|
16
|
Harada N, Nakayama M, Nakano H, Fukuchi Y,
Yagita H and Okumura K: Pro-inflammatory effect of TWEAK/Fn14
interaction on human umbilical vein endothelial cells. Biochem
Biophys Res Commun. 299:488–493. 2002. View Article : Google Scholar
|
|
17
|
Zhang F, Zhang M, Wang A, Xu M, Wang C, Xu
G, Zhang B, Zou X and Zhuge Y: TWEAK increases SIRT1 expression and
promotes p53 deacetylation affecting human hepatic stellate cell
senescence. Cell Biol Int. 41:147–154. 2017. View Article : Google Scholar
|
|
18
|
Wu YH, Li Q, Li P and Liu B: GSK621
activates AMPK signaling to inhibit LPS-induced TNFα production.
Biochem Biophys Res Commun. 480:289–295. 2016. View Article : Google Scholar
|
|
19
|
Chen L, Chen Q, Deng G, Kuang S, Lian J,
Wang M and Zhu H: AMPK activation by GSK621 inhibits human melanoma
cells in vitro and in vivo. Biochem Biophys Res Commun.
480:515–521. 2016. View Article : Google Scholar
|
|
20
|
Ballinger SW, Van Houten B, Jin GF,
Conklin CA and Godley BF: Hydrogen peroxide causes significant
mitochondrial DNA damage in human RPE cells. Exp Eye Res.
68:765–772. 1999. View Article : Google Scholar
|
|
21
|
Handschin C, Rhee J, Lin J, Tarr PT and
Spiegelman BM: An autoregulatory loop controls peroxisome
proliferator-activated receptor gamma coactivator 1alpha expression
in muscle. Proc Natl Acad Sci USA. 100:7111–7116. 2003. View Article : Google Scholar :
|
|
22
|
Münzel T, Sinning C, Post F, Warnholtz A
and Schulz E: Pathophysiology, diagnosis and prognostic
implications of endothelial dysfunction. Ann Med. 40:180–196. 2008.
View Article : Google Scholar
|
|
23
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109 23 Suppl
1:III27–III32. 2004. View Article : Google Scholar
|
|
24
|
Madrigal-Matute J, Fernandez-Laso V,
Sastre C, Llamas-Granda P, Egido J, Martin-Ventura JL, Zalba G and
Blanco-Colio LM: TWEAK/Fn14 interaction promotes oxidative stress
through NADPH oxidase activation in macrophages. Cardiovasc Res.
108:139–147. 2015. View Article : Google Scholar
|
|
25
|
Dikalov S: Cross talk between mitochondria
and NADPH oxidases. Free Radic Biol Med. 51:1289–1301. 2011.
View Article : Google Scholar :
|
|
26
|
Madamanchi NR and Runge MS: Mitochondrial
dysfunction in atherosclerosis. Circ Res. 100:460–473. 2007.
View Article : Google Scholar
|
|
27
|
Krzywanski DM, Moellering DR, Westbrook
DG, Dunham-Snary KJ, Brown J, Bray AW, Feeley KP, Sammy MJ, Smith
MR, Schurr TG, et al: Endothelial cell bioenergetics and
mitochondrial DNA damage differ in humans having African or West
Eurasian maternal ancestry. Circ Cardiovasc Genet. 9:26–36. 2016.
View Article : Google Scholar :
|
|
28
|
Panth N, Paudel KR and Parajuli K:
Reactive oxygen species: A key hallmark of cardiovascular disease.
Adv Med. 2016:91527322016. View Article : Google Scholar :
|
|
29
|
Yu EP and Bennett MR: The role of
mitochondrial DNA damage in the development of atherosclerosis.
Free Radic Biol Med. 100:223–230. 2016. View Article : Google Scholar
|
|
30
|
Shi J, Jiang B, Qiu Y, Guan J, Jain M, Cao
X, Bauer M, Su L, Burkly LC, Leone TC, et al: PGC1α plays a
critical role in TWEAK-induced cardiac dysfunction. PLoS One.
8:e540542013. View Article : Google Scholar :
|
|
31
|
Hindi SM, Mishra V, Bhatnagar S, Tajrishi
MM, Ogura Y, Yan Z, Burkly LC, Zheng TS and Kumar A: Regulatory
circuitry of TWEAK-Fn14 system and PGC-1α in skeletal muscle
atrophy program. FASEB J. 28:1398–1411. 2014. View Article : Google Scholar :
|
|
32
|
Macmillan-Crow LA and Cruthirds DL:
Invited review: Manganese superoxide dismutase in disease. Free
Radic Res. 34:325–336. 2001. View Article : Google Scholar
|
|
33
|
Bresciani G, da Cruz IB and
González-Gallego J: Manganese superoxide dismutase and oxidative
stress modulation. Adv Clin Chem. 68:87–130. 2015. View Article : Google Scholar
|