Introduction
Cardiac arrest (CA), also termed cardiopulmonary
arrest, is a sudden stop in effective blood circulation, due to the
failure of the heart to effectively contract or to contract at all.
CA remains a major public health issue and is the most common
direct cause of mortality in Western and developing countries
(1,2). Overall survival of cardiac arrest
patients outside of a hospital setting is only ~6.4% (3). This low rate is due to the initial
difficulty in restoring hemodynamic stability (4), as well as to a high incidence of
severe neurologic deficits caused by the CA (5). Therefore, <30% of survivors are
able to return to a normal functioning lifestyle (6).
The brain injury that results from the return of
spontaneous circulation (ROSC) is a complicated process. The
overall mechanisms remain elusive, but include altered
Ca2+ homeostasis, free radical formation, mitochondrial
dysfunction, protease activation, altered gene expression and
inflammation (7,8). These then lead to neuronal death in
the central nervous system. Among all the potential mechanisms,
inflammation stands out, as it is correlated with postischemic
neuronal apoptosis (9). NACHT,
LRR, and PYD domains-containing protein 3 (NLRP3), a component of
the inflammasome, has been reported to function as a pathogen
recognition receptor that recognizes pathogen-associated molecular
patterns (10,11). The inflammasome complex is a
central component of the innate immune response via regulation of
interleukin (IL)-1β, IL-18 and pyroptosis (12). NLPR3 is significant in
ischemia-reperfusion injuries in many tissue types, including renal
(13), brain (14) and spinal cord (15). In the cerebral cortex, activation
of the NLRP3-inflammasome induces the processing and release of
IL-1β and IL-18 (16). Deletion of
NLRP3 may ameliorate the neurovascular damage in ischemic stroke
mice (17). Therefore, ischemic
neural deficits caused by CA may also be mitigated via inhibition
of NLRP3.
Hypothermia is defined as a body core temperature
<35.0°C and has been known to be a potent putative
neuroprotectant (18). Mild
hypothermia (33°C) inhibits ischemia-induced promotion of
mitochondrial membrane permeability, which may provide
neuroprotection against cerebral injury following CA (19). Spontaneous hypothermia (SH) is
clinically associated with the risk of mortality (20). Hickey et al (21) demonstrated that rats resuscitated
from asphyxial CA developed neuroprotective SH (21). However, whether the neural deficit
resulting from CA was correlated with activation of the
inflammasome and was ameliorated by SH remains unclear. In the
present study, the neural damage alterations and inflammasome
component expression levels were elucidated following ROSC in
established CA rat models. In addition, the role of SH in the
inhibition of inflammasome component expression, and the
amelioration of the neurologic deficit and neuronal death in the
cerebral cortex were determined. The aim of the present study was
to investigate the underlying mechanisms of ROSC-induced
neurological deficits, and assess the potential strategies for the
prevention and treatment of post-CA syndrome.
Materials and methods
Grouping
In total, 84 male specific pathogen-free grade
Sprague Dawley rats (weight, 350–400 g; age, 8 weeks) were
purchased from the Laboratory Animal Center of Guangzhou University
of Chinese Medicine (Guangzhou, China). Firstly, 18 rats were
randomly separated into three groups as follows: Group I, control
group; Group II, the CA model group that received cardiopulmonary
resuscitation (CPR) 4 min after CA; and Group III, the CA model
group, which received CPR 6 min after CA. Secondly, 36 rats were
then randomly separated into two groups, including 6 control rats
and 30 CA model rats. CPR was conducted 6 min after CA. The CA rats
were equally sub-divided into five groups, according to the
different time points (6, 12, 24, 48 and 72 h) after ROSC. Thirdly,
30 rats were randomly divided into two groups, including six
control rats and 24 CA model rats. These CA rats were equally
sub-divided into SH group and controlled normothermia (CN) group.
The SH and CN groups were again divided into two, determined by 24
h (SH Group I and CN Group I) or 48 h (SH Group II and CN Group II)
after ROSC. The mortality rate of rats during the preparation of
rat models was 6.7%. The present study was approved by the ethics
committee of Sun Yat-Sen University (Guangzhou, China).
CA rat model
Rat CA and resuscitation were performed, as
previously described (21,22). Anesthesia was achieved using 45
mg/kg pentobarbital sodium. Animals were ventilated with a Harvard
Rodent Ventilator (Harvard Apparatus, Holliston, MA, USA), and the
temperature was measured and maintained at 37±0.5°C throughout the
preparation, insult, and first hour of recovery. Prior to asphyxia,
the anesthetic gases were washed out with 3 min of ventilation at
100% oxygen, followed by 2 min of room air. Vecuronium was
administered prior to asphyxia, in order to prevent reflex
respiratory efforts during asphyxia. Subsequent to the washout
period, animals were asphyxiated by disconnecting the respiratory
tubing from the ventilator for 6 min, resulting in ~5 min of
CA.
Resuscitation
Following exactly 5 min, the ventilator was
reconnected and ventilation was resumed with oxygen at a rate of 60
breaths/min. Intravenous epinephrine (0.005 mg/kg) and bicarbonate
(1.0 mEq/kg) were administered, and external chest compressions
were performed at a rate of 250–300 compressions/min. Rats
generally experienced a ROSC within 2 min. If not, an additional
dose of epinephrine was administered. Following stabilization for
at least 60 min and confirmation of adequate spontaneous
respirations, rats were extubated and weaned from oxygen back to
room air.
Temperature control
In the CN group, rats were maintained at 37±0.5°C
during CA and for 4 h after resuscitation. In the SH group, rats
were maintained at 37°C during CA, but following resuscitation were
allowed to regulate their own temperature.
Neurologic deficit scale (NDS) score
measurement
General neurologic status was assessed using the
validated NDS at 24, 48, and 72 h after CA (23). The score includes an assessment of
consciousness, respiration, cranial nerve activity, motor and
sensory function, and coordination. Normal rats have an NDS of
zero.
Enzyme-linked immunosorbent assay
(ELISA)
Arterial blood samples (10 ml) were drawn at
different time points after ROSC. Concentration changes in serum
S100 calcium-binding protein B [S100B; cat. no. H6-KA0037, Multi
Sciences (Lianke) Biotech Co., Ltd., Hangzhou, China], IL-1β [cat.
no. 70-EK201B1/2, Multi Sciences (Lianke) Biotech Co., Ltd.] and
IL-18 (cat. no. RK-KOA0362; Rockland Immunochemicals, Inc.,
Limerick, PA, USA) in the cerebral cortex were monitored by ELISA
for the different groups. After coating with coating buffer (50
mmol/l sodium carbonate buffer, pH=9.6), plates were sequentially
washed with phosphate buffered saline with 0.05% Tween-20 (PBST)
buffer (cat. no. A100235-0001; Sangon Biotech Co., Ltd., Shanghai,
China), blocked with 1% bovine serum albumin (BSA; cat. no.
A602440-0050, Sangon Biotech Co., Ltd.), and incubated for 1 h at
37°C. Anti-S100B (1:200), anti-IL-1β (1:200) or anti-IL-18
antibodies (1:100), and horseradish peroxidase-conjugated antibody
(1:1,000; all included in the corresponding ELISA kit) were
sequentially added and incubated for 1 h at 37°C. The chromogenic
substrate 3,3′,5,5′-Tetramethylbenzidine was added for detection.
Absorbance was measured at a wavelength of 450 nm using an EnSpire
multimode plate reader (Perkin Elmer, Waltham, Massachusetts).
Western blotting
Tissue samples were lysed in
radioimmunoprecipitation buffer (50 mM Tris-HCl buffer pH 7.4, 150
mM NaCl, 5 mM EDTA, 1% NP-40 and 0.25% sodium deoxycholate). Total
protein was extracted from tissue lysate by centrifugation at
12,000 × g for 10 min at 4°C and protein concentrations were
measured using a Bicinchoninic Acid assay (Sangon Biotech Co.,
Ltd.) according to the manufacturer's instructions. A total of 2 µg
protein was loaded onto each lane of 10% polyacrylamide gel (250V
voltage for 2 h) and blotted onto a polyvinylidene difluoride
(PVDF) membrane. After blocking with PBST containing 5% nonfat dry
milk, the membrane was incubated with antibodies against NLRP3
(cat. no. 13158; 1:500), apoptosis-associated speck-like protein
containing a CARD (ASC; cat. no. 67824; 1:500), caspase-1 (cat. no.
2225; 1:400), caspase-3 (cat. no. 9662; 1:500), and β-tubulin (cat.
no. 2146; 1:500; all Cell Signaling Technology, Inc., Danvers, MA,
USA). Peroxidase-linked anti rabbit IgG (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) served as a secondary antibody. These
proteins were visualized using an ECL western blotting detection
kit (GE Healthcare, Chicago, IL, USA).
Immunohistochemistry
The rats were anesthetized with an overdose of 150
mg/kg pentobarbital and then sacrificed. The whole brain was
immediately removed and frozen on dry ice. Then, the cerebral
cortex site was separated from the whole brain. The sections of the
cerebral cortex (~1.5 cm3) were sequentially washed in
dimethylbenzene and ethanol, before being blocked in 3%
H2O2. All nonspecific binding sites were
blocked for 30 min in PBS with 5% BSA. The sections were then
incubated at 4°C with anti-NLRP3, anti-ASC, anti-caspase-1, or
anti-caspase-3 antibodies (Cell Signaling Technology, Inc.)
overnight. Biotin-conjugated secondary antibody (cat. no. ab97044;
Abcam, Cambridge, MA, USA) was applied to the slides and incubated
for 1 h at room temperature. Finally, 3,3′-diaminobenzidine
(DAB)/H2O2 was added to the surface of the
slide to develop the color at room temperature for 10 min. The
slides were visualized using a Nikon ECLIPSE 90i.
TUNEL assay
Sections were perfused with dimethylbenzene and
sequentially washed with different concentrations of ethanol and
blocked in 3% H2O2. These sections were
incubated in 5% BSA for 30 min and the fragmented DNA was labeled
with the TUNEL reaction solution at 37°C for 1 h.
Converter-peroxidase was added to the sections at 37°C for 30 min
before the TUNEL-positive nuclei were visualized by adding the DAB
staining solution. All images were captured using a Nikon ECLIPSE
90i.
Statistical analysis
Data are presented as means ± standard error of the
mean and analysis was performed with GraphPad Prism 6 software
(GraphPad Software, Inc., La Jolla, CA, USA). The normalized
intensity for western blotting bands was measured with Image J
software version 1.45 (National Institutes of Health, Bethesda, MD,
USA). Unpaired Student's t-tests and one-way ANOVA with a Tukey
post hoc test were used to determine significant differences.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Increasing duration of CA aggravated
neural defects
In order to elucidate the role of CA in neural
defects, physiological parameters were compared between the control
group and the two asphyxia groups, determining that there was no
significant difference in physiological parameters among the three
groups (Table I). Similarly, no
apparent alteration in the majority of physiological parameters was
observed between the two asphyxia groups following resuscitation.
However, the duration of ROSC and the duration without blood flow
in the asphyxia group that received CPR at 6 min after CA (Group
III) was significantly longer than in the asphyxia group that
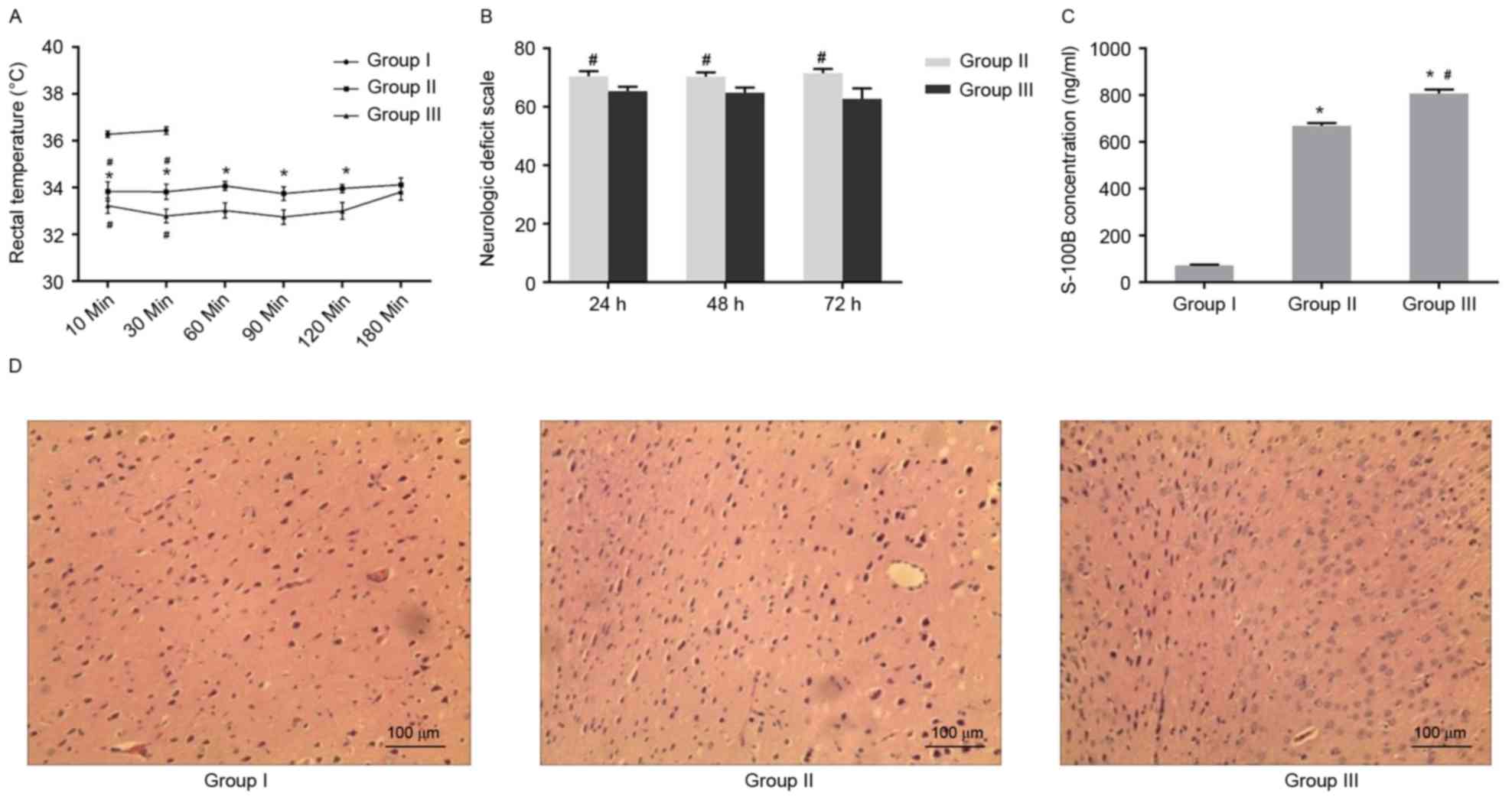
received CPR at 4 min after CA (Group II; Table II). In addition, it was found that
anal temperature was lower in the two asphyxia groups when compared
with the control group. In addition, the rectal temperature in
Group III was lower than that in Group II at five consecutive time
points (from 10–120 min), but not at 180 min after ROSC (Fig. 1A).
 | Table I.Comparison of physiological parameters
between Group I (Sham) and Groups II and III (the asphyxia
groups). |
Table I.
Comparison of physiological parameters
between Group I (Sham) and Groups II and III (the asphyxia
groups).
| Parameter | Group I (n=6) | Group II (n=6) | Group III (n=6) | P-value |
|---|
| Body weight
(g) |
379.0±18.0 |
376.0±16.0 |
370.0±14.0 | 0.7901 |
| Rectal temperature
(°C) |
36.9±0.3 |
37.0±0.2 |
37.0±0.2 | 0.7008 |
| Mean arterial blood
pressure (mmHg) |
110.0±7.0 |
119.0±8.0 |
109.0±9.0 | 0.1033 |
| Heart rate
(bpm) |
314.0±29.0 |
310.0±29.0 |
313.0±25.0 | 0.9599 |
| Partial pressure of
carbon dioxide in end expiratory gas (mmHg) |
36.3±2.8 |
37.1±2.1 |
37.9±2.4 | 0.6253 |
| Table II.Comparison of physiological
parameters between the two asphyxia groups prior to CPR. |
Table II.
Comparison of physiological
parameters between the two asphyxia groups prior to CPR.
| Groups | Group II (n=6) | Group III
(n=6) | P-value |
|---|
| Duration of
asphyxia before CA (sec) |
190±14 |
199±30 | 0.5334 |
| CPR time to ROSC
(sec) |
94±27 |
163±10a | <0.001 |
| No flow time
(sec) |
214±27 |
540±15a | <0.001 |
| Mean arterial blood
pressure (mmHg) |
| 10 min
post ROSC |
118±12 |
109±18 | 0.3947 |
| 30 min
post ROSC |
102±8 |
117±16 | 0.0561 |
| 60 min
post ROSC |
114±18 |
104±16 | 0.3558 |
| 90 min
post ROSC |
100±16 |
113±10 | 0.0555 |
| 120 min
post ROSC |
104±13 |
105±19 | 0.8802 |
| Heart rate
(bpm) |
| 10 min
post ROSC |
311±41 |
306±32 | 0.7353 |
| 30 min
post ROSC |
295±18 |
308±39 | 0.4737 |
| 60 min
post ROSC |
313±28 |
298±30 | 0.3682 |
| 90 min
post ROSC |
288±22 |
300±31 | 0.4599 |
| 120 min
post ROSC |
306±27 |
316±43 | 0.625 |
| Partial pressure of
carbon dioxide in end expiratory gas (mmHg) |
| 10 min
post ROSC |
44.5±27 |
49±4.6 | 0.0731 |
| 30 min
post ROSC |
65.1±5.7 |
71.8±4.5b | 0.0454 |
| 60 min
post ROSC |
58.0±4.1 |
57.3±4.7 | 0.7632 |
| 90 min
post ROSC |
43.7±3.3 |
45.6±3.2 | 0.3342 |
| 120 min
post ROSC |
40.4±3.4 |
40.3±2.4 | 0.9616 |
Subsequently, it was determined whether the various
neural defects were present in the asphyxia models. As shown in
Fig. 1B, the NDS score was
significantly lower in Group III than in Group II at 24, 48 and 72
h after CPR. Additionally, the S100B serum concentration of Group
II and III was significantly greater than that in Group I at 72 h
after CPR (Fig. 1C). The
immunohistochemistry results indicated that cell morphology in the
cerebral cortex was normal in the control (Group I; Fig. 1D). By contrast, morphological
damage was observed in the cerebral cortex of Group II, including a
vacuole and swelling in the cytoplasm (Fig. 1E). Further severe damage was
observed in Group III, where necrosis was observed in the cells of
cerebral cortex (Fig. 1F). These
data demonstrated that increasing duration of CA increased the
severity of neural damage in the cerebral cortex and enhanced the
incidence of inflammation.
Dynamic expression patterns of NLRP3,
ASC and caspase-1 following ROSC
As the observed neural defects were associated with
inflammation, whether the expression levels of certain key
inflammatory factors were altered following resuscitation in the CA
models were investigated. The potential changes in physiological
parameters between the control and five asphyxia groups that were
resuscitated at different time points after ROSC were detected. No
apparent changes in physiological parameters were observed among
the six groups, as presented in Table III. Furthermore, no significant
difference in duration of asphyxia and resuscitation among the six
groups was observed (Table IV).
Whether the expression levels of key components of the
inflammasome, including NLRP3, ASC and caspase-1 were altered
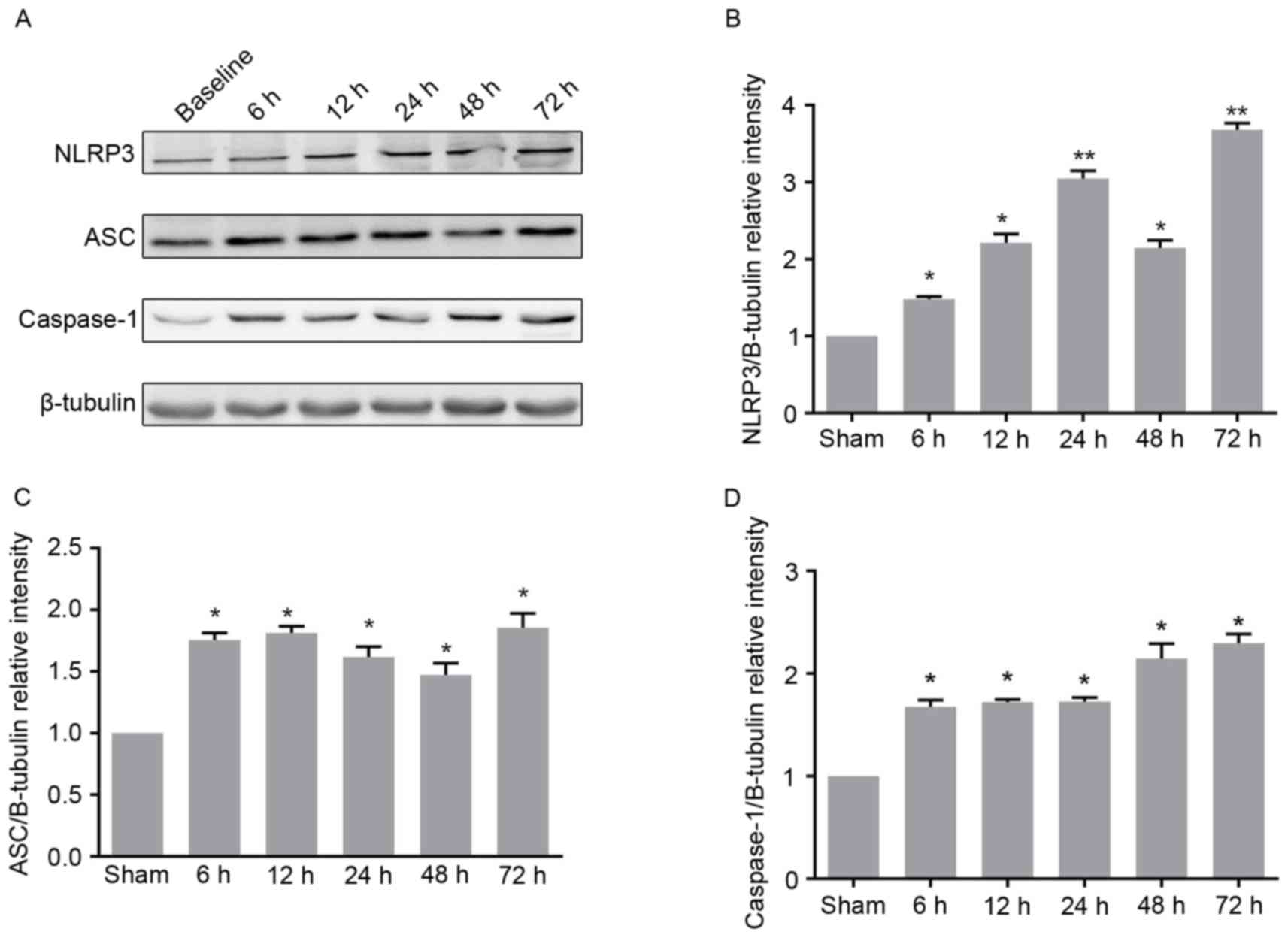
following ROSC were determined. The expression level of NLRP3 was
constantly demonstrated to increase at different time points,
except for 48 h after ROSC (Fig. 2A
and B). In addition, it was found that the expression level of
ASC significantly increased at 6 h after ROSC, but did not exhibit
any subsequent further increase (Fig.
2A and C). The expression level of caspase-1 increased at 6 and
48 h after ROSC, but showed no further increase at 12, 24 or 72 h
after ROSC compared with the previous time points (Fig. 2A and D). These findings were
confirmed by immunohistochemical analysis. The number of ASC, NLRP3
and caspase-1 positive cells significantly increased at different
time points after ROSC, compared with what was observed in the
control (Fig. 3A). Collectively,
these results indicate that the expression levels of NLRP3, ASC and
caspase-1 increase with increasing time following ROSC, further
enhancing inflammation.
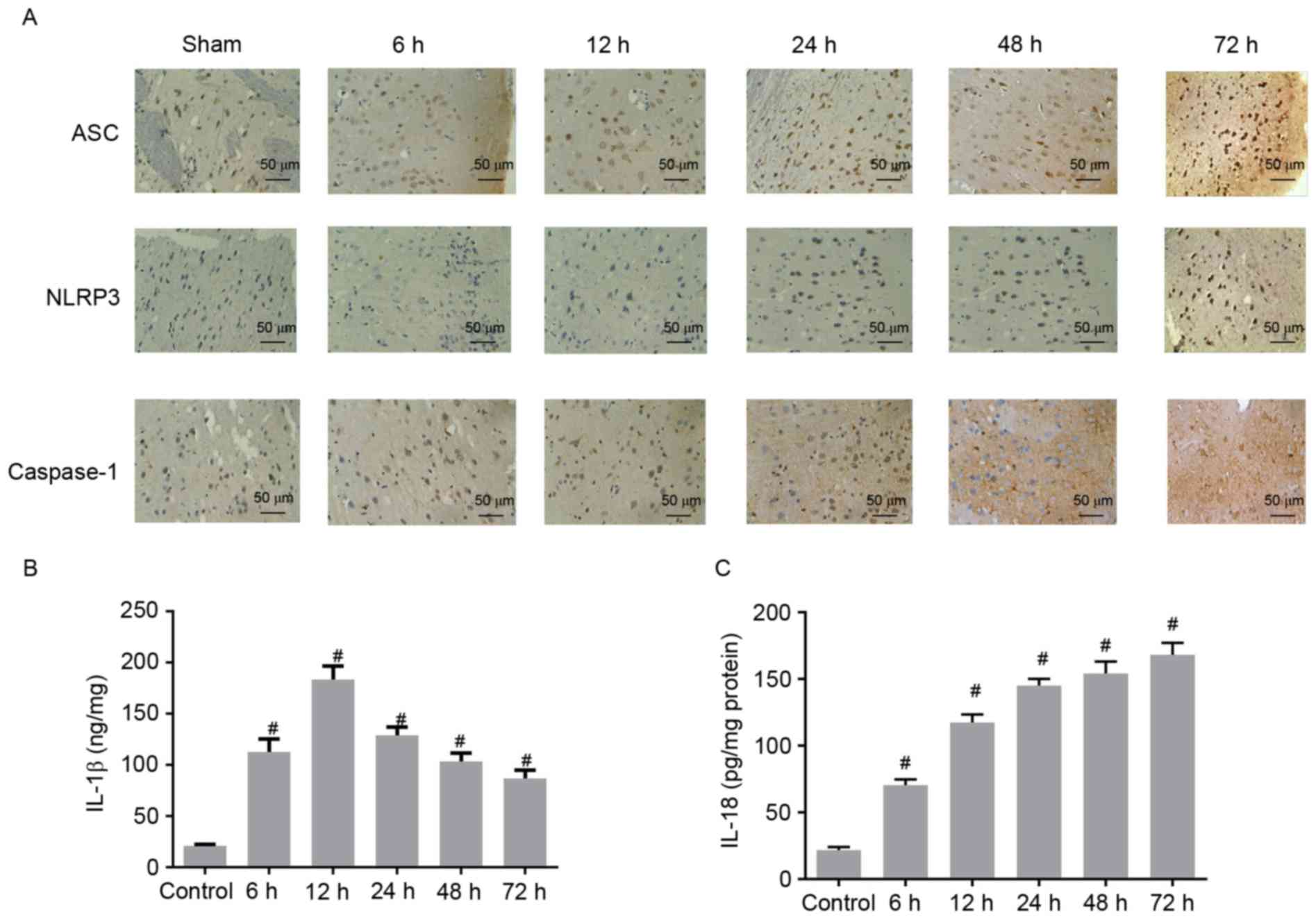 | Figure 3.(A) Immunostaining analysis of ASC,
NLRP3 and caspase-1 in the cerebral cortex of control rats and CA
models at 6, 12, 24, 48, and 72 h after ROSC. Scale bar, 50 µm. The
concentrations of (B) IL-1β and (C) IL-18 increased in the cerebral
cortex of CA models following ROSC. #P<0.01 vs. the
control group (unpaired Student's t-test). ASC,
apoptosis-associated speck-like protein containing a CARD; NLRP3,
NACHT, LRR, and PYD domains-containing protein 3; CA, cardiac
arrest; ROSC, return of spontaneous circulation; IL,
interleukin. |
| Table III.Comparison of physiological
parameters among the sham group and five asphyxia groups at
different time points after ROSC. |
Table III.
Comparison of physiological
parameters among the sham group and five asphyxia groups at
different time points after ROSC.
| Group (n) | Body weight
(g) | Rectal temperature
(°C) | Mean arterial blood
pressure (mmHg) | Heart rate
(bpm) |
PETCO2 (mmHg) |
|---|
| Sham Group (6) | 375±18 | 36.9±0.4 | 115±12 | 320±25 | 35.73±2.0 |
| Time after
ROSC |
| 6 h
(6) | 376±16 | 36.9±0.2 | 109±16 | 317±25 | 37.0±2.1 |
| 12 h
(6) | 374±16 | 37.0±0.2 | 110±10 | 314±36 | 37.7±2.5 |
| 24 h
(5) | 368±19 | 36.8±0.2 | 106±14 | 312±26 | 38.0±2.7 |
| 48 h
(6) | 368±15 | 36.9±0.3 | 113±10 | 305±30 | 38.3±2.1 |
| 72 h
(6) | 383±15 | 37.1±0.2 | 109±13 | 312±34 | 38.2±3.2 |
| P-value | 0.5976 | 0.717 | 0.9287 | 0.9670 | 0.4426 |
| Table IV.Comparison of asphyxia and CPR
duration among the five asphyxia groups with different time points
after ROSC (n=6 per group). |
Table IV.
Comparison of asphyxia and CPR
duration among the five asphyxia groups with different time points
after ROSC (n=6 per group).
| Time after ROSC
(h) | Asphyxia (sec) | CPR (sec) |
|---|
| 6 | 189±32 | 166±26 |
| 12 | 204±30 | 175±33 |
| 24 | 215±27 | 179±33 |
| 48 | 194±24 | 168±27 |
| 72 | 182±26 | 164±17 |
| P-value | 0.4245 | 0.8821 |
Concentrations of IL-1β and IL-18
increased in the cerebral cortex after ROSC
ELISA assays were performed to determine whether the
concentrations of IL-1β and IL-18 in the cerebral cortex increased
after ROSC. Compared with the control group, the concentration of
IL-1β was significantly higher at 6 h and reached the maximal level
at 12 h after ROSC (Fig. 3B). The
concentration of IL-1β concentration gradually decreased at 24, 48
and 72 h after ROSC, but continued to be higher than that of the
control group (Fig. 3B).
Similarly, the IL-18 concentration was significantly higher after 6
h, and consistently increased with increasing time after ROSC
(Fig. 3C). These data demonstrated
that IL-1β and IL-18 concentrations were elevated in CA rat models
after ROSC, indicating the aggravation of inflammation in the
cerebral cortex.
SH alleviated neurological deficiency,
apoptosis and inflammation in the CA rat model
Subsequently, the effects of SH on the neurological
deficiency and inflammation observed in CA models were elucidated.
No significant differences in physiological parameters were
observed between the control, SH and CN groups (Table V). In the CA models, no apparent
alteration in asphyxia time and CPR to ROSC time was identified,
nor was any change observed in the tested physiological parameters
among all four asphyxia groups following ROSC, as presented in
Table VI. By contrast, the rectal
temperature was significantly lower in the SH group than in the CN
group (Fig. 4A). The NDS score was
significantly higher in the SH group compared with the CN group at
24 and 48 h after ROSC (Fig. 4B).
Additionally, the S100B concentration was shown to be reduced in
the SH group compared with the CN group at 24 and 48 h after ROSC
(Fig. 4C). Western blot analysis
demonstrated that the expression levels of NLRP3, ASC, caspase-1
and caspase-3 markedly decreased in the SH groups compared with the
CN group (Fig. 4D-H). Similar
results were obtained via immunohistochemical analysis. The number
of NLRP3, ASC, caspase-1 and caspase-3 positive cells in the SH
group significantly decreased compared with the CN group (Fig. 5A). In addition, apoptosis was
partially inhibited in the SH group, as the ratio of TUNEL-positive
cells in this group was markedly reduced when compared with the CN
group, while there was a limited number of apoptotic cells in the
control group (Fig. 5B).
Furthermore, SH after ROSC markedly decreased the concentrations of
IL-1β and IL-18 (Fig. 5C).
Finally, the cells in the cerebral cortex exhibited more severe
damage in the CN group than did the cells in the SH group, while
the cell morphology in the control group was normal (Fig. 5D). Thus, these data demonstrate
that SH alleviated neural defects, apoptosis, and inflammation in
the cerebral cortex, when compared with the CN group.
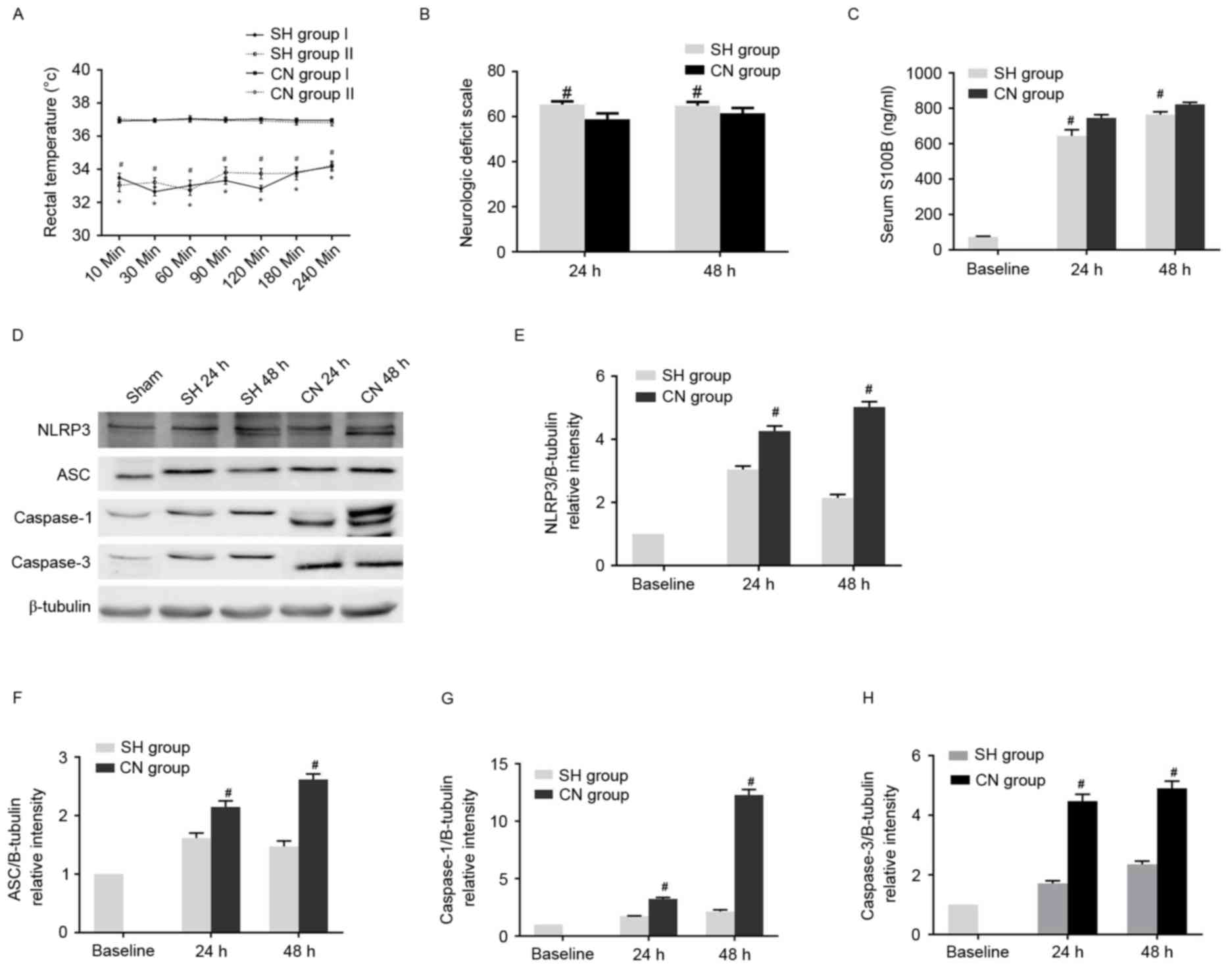 | Figure 4.(A) Rectal temperature was lower in
the SH group than in the CN group. *P<0.05 vs. CN Group I;
#P<0.05 vs. CN Group II. (B) Neurologic deficit scale
was higher in the SH group than in the CN group.
#P<0.05 vs. CN group. (C) Serum S100B concentration
was lower in the SH group than in the CN group.
#P<0.05 vs. CN group. (D) Expression levels of NLRP3,
ASC, caspase-1 and caspase-3 were observed to significantly
decrease in the SH compared with the CN groups. Statistical
analysis of the relative intensity of (E) NLRP3, (F) ASC, (G)
caspase-1 and (H) caspase-3 expression. #P<0.05 vs.
Baseline control group (unpaired Student's t-test). SH, spontaneous
hypothermia; CN, controlled normothermia; S100B, S100
calcium-binding protein B; NLRP3, NACHT, LRR, and PYD
domains-containing protein 3; ASC, apoptosis-associated speck-like
protein containing a CARD. |
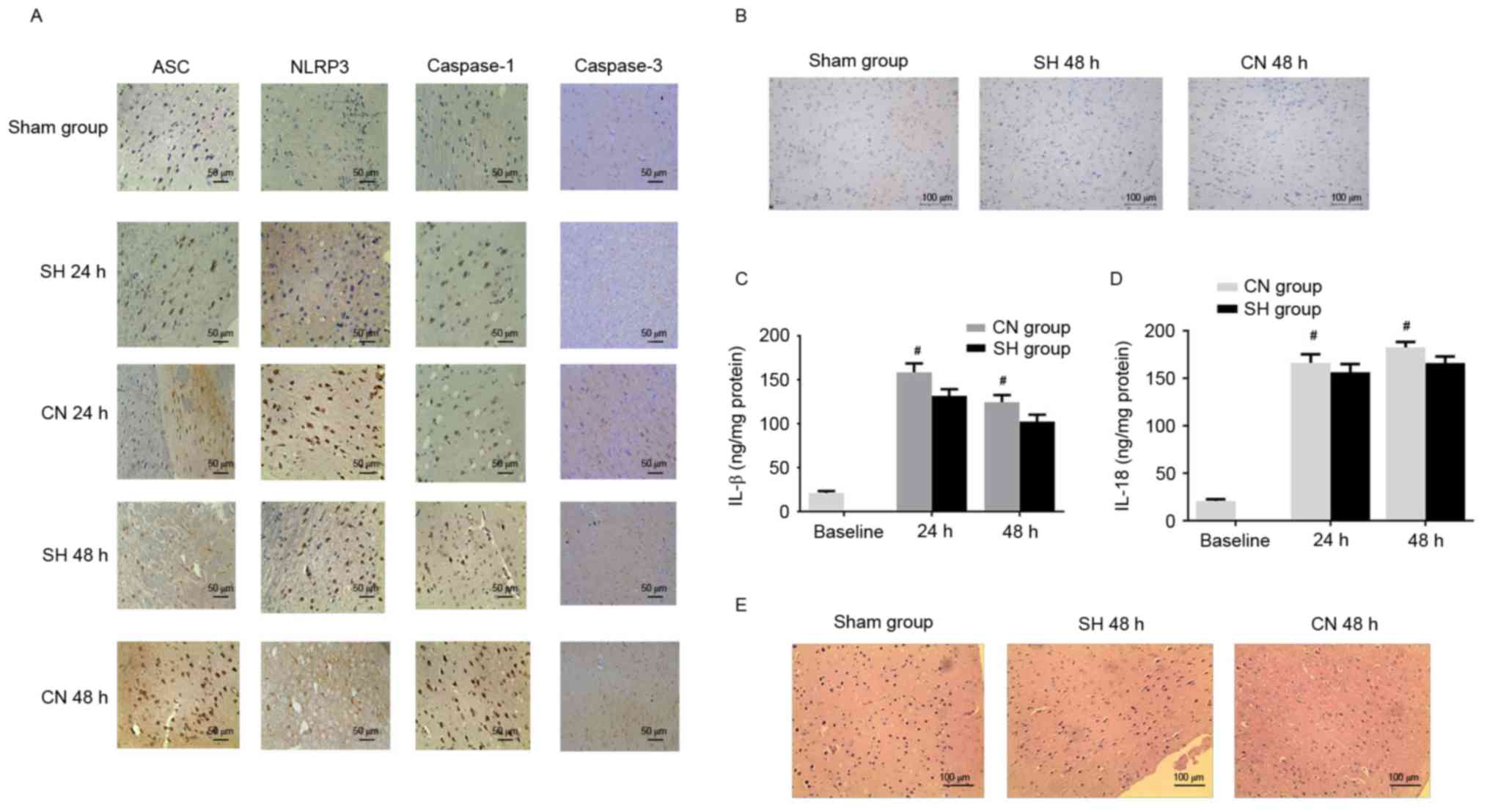 | Figure 5.(A) Immunohistochemistry indicated
that the number of ASC, NLRP3, caspase-1 and caspase-3 positive
cells in the cerebral cortex increased in the CN group compared
with the SH group. Scale bar, 50 µm. (B) A small number of
TUNEL-positive cells was observed in the cerebral cortex of the
control (Sham) group. The number of TUNEL positive cells was lower
in the SH group compared with the CN group. Scale bar, 100 µm. The
(C) IL-1β and (D) IL-18 concentrations were decreased in the SH
group compared with the CN group. (E) Mild damage was observed in
the control (Sham) group, while the damage was more severe in the
CN group compared with the SH group. Scale bar, 100 µm.
#P<0.05 vs. CN group (unpaired Student's t-test).
ASC, apoptosis-associated speck-like protein containing a CARD;
NLRP3, NACHT, LRR, and PYD domains-containing protein 3; CN,
controlled normothermia; SH, spontaneous hypothermia; IL,
interleukin. |
| Table V.Comparison of physiological
parameters among the sham group and asphyxia groups with SH and
CN. |
Table V.
Comparison of physiological
parameters among the sham group and asphyxia groups with SH and
CN.
| Group | Body weight
(g) | Rectal temperature
(°C) | Mean arterial blood
pressure (mmHg) | Heart rate
(bpm) |
PETCO2 (mmHg) |
|---|
| Sham (6) | 378±19 | 37.0±0.3 | 105±13 | 320±22 | 37.1±3.3 |
| SH Group I (6) | 385±10 | 37.0±0.3 | 113±13 | 313±28 | 36.7±1.9 |
| CN Group I (6) | 376±16 | 36.9±0.3 | 111±12 | 312±25 | 37.1±2.5 |
| SH Group II
(6) | 376±21 | 37.0±0.4 | 107±17 | 304±28 | 36.4±1.9 |
| CN Group II
(6) | 377±18 | 37.0±0.3 | 109±12 | 310±32 | 37.6±2.6 |
| P-value | 0.8626 | 0.8268 | 0.9287 | 0.8774 | 0.7148 |
| Table VI.Comparison of asphyxia and
resuscitation duration, as well as physiological parameters among
the four asphyxia groups (n=6). |
Table VI.
Comparison of asphyxia and
resuscitation duration, as well as physiological parameters among
the four asphyxia groups (n=6).
|
| Group I | Group II |
|
|---|
|
|
|
|
|
|---|
| Group | SH | CN | SH | CN | P-value |
|---|
| Asphyxia time
(sec) |
190±30 |
198±33 |
206±38 |
196±40 | 0.8816 |
| Cardiopulmonary
resuscitation to ROSC time (sec) |
164±18 |
180±35 |
176±27 |
175±34 | 0.7839 |
| Mean arterial blood
pressure (mmHg) |
| 30 min
post ROSC |
110±10 |
125±10 |
117±11 |
116±15 | 0.2102 |
| 60 min
post ROSC |
108±11 |
115±12 |
109±12 |
119±14 | 0.3488 |
| 90 min
post ROSC |
106±10 |
105±13 |
102±13 |
110±16 | 0.7597 |
| 120 min
post ROSC |
107±12 |
108±11 |
114±13 |
117±18 | 0.5464 |
| Heart rate
(bpm) |
| 30 min
post ROSC |
310±34 |
317±29 |
316±32 |
308±33 | 0.9530 |
| 60 min
post ROSC |
310±30 |
316±29 |
307±25 |
322±27 | 0.7792 |
| 90 min
post ROSC |
309±32 |
318±33 |
320±29 |
312±23 | 0.9069 |
| 120 min
post ROSC |
317±40 |
327±34 |
306±27 |
313±28 | 0.7242 |
|
PETCO2 (mmHg) |
| 30 min
post ROSC |
62.9±6.2 |
68.2±4.3 |
66.03±4.6 |
70.00±4.2 | 0.1060 |
| 60 min
post ROSC |
48.7±5.7 |
46.1±6.8 |
45.9±5.5 |
47.9±6.7 | 0.8226 |
| 90 min
post ROSC |
37.9±2.5 |
36.9±2.8 |
36.8±2.3 |
39.6±3.6 | 0.2945 |
| 120 min
post ROSC |
36.6±2.9 |
38.60±3.2 |
34.5±5.7 |
38.1±3.0 | 0.2784 |
Discussion
The present study demonstrates that the expression
levels of inflammasome components changed in CA rat models
following ROSC, potentially indicating the participation of the
inflammasome in post-CA syndrome. In addition, it was found that
spontaneous hypothermia mitigated the neural defects and
inflammation induced by CPR and ROSC in CA models compared with the
CN model, providing mechanistic exploration of the effect of SH on
post-CA syndrome and its potential correlation with the
inflammasome.
Hypothermia is common in patients with neurologic
disorders and those in a critical condition. In the present study,
hypothermia was demonstrated in the established CA rat models. This
is consistent with previous findings showing that rats resuscitated
from asphyxial CA developed mild to moderate hypothermia (21). An important factor leading to
hypothermia is the duration of the perfusion without blood flow. It
was identified that a longer duration of asphyxia prior to
resuscitation caused a lower body temperature and thus a more
serious neural deficit. Hypothermia is a double-edged sword,
however, as SH exerted a protective effect against neurologic
damage and inflammation in the CA models. Until now, hypothermia
has only been a validated effective treatment for brain
resuscitation following CA. There is no consensus on the specific
duration of hypothermia in the treatment, but the generally
accepted time is 12–24 h (24,25).
Theoretically, longer durations of hypothermia are more beneficial
to the neural tissues. However, there are side effects during the
treatment, such as prolonged clotting time and pulmonary infection.
Consistent with the current finding in rat models, Vijlbrief et
al (26) identified that
hypothermia following perinatal asphyxia exerted a beneficial
effect on cardiac function in infants, indicating its potential
clinical application.
CA induced increased expression levels of
pro-inflammatory cytokines, therefore, aggravating the inflammatory
reaction. In the hypothermia treatment, however, expression levels
of inflammation-associated components are altered. Callaway et
al (27) identified that SH
alleviated the NDS in CA models, but concluded that altering the
inflammatory response subsequent to CA is not necessary to achieve
the beneficial effects of hypothermia (27). By contrast, NLRP3 expression was
demonstrated to be lower in the SH group compared with the CN
group, and the secretion of IL-18 and IL-1β was decreased in the SH
group. This discrepancy is likely due to the different techniques
used to initiate the asphyxia and trigger the activation of the
NLRP3 inflammasome. Activation of the NLRP3 inflammasome depends on
an increase in ATP provision and calcium load, while hypothermia
may inhibit the calcium influx and upload (28). Therefore, SH following
resuscitation may work via the above-mentioned mechanisms to
inhibit the NLRP3 inflammasome, and potentially downstream IL-18
and IL-1β expression, to exert a neuroprotective effect. However,
the specific mechanisms involved require further investigation.
Cell death or apoptosis occurs following ischemia or
reperfusion. Previous reports have indicated that hypothermia
inhibits caspase-3 mRNA expression (29). In the caspase family, caspase-1
participates in the inflammatory reaction and is responsible for
the activation of pro-inflammatory cytokines, while caspase-3
mediates apoptosis (30). In the
present study, CA caused upregulation of the expression level of
caspase-1 and caspase-3; further demonstrating that inflammation
and apoptosis were promoted by CA. The initiation of apoptosis
occurs subsequent to inflammation. There is, therefore, enough time
to intervene before CA induces apoptosis. Various types of
treatments such as hypothermia may inhibit apoptosis via caspase-3
inhibition. The current study identified that expression levels of
caspase-1 and caspase-3 decreased with SH in CA models. This result
was similar to that observed in the hippocampus, where caspase-1
and caspase-3 expression increased following asphyxia. In recent
years, combination therapy has been used to ameliorate neural
disorders (31). Therefore, the
present study provides solid evidence to support the potential
clinical application of hypothermia. Future investigations will
focus on survival prognosis, in order to determine whether
hypothermia completely replaces the standard treatment for
CA-induced neurologic deficits.
In conclusion, the present study demonstrates that
SH ameliorated inflammation and neurologic deficit in CA models
following resuscitation. The findings are important to increase
understanding of the underlying mechanisms in CA-induced
inflammation and neurologic damage following ROSC. Furthermore, the
current findings promote a potential novel therapeutic strategy,
which may be a promising candidate for increasing the survival rate
and quality of life for patients suffering from post-CA
syndrome.
Acknowledgements
The current study was supported by a research grant
from project of Leading Talents in the Pearl River Talent Plan of
Guangdong Province (grant no. 81000-42020004).
References
|
1
|
Berdowski J, Berg RA, Tijssen JG and
Koster RW: Global incidences of out-of-hospital cardiac arrest and
survival rates: Systematic review of 67 prospective studies.
Resuscitation. 81:1479–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hua W, Zhang LF, Wu YF, Liu XQ, Guo DS,
Zhou HL, Gou ZP, Zhao LC, Niu HX, Chen KP, et al: Incidence of
sudden cardiac death in China: Analysis of 4 regional populations.
J Am Coll Cardiol. 54:1110–1118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nichol G, Stiell IG, Laupacis A, Pham B,
Maio VJ and Wells GA: A cumulative meta-analysis of the
effectiveness of defibrillator-capable emergency medical services
for victims of out-of-hospital cardiac arrest. Ann Emerg Med.
34:517–525. 1999. View Article : Google Scholar
|
|
4
|
Wang HE, Min A, Hostler D, Chang CC and
Callaway CW: Differential effects of out-of-hospital interventions
on short- and long-term survival after cardiopulmonary arrest.
Resuscitation. 67:69–74. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laver S, Farrow C, Turner D and Nolan J:
Mode of death after admission to an intensive care unit following
cardiac arrest. Intensive Care Med. 30:2126–2128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nolan JP, Laver SR, Welch CA, Harrison DA,
Gupta V and Rowan K: Outcome following admission to UK intensive
care units after cardiac arrest: A secondary analysis of the ICNARC
Case Mix Programme Database. Anaesthesia. 62:1207–1216. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Neumar RW: Molecular mechanisms of
ischemic neuronal injury. Ann Emerg Med. 36:483–506. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson EM Jr, Greenlund LJ, Akins PT and
Hsu CY: Neuronal apoptosis: Current understanding of molecular
mechanisms and potential role in ischemic brain injury. J
Neurotrauma. 12:843–852. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mizushima H, Zhou CJ, Dohi K, Horai R,
Asano M, Iwakura Y, Hirabayashi T, Arata S, Nakajo S, Takaki A, et
al: Reduced postischemic apoptosis in the hippocampus of mice
deficient in interleukin-1. J Comp Neurol. 448:203–216. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu A and Wu H: Structural mechanisms of
inflammasome assembly. FEBS J. 282:435–444. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinon F: Detection of immune danger
signals by NALP3. J Leukoc Biol. 83:507–511. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rathinam VA, Vanaja SK and Fitzgerald KA:
Regulation of inflammasome signaling. Nat Immunol. 13:333–342.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shigeoka AA, Mueller JL, Kambo A, Mathison
JC, King AJ, Hall WF, Correia Jda S, Ulevitch RJ, Hoffman HM and
McKay DB: An inflammasome-independent role for epithelial-expressed
Nlrp3 in renal ischemia-reperfusion injury. J Immunol.
185:6277–6285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu Y, Sheng H, Bao Q, Wang Y, Lu J and Ni
X: NLRP3 inflammasome activation mediates estrogen
deficiency-induced depression- and anxiety-like behavior and
hippocampal inflammation in mice. Brain Behav Immun. 56:175–186.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zendedel A, Johann S, Mehrabi S, Joghataei
MT, Hassanzadeh G, Kipp M and Beyer C: Activation and regulation of
NLRP3 Inflammasome by Intrathecal application of SDF-1a in a spinal
cord injury model. Mol Neurobiol. 53:3063–3075. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu HD, Li W, Chen ZR, Hu YC, Zhang DD,
Shen W, Zhou ML, Zhu L and Hang CH: Expression of the NLRP3
inflammasome in cerebral cortex after traumatic brain injury in a
rat model. Neurochem Res. 38:2072–2083. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang F, Wang Z, Wei X, Han H, Meng X,
Zhang Y, Shi W, Li F, Xin T, Pang Q and Yi F: NLRP3 deficiency
ameliorates neurovascular damage in experimental ischemic stroke. J
Cereb Blood Flow Metab. 34:660–667. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu L and Yenari MA: Therapeutic
hypothermia: Neuroprotective mechanisms. Front Biosci. 12:816–825.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gong P, Hua R, Zhang Y, Zhao H, Tang Z,
Mei X, Zhang M, Cui J and Li C: Hypothermia-induced neuroprotection
is associated with reduced mitochondrial membrane permeability in a
swine model of cardiac arrest. J Cerebr Blood Flow Metad.
33:928–934. 2013. View Article : Google Scholar
|
|
20
|
Rubiano AM, Sanchez AI, Estebanez G,
Peitzman A, Sperry J and Puyana JC: The effect of admission
spontaneous hypothermia on patients with severe traumatic brain
injury. Injury. 44:1219–1225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hickey RW, Ferimer H, Alexander HL, Garman
RH, Callaway CW, Hicks S, Safar P, Graham SH and Kochanek PM:
Delayed, spontaneous hypothermia reduces neuronal damage after
asphyxial cardiac arrest in rats. Crit Care Med. 28:3511–3516.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
D'Cruz BJ, Logue ES, Falke E, DeFranco DB
and Callaway CW: Hypothermia and ERK activation after cardiac
arrest. Brain Res. 1064:108–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jia X, Koenig MA, Shin HC, Zhen G, Pardo
CA, Hanley DF, Thakor NV and Geocadin RG: Improving neurological
outcomes post-cardiac arrest in a rat model: Immediate hypothermia
and quantitative EEG monitoring. Resuscitation. 76:431–442. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Drabek T, Janata A, Wilson CD, Stezoski J,
Janesko-Feldman K, Tisherman SA, Foley LM, Verrier JD and Kochanek
PM: Minocycline attenuates brain tissue levels of TNF-α produced by
neurons after prolonged hypothermic cardiac arrest in rats.
Resuscitation. 85:284–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kida K, Shirozu K, Yu B, Mandeville JB,
Bloch KD and Ichinose F: Beneficial effects of nitric oxide on
outcomes after cardiac arrest and cardiopulmonary resuscitation in
hypothermia-treated mice. Anesthesiology. 120:880–889. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vijlbrief DC, Benders MJ, Kemperman H, van
Bel F and de Vries WB: Cardiac biomarkers as indicators of
hemodynamic adaptation during postasphyxial hypothermia treatment.
Neonatology. 102:243–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Callaway CW, Rittenberger JC, Logue ES and
McMichael MJ: Hypothermia after cardiac arrest does not alter serum
inflammatory markers. Crit Care Med. 36:2607–2612. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ding L, Gao X, Yu S and Yang J: Effects of
mild and moderate hypothemia therapy on expression of cerebral
neuron apoptosis related proteins and glial fiber acidic protein
after rat cardio-pulmonary resuscitation. Cell Biochem Biophys.
70:1519–1525. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu J, Shen Y, Qian HY, Liu LJ, Zhou BC,
Xiao Y, Mao JN, An GY, Rui MZ, Wang T and Zhu CL: Effects of mild
hypothermia on the ROS and expression of caspase-3 mRNA and LC3 of
hippocampus nerve cells in rats after cardiopulmonary
resuscitation. World J Emerg Med. 5:298–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thornberry NA and Lazebnik Y: Caspases:
Enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wiklund L, Zoerner F, Semenas E, Miclescu
A, Basu S and Sharma HS: Improved neuroprotective effect of
methylene blue with hypothermia after porcine cardiac arrest. Acta
Anaesth Scand. 57:1073–1082. 2013. View Article : Google Scholar : PubMed/NCBI
|