Introduction
Atherosclerosis is a major cause of morbidity and
mortality in Western societies (1,2). It
is a chronic inflammatory disease of the arterial wall, which is
characterized by lipid accumulation, leukocyte infiltration and
smooth muscle cell proliferation. Atherosclerosis is considered to
be a risk factor for cognitive deterioration in the elderly,
including Alzheimer's disease (AD) (3,4). The
latter is a neurodegenerative disease, which is characterized by
dementia, along with a dense deposition of amyloid-β (Aβ) protein
in senile plaques in the brain, hyperphosphorylated tau protein and
neuron loss. Several findings suggest that atherosclerosis and AD
are linked: i) Atherosclerotic vascular disease and AD share common
risk factors, such as hypertension, diabetes, hypercholesterolemia,
and apolipoprotein ε4 allele (5).
In addition, old age is a major risk factor for both
atherosclerosis and AD (6). ii)
There are reports of a correlation between carotid atherosclerosis
and AD (7) as well as
atherosclerosis of the circle of Willis and AD (8,9).
Moreover, coronary artery disease is increased in AD patients
(10). iii) In a transgenic mouse
model of AD (B6Tg2576), early atherosclerosis lesions were detected
and were positively correlated with cerebral β amyloid deposits
when mice were fed a normal diet (11) or atherogenic diets (12). iv) Brain cholesterol affects the Aβ
formation from amyloid precursor protein (APP) (13,14).
v) APP and Aβ are present in human carotid plaques (15). vi) APP transgenic mice with
apolipoprotein E (ApoE) deficiency had increased atherosclerosis
and vascular inflammation (16).
However, whether atherosclerosis contributes to AD or they just
share similar epidemiology is still not known. ApoE is the main
lipid carrier protein in the brain, and it is released by
astrocytes in order to supply neurons with cholesterol. The human
ApoE gene has 3 allelic variants (ε2, ε3, and ε4). The ε4 allele
has been associated with a higher risk of cardiovascular disease
and AD, while the ε2 allele has a protective effect against AD. In
mice, the association between ApoE deficiency and AD is not clear
(17–26). Recent findings from our group and
others demonstrate the involvement of coagulation factors in both
atherosclerosis (27,28) and AD (29,30).
The contact activation pathway plays an essential role in
hemostasis, and also in the progression of thrombosis and
inflammation (31). In addition,
factor XII, the initiator of contact activation, has been shown to
be activated by aggregated Aβ (30), which resulted in activation of
factor XI (FXI), thus enhancing brain thrombin generation. In
humans, increased FXI is associated with increased incidence of
ischemic stroke (32), whereas
subjects with severe FXI deficiency have a reduced incidence of
ischemic stroke (33). High levels
of thrombin were found in the circulation, brain parenchyma and
micro-vessel walls of AD patients and also in AD mouse models
(30,34,35).
In addition, high levels of fibrinogen are correlated with
increased cerebral amyloid angiopathy and microglial activation
when assessed in AD mouse models (36). Procoagulant proteins, including
FXI, were found adjacent to macrophages and smooth muscle cells
inside atherosclerotic plaques (27,37).
Deprivation of FXI in ApoE knockout (KO) mice aged 24 and 42 weeks
resulted in a significant reduction of atherosclerosis in the
aortic sinus and aortic arch in comparison to ApoE KO mice
(28). Targeting FXI prevented
thrombosis on acutely ruptured atherosclerotic plaques (38). Furthermore, APP and Aβ were found
in advanced human carotid plaques, in proximity to activated
macrophages and platelets (15).
Hence, it can be suggested that coagulation factors, including FXI,
play a role in both atherosclerosis and AD. Old age is the main
risk factor for AD; therefore, in this work, we sought to study
whether FXI deficiency in elderly ApoE KO mice would decrease
pathological changes compatible with atherosclerosis and AD.
Materials and methods
Mice
Male ApoE KO mice (n=12; Jackson Laboratory, Ben
Harbor, ME, USA) and male ApoE/FXI double knock out (DKO) mice
(n=10) (28) were included in the
study. The mice fed with a chow diet and water provided ad libitum,
throughout the experiments. Mice were sacrificed at 64 weeks of age
and organs were collected for further analysis. All procedures were
approved by the Institutional Animal Care and Use Committee of the
Sheba Medical Center.
Atherosclerosis assessment
Quantification of atherosclerotic lesions was
performed by calculating the lesion area in the aortic arch. The
aorta was removed from the aortic arch to the iliac branches and
fixed in 4% formalin. The aorta was cut longitudinally and stained
with Sudan IV (Fluka). Lesion-area analysis in the thoracic aorta
was conducted blind, using NIS Elements imaging software (Nikon
Corporation, Tokyo, Japan).
Systemic inflammation
For plasma preparation, blood was drawn from the
vena cava in tubes with 10% EDTA. Plasma was separated by
centrifuge (1,000 × g) for 10 min, and stored at −80ºC until
analysis was performed. The interleukin-6 (IL-6) concentration was
measured by enzyme-linked immunosorbent assay (ELISA) kit (EMD
Millipore, Billerica, MA, USA).
Brain tissue preparation
Brain specimens were harvested, hemi-dissected, and
one hemisphere was fresh-frozen for histological and morphological
studies. The frontal and parietal cortices, hippocampus and
cerebellum were dissected from the other hemisphere. Specimens were
frozen rapidly in liquid nitrogen and stored at −80°C until
analysis. Slices of brain from 5XFAD transgenic male mice were
kindly provided by Dr Frenkel D. (Tel-Aviv University, Israel).
Cortex cytokines levels
Cortices were homogenized in
radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCL pH
7.4, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.5% sodium
deoxycholate, 0.1% SDS) supplement with protease inhibitors. The
homogenate was centrifuged at 13,000 × g for 18 min. Quantitation
of total protein in the extract was measured by Micro BCA protein
assay kit (Pierce; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Tumor necrosis factor-α (TNF-α) and IL-6 concentrations were
measured using ELISA kits (EMD Millipore).
Histology
Frozen brains were serially sectioned to slices of
10-µm thickness using a cryostat (Leica CM 1850; Leica Microsystems
GmbH, Wetzlar, Germany). The histologic sections were stained with
H&E. The Aβ deposits in the brain were detected using a
Congo-red staining kit (Ventana Medical Systems, Inc., Tucson, AZ,
USA). For microglia immunostaining, brain slices were fixed with 4%
paraformaldehyde rinsed in PBS [0.1 M (pH 7.2)]. The slices were
permeabilized and blocked with 0.1% Triton X-100/PBS (PBST)
containing 10% normal serum to reduce the non-specific adherence of
antibodies. Brain slices were incubated in primary antisera [goat
anti-ionized Iba 1 (1:100; Abcam)] for 1 h at 25°C in a humid
chamber. After incubation with the primary antisera, the slices
were rinsed with PBST and incubated with anti-goat Alexa Fluor-568
conjugated secondary antibody (Molecular Probes; Thermo Fisher
Scientific, Inc.). Slices were rinsed with PBST 3 times. Sections
were then rinsed 3 times with PBST and cover slipped with
fluoromount mounting medium (Sigma-Aldrich Israel, Ltd., Rehovot,
Israel). The sections were observed with ×20 magnification under an
aBX-43 fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Color pictures were acquired and analyzed using a digital camera
system coupled to imaging software (cellSens Entry digital imaging
software; Olympus Corporation) under a constant exposure time, gain
and offset, which were chosen in order to increase the threshold
for fluorescence.
Results
Atherosclerosis
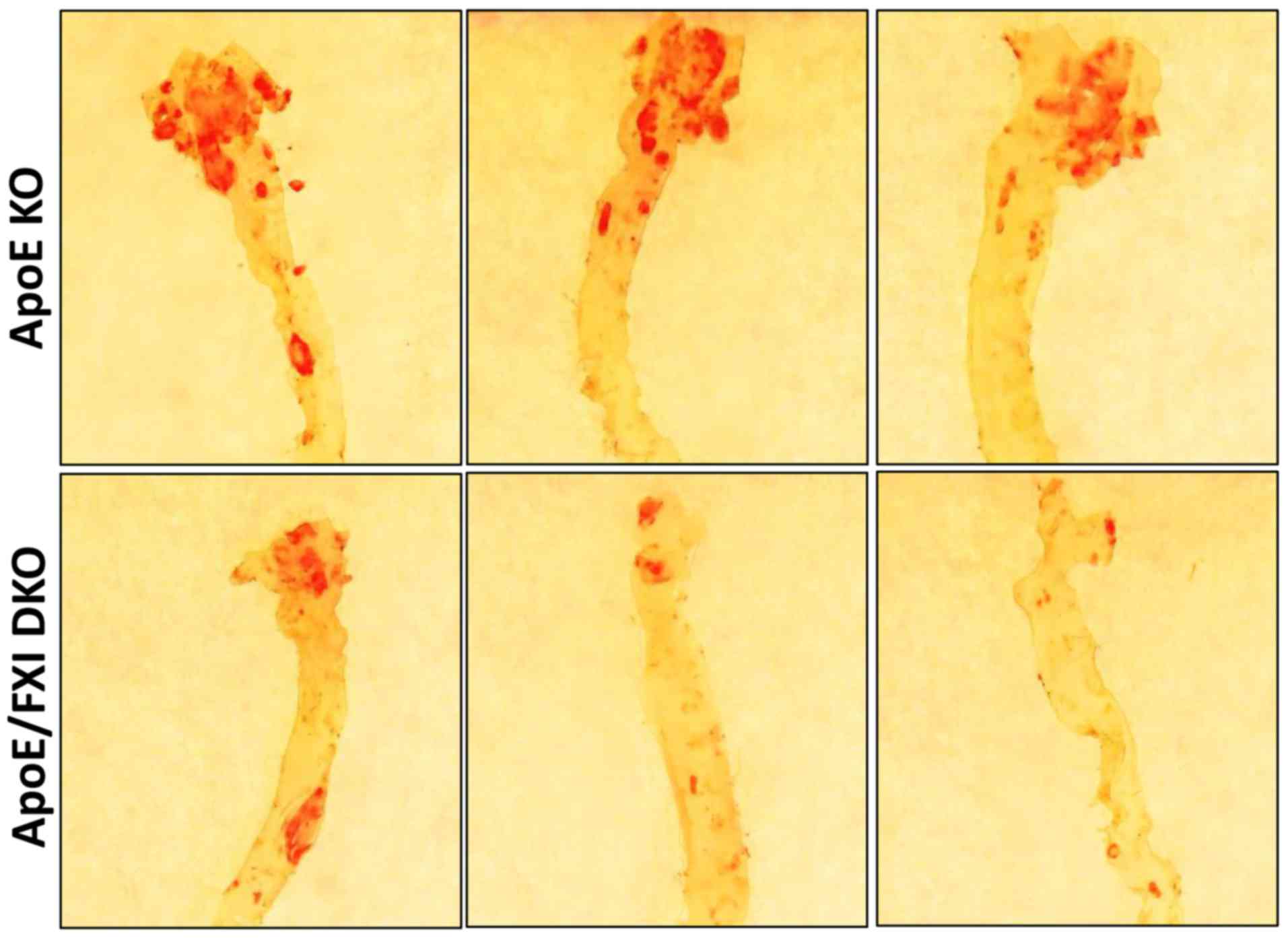
To investigate the effect of FXI deficiency on
atherosclerosis in elderly mice, we measured the atherosclerotic
lesion area at the aortic arch of 64-week-old ApoE KO and
FXI-deficient DKO mice. As expected, in the aortic arch, the
elderly ApoE KO mice had a higher proportion of atherosclerotic
lesion area (~30%, n=10) compared to the FXI-deficient DKO mice
(~20%, n=5) (Fig. 1). As the
elderly ApoE KO mice presented advanced aortic atherosclerosis, we
assumed that they would develop pathological changes in the brain
and that FXI deficiency would inhibit these pathological changes.
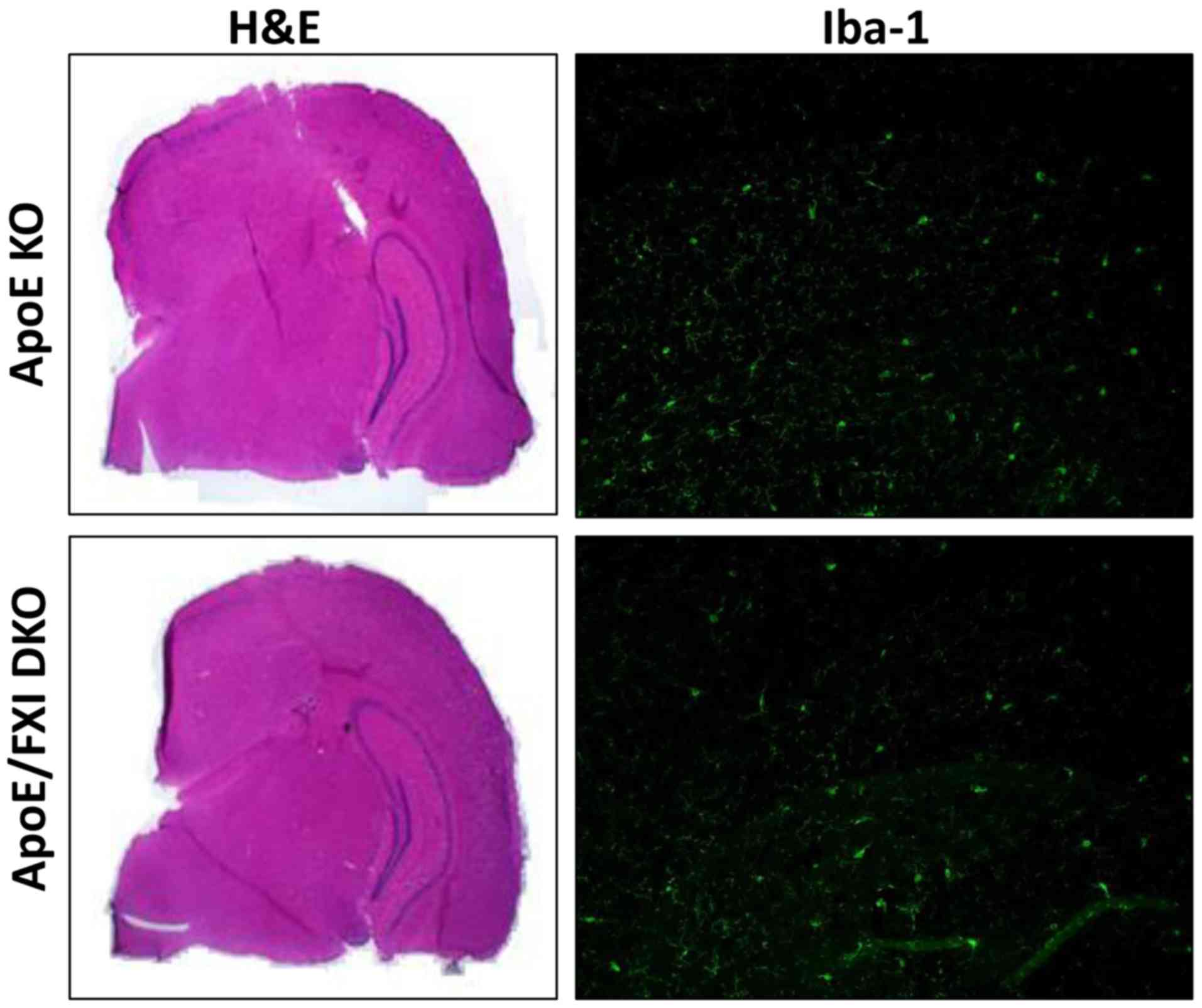
Therefore, we analyzed whether the ApoE KO mice had suffered from
brain atrophy and modification of hippocampus size in comparison to
DKO mice. Unexpectedly, hemotoxylin and eosin (H&E) staining
showed no pathologic damage to the brain in these very elderly ApoE
KO mice (Fig. 2), and there was a
histological similarity between the ApoE KO and DKO mice
brains.
Inflammation
To investigate neuro-inflammation, cortex cytokines
and microglia levels were measured. ApoE KO and DKO mice had
similar levels of IL-6 (1.3 vs. 1.7 pg/mg protein) and TNF-α (2.21
vs. 2.07 pg/mg protein) in the brain cortex. Similar densities of
microglia were detected in the brain sections from ApoE KO and DKO
as demonstrated by ionized calcium binding adapter molecule 1
(Iba-1) staining (Fig. 2).
Changes compatible with AD
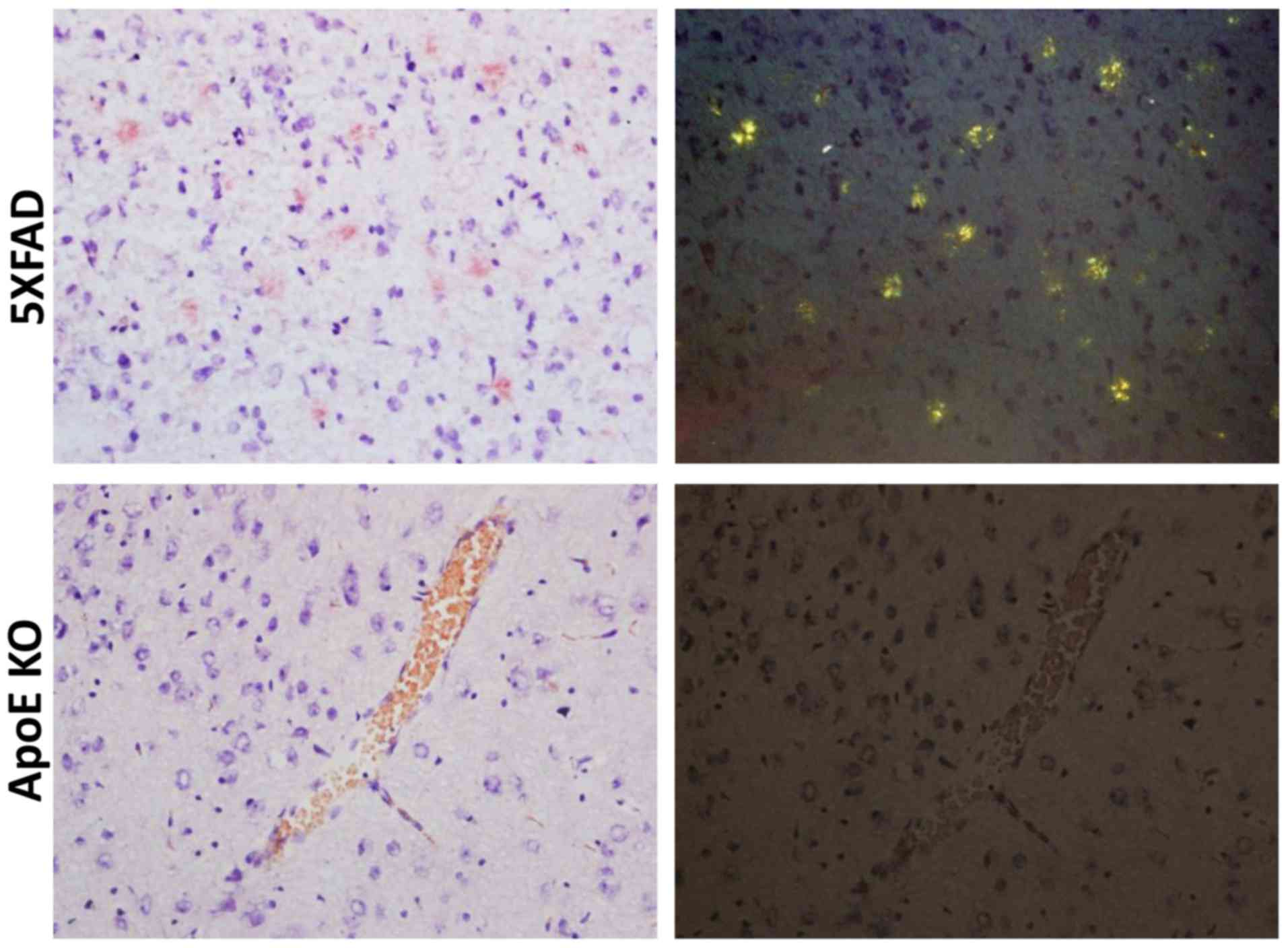
Congo red staining was used to measure the
deposition of Aβ in the brain. Compared to 5XFAD transgenic male
mice, which showed patchy deposits of Aβ in the brain tissue,
elderly ApoE KO mice had no Aβ lesions in the brain (Fig. 3).
Discussion
Previous studies have suggested that the coagulation
pathway is involved in both atherosclerosis and AD (28,30).
This motivated us to investigate the influence of FXI deficiency on
atherosclerosis and pathological brain changes associated with AD
in very old ApoE KO mice. Our results show that: I. although
elderly ApoE KO mice develop advanced aortic atherosclerotic
lesions, they do not develop senile Aβ plaques in the brain, and
II. FXI deficiency reduces the aortic atherosclerotic burden, and
similar to ApoE KO mice, ApoE/factor XI double KO (ApoE/FXI DKO)
mice do not develop Aβ plaques.
We first asked whether FXI deficiency would inhibit
atherogenesis in very old mice, similar to its effects in younger
mice (28). The results show that
elderly mice develop advanced atherosclerosis, and 30% of the
aortic arch is covered with atherosclerotic lesions. This is in
accord with previous reports showing the development of advanced
atherosclerotic lesions in ApoE KO mice that have been fed a normal
diet (39). FXI deficiency reduced
the lesion area by 33%, similar to its effect in 42-week-old ApoE
KO mice (28). Congenital FXI
deficiency or targeting FXI do not cause spontaneous bleeding
(40–42), in contrast to targeting factor VIII
or factor IX. Furthermore, it was recently shown that inhibition of
FXI synthesis by antisense oligonucleotide reduces blood pressure
in mice and rats (43). Therefore,
it is conceivable to use FXI KO mice to study the effect of the
coagulation pathway on pathological changes in the brain in elderly
mice.
As we used very old ApoE KO mice, we anticipated
that these mice would develop pathological changes compatible with
AD, including senile Aβ plaques. Unexpectedly, no lesions
compatible with AD (i.e., Aβ plaques, brain atrophy, increased
glial cells or increased levels of cortex inflammatory cytokines,
such as IL-6 and TNF-α) were detected in the ApoE KO mice or in the
FXI-deficient mice. There could be several explanations for such
findings, which may all relate to ApoE's role in the brain. In
contrast to ApoE4 transgenic mice that clearly develop the typical
symptoms of AD, the effect of ApoE deficiency on pathological
changes compatible with AD is still unknown. Several studies show
that ApoE KO mice are associated with an increased risk of
developing AD-related pathologies, that is, memory deficit
(20), tau phosphorylation
(19,26), a leaky blood-brain barrier (BBB)
(24), higher levels of protein
oxidation (18), age-dependent
synaptic loss (23) and even Aβ
deposits (26). In addition, a
recent study shows synaptic loss and dysfunction in mice that
express ApoE in peripheral tissues, but that have severely reduced
ApoE in the brain (22). Notably,
no pathologic brain changes were observed in 9-month-old
ApoE-deficient mice (25). Bales
et al (17) claimed that
the ApoE KO mouse is not a suitable model to study AD, since ApoE
facilitates Aβ deposition, while completely ablation of ApoE,
decreased cerebral Aβ sedimentation. It is important to note that
Bales et al demonstrated their findings in mice aged 6–11
months and studied only Aβ deposition in the brain, while we
studied 15-month-old mice and also investigated
atherosclerosis-related pathologies in the brain. Taken together,
ApoE KO and ApoE/FXI DKO cannot serve as a model to study AD or
pathologic brain changes related to atherosclerosis. It appears
that there is a dichotomy between the brain and peripheral blood
vessels. The mechanism by which ApoE KO protects against brain
pathology should be further studied as it may prove helpful for
future treatment for senile dementia. Yet, targeting FXI can still
serve as an important therapy to attenuate atherosclerosis. As
such, it is conceivably that elderly patients might benefit from
FXI target therapy to reduce the process of peripheral
atherosclerosis, though more studies are needed.
Acknowledgements
Professor Gailani D. (Vanderbilt University,
Nashville, TN, USA) for providing the FXI KO mice, Dr Frenkel D.
(Tel-Aviv University, Israel) for providing 5XFAD transgenic mouse
brains and for his constructive comments to the manuscript. We also
thank the Elsa and Leo foundation, Tel Aviv University and the
Neuhar family for financially supporting this research.
References
|
1
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roher AE, Tyas SL, Maarouf CL, Daugs ID,
Kokjohn TA, Emmerling MR, Garami Z, Belohlavek M, Sabbagh MN, Sue
LI and Beach TG: Intracranial atherosclerosis as a contributing
factor to Alzheimer's disease dementia. Alzheimers Dement.
7:436–444. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Casserly I and Topol E: Convergence of
atherosclerosis and Alzheimer's disease: Inflammation, cholesterol,
and misfolded proteins. Lancet. 363:1139–1146. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Honig LS, Kukull W and Mayeux R:
Atherosclerosis and AD: Analysis of data from the US national
Alzheimer's coordinating center. Neurology. 64:494–500. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luoto TM, Haikonen S, Haapasalo H,
Goebeler S, Huhtala H, Erkinjuntti T and Karhunen PJ: Large vessel
cerebral atherosclerosis is not in direct association with
neuropathological lesions of Alzheimer's disease. Eur Neurol.
62:93–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van Exel E, Gussekloo J, Houx P, de Craen
AJ, Macfarlane PW, Bootsma-van der Wiel A, Blauw GJ and Westendorp
RG: Atherosclerosis and cognitive impairment are linked in the
elderly. The Leiden 85-plus Study. Atherosclerosis. 165:353–359.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dolan D, Troncoso J, Resnick SM, Crain BJ,
Zonderman AB and O'Brien RJ: Age, Alzheimer's disease and dementia
in the baltimore longitudinal study of ageing. Brain.
133:2225–2231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beach TG, Wilson JR, Sue LI, Newell A,
Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, et al:
Circle of Willis atherosclerosis: Association with Alzheimer's
disease, neuritic plaques and neurofibrillary tangles. Acta
Neuropathol. 113:13–21. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yarchoan M, Xie SX, Kling MA, Toledo JB,
Wolk DA, Lee EB, Van Deerlin V, Lee VM, Trojanowski JQ and Arnold
SE: Cerebrovascular atherosclerosis correlates with Alzheimer
pathology in neurodegenerative dementias. Brain. 135:3749–3756.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan J, Wen G, Li Y and Liu C: The
occurrence of cerebrovascular atherosclerosis in Alzheimer's
disease patients. Clin Interv Aging. 8:581–584. 2013.PubMed/NCBI
|
|
11
|
Li L, Cao D, Garber DW, Kim H and Fukuchi
K: Association of aortic atherosclerosis with cerebral
beta-amyloidosis and learning deficits in a mouse model of
Alzheimer's disease. Am J Pathol. 163:2155–2164. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Franciosi S, Gama Sosa MA, English DF,
Oler E, Oung T, Janssen WG, De Gasperi R, Schmeidler J, Dickstein
DL, Schmitz C, et al: Novel cerebrovascular pathology in mice fed a
high cholesterol diet. Mol Neurodegener. 4:422009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Frears ER, Stephens DJ, Walters CE, Davies
H and Austen BM: The role of cholesterol in the biosynthesis of
beta-amyloid. Neuroreport. 10:1699–1705. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Simons M, Keller P, De Strooper B,
Beyreuther K, Dotti CG and Simons K: Cholesterol depletion inhibits
the generation of beta-amyloid in hippocampal neurons. Proc Natl
Acad Sci USA. 95:pp. 6460–6464. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
De Meyer GR, De Cleen DM, Cooper S,
Knaapen MW, Jans DM, Martinet W, Herman AG, Bult H and Kockx MM:
Platelet phagocytosis and processing of beta-amyloid precursor
protein as a mechanism of macrophage activation in atherosclerosis.
Circ Res. 90:1197–1204. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tibolla G, Norata GD, Meda C, Arnaboldi L,
Uboldi P, Piazza F, Ferrarese C, Corsini A, Maggi A, Vegeto E and
Catapano AL: Increased atherosclerosis and vascular inflammation in
APP transgenic mice with apolipoprotein E deficiency.
Atherosclerosis. 210:78–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bales KR, Verina T, Dodel RC, Du Y,
Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins
DJ, et al: Lack of apolipoprotein E dramatically reduces amyloid
beta-peptide deposition. Nat Genet. 17:263–264. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi J, Forster MJ, McDonald SR, Weintraub
ST, Carroll CA and Gracy RW: Proteomic identification of specific
oxidized proteins in ApoE-knockout mice: Relevance to Alzheimer's
disease. Free Radic Biol Med. 36:1155–1162. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Genis I, Gordon I, Sehayek E and
Michaelson DM: Phosphorylation of tau in apolipoprotein E-deficient
mice. Neurosci Lett. 199:5–8. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gordon I, Grauer E, Genis I, Sehayek E and
Michaelson DM: Memory deficits and cholinergic impairments in
apolipoprotein E-deficient mice. Neurosci Lett. 199:1–4. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tai LM, Youmans KL, Jungbauer L, Yu C and
Ladu MJ: Introducing human APOE into Aβ transgenic mouse models.
Int J Alzheimer's Dis. 2011:8109812011.PubMed/NCBI
|
|
22
|
Lane-Donovan C, Wong WM, Durakoglugil MS,
Wasser CR, Jiang S, Xian X and Herz J: Genetic restoration of
plasma ApoE improves cognition and partially restores synaptic
defects in ApoE-deficient mice. J Neurosci. 36:10141–10150. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Masliah E, Mallory M, Ge N, Alford M,
Veinbergs I and Roses AD: Neurodegeneration in the central nervous
system of apoE-deficient mice. Exp Neurol. 136:107–122. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Methia N, André P, Hafezi-Moghadam A,
Economopoulos M, Thomas KL and Wagner DD: ApoE deficiency
compromises the blood brain barrier especially after injury. Mol
Med. 7:810–815. 2001.PubMed/NCBI
|
|
25
|
Moghadasian MH, McManus BM, Nguyen LB,
Shefer S, Nadji M, Godin DV, Green TJ, Hill J, Yang Y, Scudamore CH
and Frohlich JJ: Pathophysiology of apolipoprotein E deficiency in
mice: Relevance to apo E-related disorders in humans. FASEB J.
15:2623–2630. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park SH, Kim JH, Choi KH, Jang YJ, Bae SS,
Choi BT and Shin HK: Hypercholesterolemia accelerates amyloid
β-induced cognitive deficits. Int J Mol Med. 31:577–582. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Borissoff JI, Spronk HM and ten Cate H:
The hemostatic system as amodulator of atherosclerosis. N Engl J
Med. 364:1746–1760. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shnerb Ganor R, Harats D, Schiby G,
Gailani D, Levkovitz H, Avivi C, Tamarin I, Shaish A and Salomon O:
Factor XI deficiency protects against atherogenesis in
apolipoprotein E/Factor XI double knockout mice. Arterioscler
Thromb Vasc Biol. 36:475–481. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arai T, Miklossy J, Klegeris A, Guo JP and
McGeer PL: Thrombin and prothrombin are expressed by neurons and
glial cells and accumulate in neurofibrillary tangles in Alzheimer
disease brain. J Neuropathol Exp Neurol. 65:19–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zamolodchikov D, Renné T and Strickland S:
The Alzheimer's disease peptide β-amyloid promotes thrombin
generation through activation of coagulation factor XII. J Thromb
Haemost. 14:995–1007. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mackman N: The clot thickens in
atherosclerosis. Arterioscler Thromb Vasc Biol. 36:425–426. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang DT, Flanders MM, Kim H and Rodgers
GM: Elevated factor XI activity levels are associated with an
increased odds ratio for cerebrovascular events. Am J Clin Pathol.
126:411–415. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Salomon O, Steinberg DM, Koren-Morag N,
Tanne D and Seligsohn U: Reduced incidence of ischemic stroke in
patients with severe Factor XI deficiency. Blood. 111:4113–4117.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grammas P, Samany PG and Thirumangalakudi
L: Thrombin and inflammatory proteins are elevated in Alzheimer's
disease microvessels: Implications for disease pathogenesis. J
Alzheimers Dis. 9:51–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tripathy D, Sanchez A, Yin X, Luo J,
Martinez J and Grammas P: Thrombin, a mediator of cerebrovascular
inflammation in AD and hypoxia. Front Aging Neurosci. 5:192013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cortes-Canteli M, Zamolodchikov D, Ahn HJ,
Strickland S and Norris EH: Fibrinogen and altered hemostasis in
Alzheimer's disease. J Alzheimers Dis. 32:599–608. 2012.PubMed/NCBI
|
|
37
|
Loeffen R, Spronk HM and ten Cate H: The
impact of blood coagulability on atherosclerosis and cardiovascular
disease. J Thromb Haemost. 10:1207–1216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
van Montfoort ML, Kuijpers MJ, Knaup VL,
Bhanot S, Monia BP, Roelofs JJ, Heemskerk JW and Meijers JC: Factor
XI regulates pathological thrombus formation on acutely ruptured
atherosclerotic plaques. Arterioscler Thromb Vasc Biol.
34:1668–1673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nakashima Y, Plump AS, Raines EW, Breslow
JL and Ross R: ApoE-deficient mice develop lesions of all phases of
atherosclerosis throughout the arterial tree. Arterioscler Thromb.
14:133–140. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gailani D and Gruber A: Factor XI as a
therapeutic target. Arterioscler Thromb Vasc Biol. 36:1316–1322.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Büller HR, Bethune C, Bhanot S, Gailani D,
Monia BP, Raskob GE, Segers A, Verhamme P and Weitz JI; FXI-ASO TKA
Investigators, : Factor XI antisense oligonucleotide for prevention
of venous thrombosis. N Engl J Med. 372:232–240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Duga S and Salomon O: Congenital factor XI
deficiency: An update. Semin Thromb Hemost. 39:621–631. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kossmann S, Lagrange J, Jäckel S, Jurk K,
Ehlken M, Schönfelder T, Weihert Y, Knorr M, Brandt M, Xia N, et
al: Platelet-localized FXI promotes a vascular
coagulation-inflammatory circuit in arterial hypertension. Sci
Transl Med. 9:pii:eaah49232017. View Article : Google Scholar
|