Introduction
Breast cancer is the most common type of invasive
cancer in women. Approximately 1.7 million women are diagnosed with
breast cancer annually, and >500,000 succumb to it worldwide
(1). While surgery, traditional
chemotherapy and radiotherapy are commonly used to treat breast
cancer; targeted therapy has drawn the attention of clinicians and
researchers in the past decade for its improved therapeutic effect
in metastatic cancer, as compared with traditional chemotherapy.
Thus, numerous multi-kinase inhibitors have been adopted in the
targeted therapy of metastatic breast cancer (2–4).
However, the resistance of breast cancer to these inhibitors and
drugs remains challenging in chemotherapy and targeted therapy.
Therefore, continuing to develop novel anti-cancer drugs is
necessary in cancer therapy.
In tumorigenesis of breast and other types of
tissue, protein kinases are important in the regulation of
proliferation, apoptosis and migration (5). For example, platelet-derived growth
factor receptor-β (PDGFRβ), a member of the tyrosine kinase
receptors type III family, is associated with the malignancy of
breast carcinoma (6–8). In response to survival signals,
PDGFRβ activates Akt by upregulating phosphoinositide 3-kinase
(PI3K) and phosphoinositide-dependent protein kinase-1 (PDK1)
(9). Thus, inhibition of PDGFRβ
activity by TKI inhibitor(s) may suppress breast tumor growth
(10).
In addition, the Ras/Raf/mitogen-activated protein
kinases (MAPK) signaling pathway is critical in breast
tumorigenesis (11). Studies have
demonstrated that constitutive activation of the MAPK signaling
pathway is associated with the progression of breast cancer via the
induction of chemoresistance and distant metastases (12–16).
Thus, the MAPK signaling pathway may be a potent target for breast
cancer chemotherapy (17).
Currently, certain drugs, such as Trastuzumab,
Lapatinib, Bevacizumab and Taxol have been identified for breast
cancer targeted therapy. Of them, Taxol is the commonly
administered drug for the treatment of breast cancer. However,
continuous use of Taxol results in acquired drug-resistance of
breast cancer (18). Therefore,
development of novel drugs is essential to improve targeted therapy
of breast cancer and Taxol-resistant breast cancer. In the current
study, a novel multi-kinase inhibitor, T03 is reported. T03 is a
novel multi-kinase inhibitor against PDGFRβ and c-Raf, and
inhibition of PDGFRβ and c-Raf by T03 may downregulate the
Raf/mitogen-activated protein kinase kinase (MEK)/extracellular
signal-regulated kinase (ERK) and PDGFR/Akt/mechanistic target of
rapamycin (mTOR) survival pathway. In the present study, the
anti-tumor activity and underlying mechanism of T03 in regular and
Taxol-resistant breast cancer were investigated in vitro and
in vivo.
Materials and methods
Cell culture
Breast cancer cell line MCF-7 was obtained from the
cell center of Chinese Academy of Medical Sciences (CAMS) and
Peking Union Medical College (PUMC; Beijing, China). Cell lines
MX-1 and MX-1/T (Taxol-resistant) A549, A549/T (Taxol-resistant)
were obtained from the Professor Yongkui Jing (Mount Sinai School
of Medicine, New York, NY, USA). The MCF-7/T (Taxol-resistant) cell
was established in our laboratory (Institute of Materia Medica,
Chinese Academy of Medical Sciences and Peking Union Medical
College, Beijing China) (19).
MCF-7/ADM (Adriamycin-resistant) was obtained from the Assistant
Professor Hongbo Wang (Yantai University, Shandong, China). The
cells were maintained in Dulbecco's modified Eagle's medium
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), 100 IU/ml penicillin and 100 µg/ml streptomycin
in a humidified incubator containing 5% CO2 at 37°C. In
the present study, four specific cells (MX-1/T, MCF-7/T, MCF-7/ADM,
A549/T) were used.
Drugs
T03, a small molecule compound containing
2-picolinylhydrazide moiety (Chinese patent application no.
201110129115.7) was synthesized by the Department of
Pharmacochemistry at the Institute of Materia Medica, CAMS and PUMC
(Purity >97%; high-performance liquid chromatography). For in
vitro experiments, T03 and Taxol (Beijing Union Pharmaceutical
Factory, Beijing, China) were dissolved in dimethyl sulfoxide
(DMSO) and stored at 4°C until use. DMSO served as a vehicle
control in all experiments at a final concentration of 0.1%. For
the in vivo experiments, T03 was dissolved in a solution of
Cremophor EL (Aladdin Industrial Corporation, Shanghai, China; cat
no. C107105)/ethanol/water (12.5:12.5:75) (20).
Cell viability assay
MX-1, MX-1/T, MCF-7 and MCF-7/T cells (2,500 cells
per well) were seeded in a 96-well plate. After 24 h of incubation
at 37°C, cells were treated with various different concentrations
of Taxol and T03. After 72 h of incubation at 37°C, the Cell
Counting kit-8 (CCK-8; cat. no. C0037; Beyotime Institute of
Biotechnology, Shanghai, China) assay was performed to evaluate
cell viability. Absorbance values which was measured using an ELISA
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 450 nm
were normalized to the values obtained for vehicle-treated cells to
determine the percentage of surviving cells. The median inhibitory
concentration (IC50) was defined as the drug
concentration at which cell growth was inhibited by 50%. Each assay
was performed in triplicate.
Colony formation assay
MCF-7 and MCF-7/T cells were trypsinized to
single-cell suspensions, and resuspended in DMEM culture medium
containing 10% FBS. Approximately 500 cells were plated in 6-well
tissue culture plates. After a 24-h incubation at 37°C, the cells
were treated with either T03, 0.1% DMSO, or nothing. Cells were
incubated in 5% CO2 at 37°C for 14 days, and the
colonies were washed, fixed and stained with 0.005% crystal violet
in methanol. The number of colonies was manually counted without a
microscope, and experiments were performed in triplicate and
repeated three times.
Apoptosis analysis
MX-1, MX-1/T, MCF-7 and MCF-7/T cells were treated
with either T03 or 0.1% DMSO for 72 h. Apoptotic cells were
measured using Annexin V-fluorescein isothiocyanate (FITC)
Apoptosis Detection kit (cat. no. 556570; BD Biosciences, Franklin
Lakes, NJ, USA). Briefly, cells were trypsinized and washed with
PBS following treatment and stained with Annexin V-FITC according
to the manufacturer's protocol, then analyzed by ACCURI C6 flow
cytometry with BD Accuri C6 software (BD Biosciences).
Cell-cycle analysis
MX-1, MX-1/T, MCF-7 and MCF-7/T cells were incubated
at 37°C with either T03 or 0.1% DMSO for 72 h. Cells were washed in
PBS and fixed with 4°C ice-cold 70% ethanol overnight. The cells
were then suspended in PBS containing RNase A (100 µg/ml; cat. no.
R1030; Beijing Solarbio Science and Technology Co., Ltd., Beijing,
China), propidium iodide (50 µg/ml; cat. no. P8080; Beijing
Solarbio Science and Technology Co., Ltd.), Triton X-100 (0.1%),
and incubated on the ice in the dark for at least 1 h (21). The cell cycle profiles were
determined by flow cytometric analysis.
Tumor implantation and growth in MX-1,
MX-1/T, MCF-7 and MCF-7/T xenografts
All animal studies were performed in compliance with
the policies of the Institute of Materia Medica Animal Care and Use
Committee. Six-week-old, female BALB/c/nu nude mice were used in
the present study (all had 20 mice/experiment). The body weight was
15–16 g for MX-1 and MX-1/T xenograft model, and 16–22 g for MCF-7
and MCF-7/T xenograft model. They were purchased from Vital River
Laboratory Animal Technology Co., Ltd., (Beijing, China) and housed
in the controlled environment at 25°C on a 12-h light/dark cycle (5
mice per group). When tumors grew to an average volume of 100–250
mm3, tumor-bearing mice were randomly separated into
four groups of five animals. A total of one group received per os
Cremophor EL/ethanol/water and served as a vehicle control; the
other groups received injections of 5 mg/kg Taxol (twice per week),
or received an oral dose of 50 or 100 mg/kg T03 six times per week
for 13 days (MX-1 and MX-1/T) and 33 days (MCF-7, MCF-7/T). Mice
were euthanized at the end of the treatment period. Tumors were
removed and weighed, and samples of all of the sections were stored
at −80°C for western blot analysis.
Kinase inhibition assay
Inhibition of kinase activity against target kinases
was measured using Caliper and Glo-ATP assays (ADP-Glo assay
buffer: 25 mM HEPES, 10 mM MgCl2, 0.01% Triton X-100,
100 µg/ml BSA, 2.5 mM DTT, (pH 7.4); Caliper assay buffer: 100 mM
HEPES, 10 mM MgCl2, 100 µl/l Brij35 (30%), 1 mM DTT, (pH
7.4); Other reagents: ATP (cat. no. A7699 Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany); ADP Gloreagent (cat. no. V9102; Promega
Corporation, Madison, WI, USA). The Biochemical assay was performed
according to the manufacturer's protocol. The assay was performed
by HD Biosciences (China) Co., Ltd. (Shanghai, China).
Western blot analysis
Lysates (portions of two or three randomly selected
tumors from MCF-7 and MCF-7/T xenograft mice) were prepared as
previously described. The protein extraction buffer was a
radioimmunoprecipitation buffer (1 mM phenylmethylsulfonyl
fluoride) (21). Protein
concentration was determined using the bicinchoninic acid method.
Total proteins (50 µg) were separated by 10.0% SDS-PAGE and
transferred to a nitrocellulose membrane by semi-wet
electrophoresis were incubated with primary antibodies overnight at
4°C following blocking with TBS containing 1% Tween-20 and 5%
skimmed dry milk for 1 h at room temperature. The antibodies were
as follows: Rabbit anti-phosphorylated (p)-c-Raf (Ser259; cat. no.
9421), c-Raf (cat. no. 9422), p-MEK (cat. no. 9127), MEK (cat. no.
9903), p-ERK (cat. no. 4370), ERK (cat. no. 9101), p-PDGFRβ (cat.
no. 3170), PDGFRβ (cat. no. 3169), p-PDK (cat. no. 3061), PDK (cat.
no. 3062), p-Akt (Thr 308; cat. no. 9275), Akt (cat. no. 4691),
p-mTOR (cat. no. 2971), mTOR (cat. no. 2983), p-AuroraA (cat. no.
2914), AuroraA (cat. no. 14475) (all from Cell Signaling
Technology, Inc., Danvers, MA, USA) and mouse anti-actin (cat. no.
3700; CST Biological Reagents Co., Ltd., Shanghai China). All the
primary antibodies were used at 1:1,000. The samples were detected
with peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:5,000,
cat. no. sc-2004, Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
for 1 h at room temperature and developed using an enjhanced
chemiluminescence system western blot detection and analysis system
(Applygen Technologies, Inc., Beijing, China). The membranes were
assessed for equal loading by probing for β-actin.
Statistical analysis
Data were expressed as means ± standard deviation.
Statistical analysis of the results was performed using one-way
analysis of variance followed by a Bonferroni post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference. Statistical software used was Microsoft Excel 2010
(Microsoft Corporation, Redmond, WA, USA) and SPSS software
(version 19.0; SPSS, Inc., Chicago, IL, USA).
Results
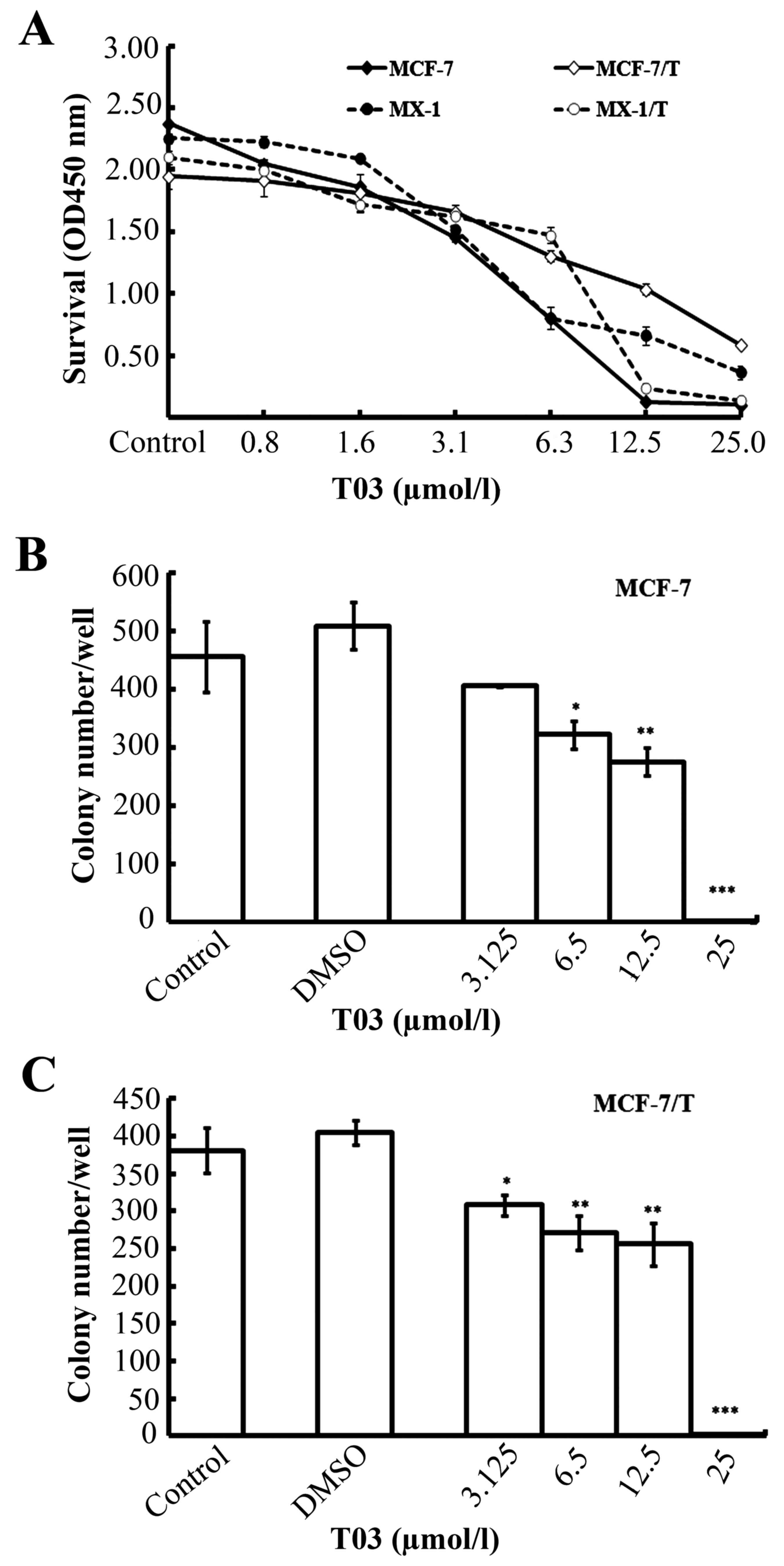
T03 inhibits the proliferation of
breast cancer cells
To evaluate the suppressive efficacy of T03 on
breast cancer cells in vitro, the regular (MX-1, MCF-7) and
Taxol-resistant (MX-1/T, MCF-7/T) breast cancer cells were treated
with various concentrations of T03 (0.8–25.0 µmol/l) for 72 h. The
CCK-8 assay results showed that T03 inhibited the growth of the
breast cancer cells in dose-dependent manner (Fig. 1A). The T03 IC50 S values
were 8.46±0.28 (MX-1), 11.65±2.19 (MX-1/T), 6.39±1.15 (MCF-7), and
10.95±0.49 µmol/l (MCF-7/T; Table
I). These results indicated that T03 effectively inhibited the
growth of breast cancer cells, as well as the Taxol-resistant
breast cancer cells. To establish whether T03 inhibits other
drug-resistant cells, similar experiments on Doxorubicin-resistant
breast cancer cells (MCF-7/ADM) were conducted. Similarly, T03
inhibited Adriamycin-resistant cells in a dose-dependent manner
(Table II).
 | Table I.Effect of T03 on proliferation in
breast cancer cells. |
Table I.
Effect of T03 on proliferation in
breast cancer cells.
|
| Median inhibitory
concentration, mol/l |
|---|
|
|
|
|---|
| Treatment | MX-1 | MX-1/T | MCF-7 | MCF-7/T |
|---|
| T03 |
8.46±0.28×10-6 |
11.65±2.19×10-6 |
6.39±1.15×10-6 |
10.95±0.49×10-6 |
| Taxol |
4.44±0.30×10-8 |
1.52±0.50×10-6 |
1.83±0.36×10-9 |
7.66±2.06×10-7 |
| Table II.Effect of T03 on proliferation in
resistant cells. |
Table II.
Effect of T03 on proliferation in
resistant cells.
|
| Median inhibitory
concentration, mol/l |
|---|
|
|
|
|---|
| Treatment | MCF-7 | MCF-7/ADM | A549 | A549/T |
|---|
| T03 | 7.91×10-6 | 1.83×10-5 | 9.83×10-6 | 2.76×10-5 |
| Taxol | 5.97×10-9 | 4.37×10-7 | 5.09×10-9 | 2.30×10-7 |
T03 suppresses colony formation in
breast cancer cells
To confirm the suppressive ability of T03 on tumor
cell proliferation, a colony formation assay was conducted in MCF-7
and MCF-7/T cells. The data revealed that T03 effectively inhibited
the colony formation in MCF-7 and MCF-7/T colony cells in a
dose-dependent manner (Fig. 1B and
C). T03 inhibited colony formation by ~50.0% (IC50)
at a concentration of 7.61 µmol/l in MCF-7 and 7.45 µmol/l in
MCF-7/T cells. The data were consistent with the results of the
CCK-8 assay.
T03 treatment led to cell cycle arrest
in breast cancer cells
T03 was observed to cause cell cycle arrest in
breast cancer cells. T03 treatment led to elevated numbers of
G2-phase cells and decreased G1-phase cells
in a dose-dependent manner in MX-1 and MX-1/T cells (Fig. 2A and B). With a treatment of 5.0
µmol/l T03, the percentage of G2-phase cells increased
to 15.23±0.83% (MX-1) and 29.30±1.37% (MX-1/T), while it was
11.37±0.90 and 11.47±1.26% in the untreated controls.
Concomitantly, the percentage of G1 phase cells reduced
to 60.37±1.01% (MX-1) and 40.60±1.06% (MX-1/T) compared with
68.10±1.65 and 56.33±0.86% in the untreated controls. Furthermore,
while T03 treatment induced cell accumulation at the G2
and G1 phases, it caused the decrease of the S phase
cells in MCF-7 and MCF-7/T (Fig. 2C
and D). The percentage of G2 phase cells was
25.50±0.92 and 26.80±1.51% in MCF-7 and MCF-7/T cells,
respectively, when they were treated with 5.0 µmol/l T03. By
contrast, the percentages were 20.20±1.25% (MCF-7) and 21.73±1.66%
(MCF-7/T) in the controls. The percentages of S phase cells
decreased from 33.43±1.45 and 30.77±0.46% to 16.90±1.49 and
18.53±2.10% (following treatment with 5.0 µmol/l T03), respectively
(Fig. 2C and D).
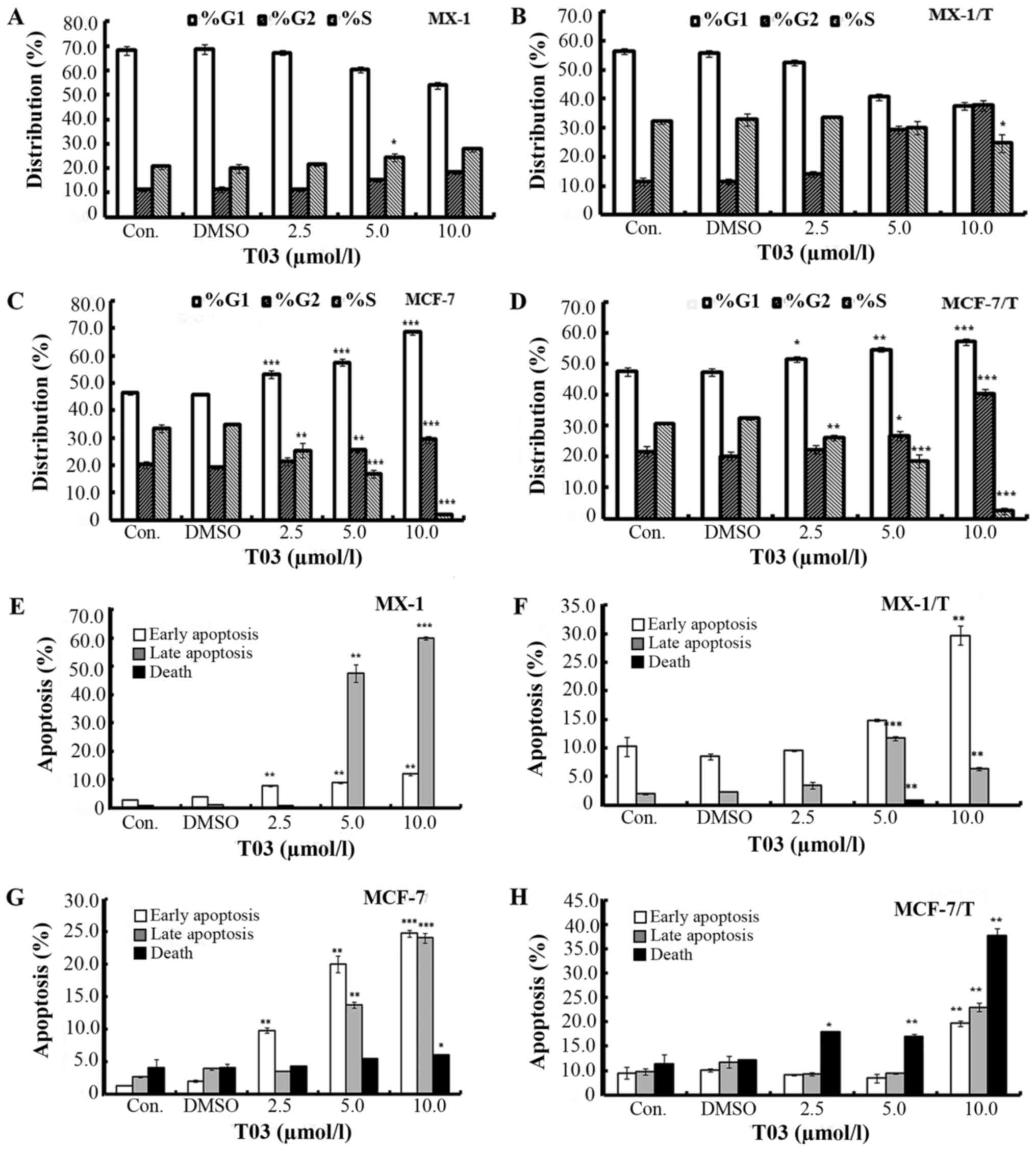 | Figure 2.T03 treatment induced cell cycle
arrest and apoptosis in breast cancer. T03 induced cell cycle
arrest in (A) MX-1, (B) MX-1/T, (C) MCF-7 and (D) MCF-7/T cells.
Cells were incubated with the indicated concentration of T03 or
DMSO (0.1%) for 72 h, stained with propidium iodide and analyzed by
flow cytometry. Error bars represent standard deviation. T03
treatment induced apoptosis in (E) MX-1, (F) MX-1/T, (G) MCF-7 and
(H) MCF-7/T cells. Cells were incubated with the indicated
concentration of T03 or DMSO for 72 h. Cells of early apoptosis,
late apoptosis and death were stained with Annexin V-FITC and
determined by flow cytometry. Quantization of the Annexin V-FITC
staining data was presented based on the percentages of the cells
for early apoptosis, late apoptosis, and death. Each graph
represents the mean, and the error bars represent standard
deviation. *P<0.05, **P<0.01, ***P<0.001 vs. the control
cells. DMSO, dimethyl sulfoxide; FITC, fluorescein isothiocyanate;
Con., control. |
T03 induced apoptosis in breast cancer
cells
T03 treatment was demonstrated to induce apoptosis
in MX-1, MX-1/T, MCF-7, and MCF-7/T breast cancer cells. Upon
treatment with 10.0 µmol/l T03, the early apoptosis increased by
4.16- and 2.90-fold, and late apoptosis increased by 67.25- and
3.23-fold in MX-1 and MX-1/T cells (Fig. 2E and F). In MCF-7 and MCF-7/T
cells, early apoptosis increased from 1.28 and 4.51% (control) to
24.75 and 14.70%, respectively, while late apoptosis increased from
2.67 and 4.73% (control) to 24.05 and 18.00%, respectively
(Fig. 2G and H).
T03 inhibited kinases in breast cancer
cells
To investigate which kinases T03 inhibited, Caliper
and ADP-Glo assays were performed on a panel of kinases in
T03-treated breast cancer cells. The results indicated that various
oncogenic kinases were susceptible to T03 inhibition. Of these
kinases, c-Raf, PDGFRβ and RET proto-oncogene may be suppressed by
T03, and the IC50s are 0.78, 0.23 and 0.71 µmol/l,
respectively. In addition, T03 may inhibit the activity of PDGFRα,
fms related tyrosine kinase 1 (FLT1), kinase insert domain
receptor, FLT3, and c-kit with IC50 between 0.05 and 1.0
µmol/l. Furthermore, it was found that T03 downregulates FGFR1,
FGFR2 and b-Raf at the micromole level. By contrast, T03 exerted
little effect on other tested kinases, such as Aurora A, insulin
like growth factor 1 receptor, ERK1, ERK2, Src, PI3K, erb-b2
receptor tyrosine kinase 2 and AMPK (Table III).
| Table III.Inhibitory activity of T03 against
different kinases (biochemical assay). |
Table III.
Inhibitory activity of T03 against
different kinases (biochemical assay).
| A, Median
inhibitory concentration of T03 against different kinases |
|---|
|
|---|
| Kinase target | In vitro IC50
value, µmol/l | Source |
|---|
| c-Raf | 0.78 | ADP-Glo |
| PDGFRβ |
0.230 | Caliper |
| Ret
proto-oncogene | 0.71 | Caliper |
| PDGFRα | 0.37 | Caliper |
| FLT1 | 0.93 | Caliper |
| Kinase insert
domain receptor |
0.504 | Caliper |
| FLT3 |
0.046 | Caliper |
| c-kit | 0.16 | Caliper |
| FGFR1 |
3.929 | Caliper |
| FGFR2 |
2.962 | Caliper |
| b-Raf |
4.063 | ADP-Glo |
|
| B, The
inhibition rate of T03 against different kinases |
|
| Kinase
target | Inhibition, 1.0
µmo/la (%) | Source |
|
| Aurora A | 9.92 | Caliper assay |
| IGFR1 | 2.57 | ADP-Glo |
| ERK1 | 9.73 | Caliper |
| ERK2 | 0.79 | Caliper |
| SRC | – | ADP-Glo |
| PI3Kα | 2.01 | ADP-Glo |
| PI3Kγ | 19.33 | Caliper |
| ERBB2 | 8.91 | Caliper |
| AMPK
(A1/B1/G1) | 14.65 | Caliper |
| AMPK
(A2/B1/G1) | 28.76 | ADP-Glo |
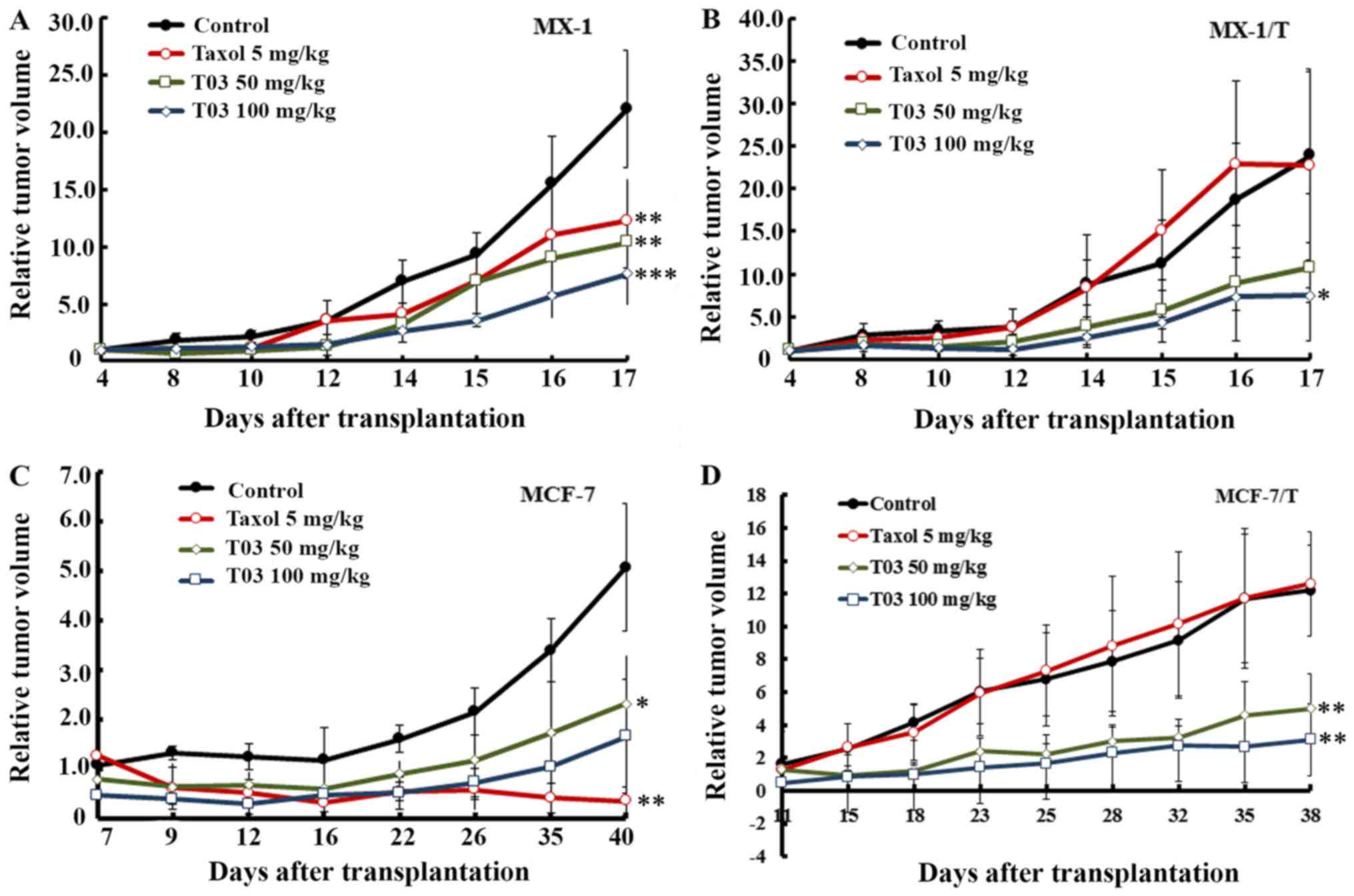
T03 inhibited xenograft tumor growth
of breast cancer cells
Based on the above results obtained in vitro,
further experiments were performed to determine whether T03
inhibits xenograft tumor growth from the breast cancer cells. To
obtain xenograft tumors, the MX-1, MX-1/T, MCF-7, and MCF-7/T human
breast cells were inoculated into BALB/c/nu nude mice. While T03
was used to treat the xenograft tumors, Taxol was adopted as a
reference compound. When 5 mg/kg Taxol was applied to MX-1 and
MX-1/T xenografts, the treated/control (T/C) ratio were 55.56 and
95.13%, respectively, according to the relative tumor volume (RTV).
Furthermore, the inhibition ratios were 41.56% in MX-1 and 0% in
MX-1/T based on the relative tumor weight, indicating that MX-1/T
was less sensitive to Taxol at a dose of 5 mg/kg. For T03, the T/C
ratio was 46.99% (50 mg/kg) and 34.68% (100 mg/kg) according to
RTV, and the inhibition ratio of tumor weight reached 50.00 and
62.90% in MX-1 xenografts (Table
IV and Fig. 3A). Furthermore,
in MX-1/T xenografts, the T/C ratio of RTV was 45.06% (50 mg/kg)
and 31.47% (100 mg/kg), and the inhibition ratio of the tumor
weight was 51.02 and 59.98%, respectively (Table V and Fig. 3B), indicating that T03 exerted more
effective inhibition on the MX-1 and MX-1/T xenografts.
| Table IV.Antitumor activity of T03 on the
breast cancer MX-1 xenograft model. |
Table IV.
Antitumor activity of T03 on the
breast cancer MX-1 xenograft model.
|
|
|
| Body weight, g | Tumor volume,
mm3 | Relative tumor
volume | Tumor weight |
|---|
|
|
|
|
|
|
|
|
|---|
| Compound | Dose, mg/kg | Animals, n | Initial | Final | Initial | Final | x±SD | Treated/control
ratio, % | x±SD, g | Inhibition, % |
|---|
| Control |
| 5/5 |
16.0±1.0 |
21.8±1.9 |
122.6±4.2 |
2,709.5±633.4 |
22.09±5.12 |
|
1.61±0.45 |
|
| Taxol |
5 | 5/5 |
16.0±0.7 |
20.8±2.2 |
140.2±13.3 |
1,714.4±308.2a |
12.27±2.33b | 55.56 |
0.94±0.20a | 41.56 |
| T03 | 50 | 5/5 |
15.0±1.0 |
19.2±1.3 |
121.2±24.9 |
1,318.2±849.5a |
10.38±5.48b | 46.99 |
0.81±0.49a | 50.00 |
|
| 100 | 5/5 |
15.2±0.5 |
18.9±1.4 |
123.7±6.7 |
945.7±35.8c |
7.66±0.48c | 34.68 |
0.60±0.03b | 62.90 |
| Table V.Antitumor activity of T03 on the
breast cancer MX-1/T xenograft model. |
Table V.
Antitumor activity of T03 on the
breast cancer MX-1/T xenograft model.
|
|
|
| Body weight, g | Tumor volume,
mm3 | Relative tumor
volume | Tumor weight |
|---|
|
|
|
|
|
|
|
|
|---|
| Compound | Dose, mg/kg | Animals, n | Initial | Final | Initial | Final | x±SD | Treated/control
ratio, % | x±SD, g | Inhibition, % |
|---|
| Control |
| 5/5 |
16.0±1.0 |
20.8±2.9 |
91.7±19.7 |
2,131.3±920.4 |
23.87±10.22 |
|
1.57±0.47 |
|
| Taxol |
5 | 5/5 |
15.6±0.6 |
20.4±0.9 |
95.7±17.1 |
2,183.1±1,091.0 |
22.71±11.09 | 95.13 |
1.58±0.79 | – |
| T03 | 50 | 5/5 |
15.2±0.5 |
19.0±2.5 |
98.5±9.8 |
1,079.1±887.6 |
10.76±8.66 | 45.06 |
0.77±0.62 | 51.02 |
|
| 100 | 5/5 |
15.8±0.8 |
18.8±2.2 |
96.7±6.4 |
728.6±78.2a |
7.51±0.86a | 31.47 |
0.63±0.06b | 59.98 |
In addition, it was observed that T03 presented even
higher inhibitory ability to MCF-7 and MCF-7/T xenografts compared
with MX-1 or MX-1/T models. While Taxol was applied to the MCF-7
xenograft, the T/C ratio of the RTV was 6.75% (5 mg/kg) and the
inhibition rate of the tumor weight was 96.36%. By contrast, Taxol
exerted almost no inhibitory effect on the MCF-7/T xenograft model.
In contrast to this, when T03 was applied to MCF-7 xenografts, the
RTV T/C ratio was 45.63% (50 mg/kg) and 32.55% (100 mg/kg), and the
inhibition rate of the tumor weight reached 57.09 and 62.60%
(Table VI and Fig. 3C). Furthermore, in MCF-7/T
xenografts, the RTV T/C ratios were 41.21% (50 mg/kg) and 25.52%
(100 mg/kg) while the inhibition rate of the tumor weight attained
44.06 and 60.22%, respectively (Table VII and Fig. 3D). These data indicated that T03
inhibits Taxol-sensitive and -resistant breast cancer tumors.
| Table VI.Antitumor activity of compounds on
the breast cancer MCF-7 xenograft model. |
Table VI.
Antitumor activity of compounds on
the breast cancer MCF-7 xenograft model.
|
|
|
| Body weight, g | Tumor volume,
mm3 | Relative tumor
volume | Tumor weight |
|---|
|
|
|
|
|
|
|
|
|---|
| Compound | Dose, mg/kg | Animals, n | Initial | Final | Initial | Final | x±SD | Treated/control
ratio, % | x±SD, g | Inhibition, % |
|---|
| Control |
| 5/5 |
21.8±0.8 |
26.0±1.2 |
237.5±32.6 |
1,163.8±503.2 |
5.06±2.46 |
|
1.02±0.46 |
|
| Taxol |
5 | 5/5 |
21.0±1.6 |
22.8±2.3 |
227.4±74.6 |
68.3±32.5a |
0.34±0.22a | 6.75 |
0.04±0.02a | 96.36 |
| T03 | 50 | 5/5 |
20.6±1.1 |
26.0±4.6 |
230.6±65.3 |
521.2±658.1 |
2.31±2.50 | 45.63 |
0.44±0.27b | 57.09 |
|
| 100 | 5/5 |
21.0±2.2 |
23.2±3.1 |
240.9±38.6 |
419.3±413.6b |
1.65±1.55b | 32.55 |
0.38±0.49 | 62.60 |
| Table VII.Antitumor activity of compounds on
the breast cancer MCF-7/T xenograft model. |
Table VII.
Antitumor activity of compounds on
the breast cancer MCF-7/T xenograft model.
|
|
|
| Body weight, g | Tumor volume,
mm3 | Relative tumor
volume | Tumor weight |
|---|
|
|
|
|
|
|
|
|
|---|
| Compound | Dose, mg/kg | Animals, n | Initial | Final | Initial | Final | x±SD | Treated/control
ratio, % | x±SD, g | Inhibition, % |
|---|
| Control |
| 5/5 |
18.0±1.4 |
18.6±2.4 |
112.0±5.8 |
1,373.5±363.1 |
12.19±2.78 |
|
1.26±0.24 |
|
| Taxol |
5 | 5/4 |
16.8±1.9 |
17.5±2.1 |
132.5±45.3 |
1,871.0±981.5 |
12.59±3.17 | – |
1.36±0.48 | – |
| T03 | 50 | 5/5 |
17.4±1.2 |
17.4±2.5 |
142.4±38.7 |
621.2±243.3a |
5.02±2.11a | 41.21 |
0.71±0.18a | 44.06 |
|
| 100 | 5/5 |
17.3±1.1 |
15.6±2.7 |
148.1±37.1 |
442.6±266.0a |
3.11±2.19a | 25.52 |
0.50±0.21b | 60.22 |
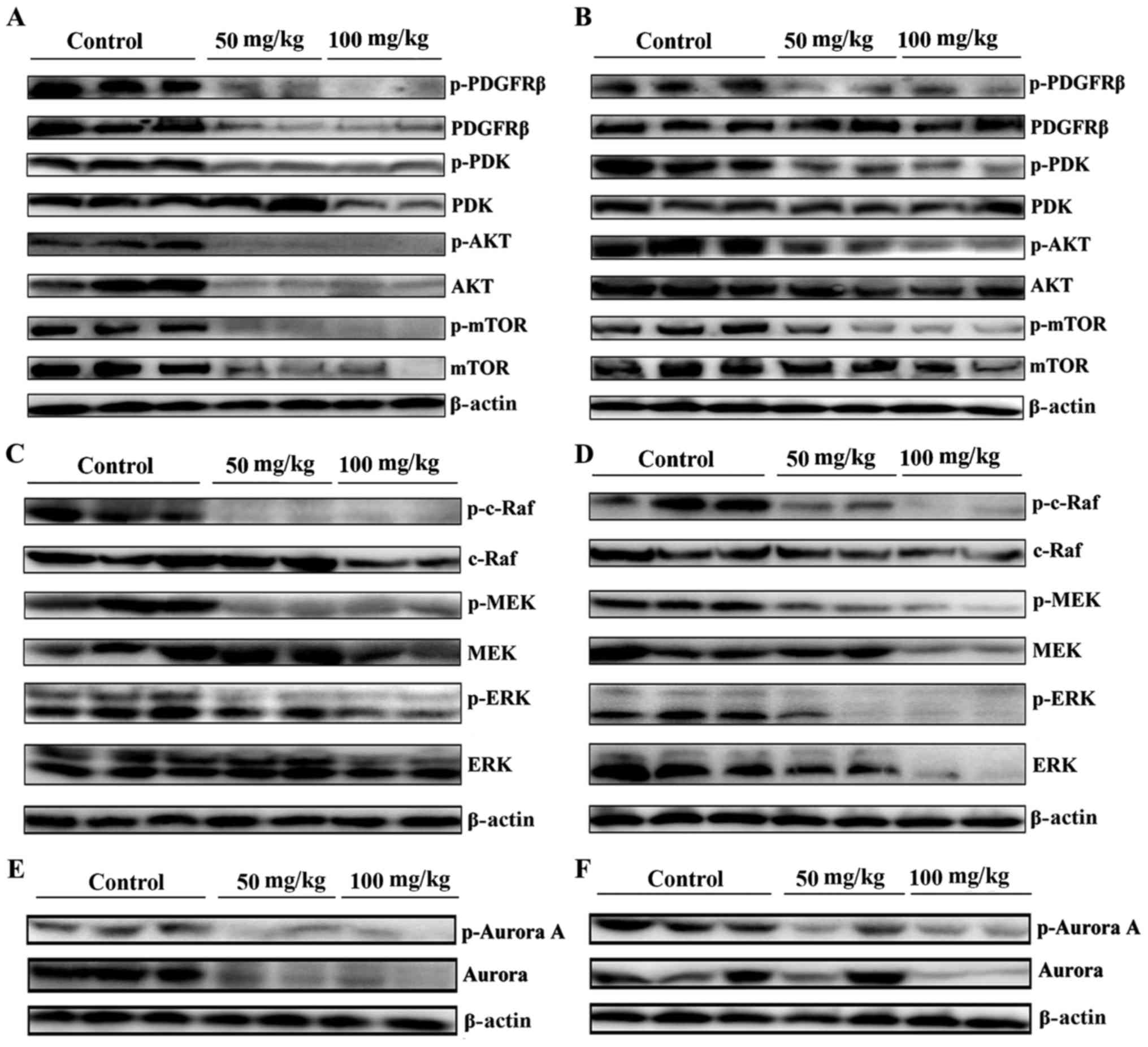
T03 downregulated the Raf/MEK/ERK and
PDGFRβ/Akt signaling pathway in MCF-7 and MCF-7/T xenograft nude
model mice
Subsequently, whether T03 inactivated the
above-mentioned kinase-associated signaling pathways was evaluated.
As expected, the phosphorylated levels of PDGFRβ decreased in
T03-treated tumors of MCF-7 and MCF-7/T xenografts (Fig. 4A and B). As PDK is an important
downstream target of PDGFRβ/PI3K signaling and a key upstream
kinase of the AKT/mTOR signaling pathway, its activation promotes
proliferation and inhibits apoptosis in numerous human cancer types
(9,22,23).
The current study observed that the activity of PDK, AKT, and mTOR
decreased in MCF-7 and MCF-7/T treated with T03 compared with the
controls (Fig. 4A and B),
indicating that T03 may downregulate PDGFRβ and PDK/AKT/mTOR and in
MCF-7 and MCF-7/T xenograft models.
As T03 inhibited c-Raf in the current study, whether
T03 downregulates the Ras/Raf/ERK signaling pathway in MCF-7 and
MCF-7/T xenografts was examined. Fig.
4C and D demonstrate that T03 treatment significantly and
dose-dependently decreased p-c-Raf, p-MEK, and p-ERK in MCF-7 and
MCF-7/T tumors, while the basal levels of c-Raf, MEK and ERK were
only reduced in the group treated with 100 mg/kg T03.
Furthermore, T03 downregulated Aurora A, a
downstream effecter of the Ras/Raf/MEK/ERK signaling pathway
(24), in MCF-7 and MCF-7/T
xenografts. Aurora A and p-Aurora A were markedly downregulated
upon T03 treatment, particularly in MCF-7 tumors, which were
consistent with the previous study (24) that Aurora A and p-Aurora A were
frequently activated by the Ras/Raf signaling pathway (Fig. 4E and F).
Discussion
Despite improvements in prevention, early detection,
and treatment, breast cancer remains one of the most common
malignant tumors affecting women in western countries (25). Drug-resistance of breast cancer to
Taxol has limited its effect and application in clinical treatment.
In the present study, the anti-tumor activity of the novel
multi-kinases inhibitor, T03 was investigated, as well as its
potential in breast cancer treatment.
As with Taxol, T03 displayed similar antitumor
effects on MX-1 and MCF-7 breast cancer cells. Furthermore, T03
inhibited the growth of Taxol-resistant MX-1/T and MCF-7/T breast
tumors in vitro and in vivo. It caused
G2/M-phase cell accumulation and induced apoptosis, thus
resulting in cell growth inhibition. In addition, T03 resulted in
tumor regression in MX-1- and MCF-7-derived xenografts, as well as
in Taxol-resistant MX-1/T and MCF-7/T tumors, indicating that T03
may exert effects on Taxol-sensitive and -resistant breast cancer
cells. In addition, T03 was demonstrated to exert efficient effects
on other types of drug-resistant breast cancer cells, such as
Doxorubicin (Adriamycin)-resistant cells, which indicated that T03
may be used for treatment of other types of drug-resistant breast
cancer.
Previous studies revealed that PDGFRβ was
overexpressed in breast cancer (26,27).
Highly activated PDGFRβ promoted tumor cell proliferation via
PI3K/Akt and Ras/MEK/ERK signaling pathways (28,29),
and resulted in distant metastasis and insensitivity to
chemotherapy (30). The current
study demonstrated that T03 may downregulate the Akt/mTOR and
Ras/MEK/ERK signaling pathways, as well as PDGFRβ in MCF-7 and
MCF-7/T in vivo. In MCF-7 and MCF-7/T xenograft tumors, T03
significantly reduced p-PDGFRβ, which was further confirmed by
performance of the biochemical assay. Furthermore, T03
downregulated PDK, AKT and mTOR. As PDK-1, Akt and mTOR are the
downstream components of PDGFRβ, and are involved in cell growth
and apoptosis, T03 may cause cell growth inhibition and apoptosis
via downregulation of the PDGFRβ/Akt signaling pathway. PDGFRβ
activation is known to induce Taxol-resistance in breast cancer
(31,32). Therefore, T03 may be used for
inhibiting Taxol-resistant breast cancer.
Raf kinase is an upstream member of the Raf/MEK/ERK
signaling cascade (33).
Activation of the Raf/MEK/ERK signaling pathway has been associated
with chemoresistance of breast cancer (34–36).
The present data indicated that T03 inhibited p-c-Raf, and
consequently resulted in downregulation of p-MEK and p-ERK. These
results indicate that T03 may inhibit the cell cycle and induce
apoptosis via downregulation of the Raf/MEK/ERK signaling pathway
in MCF-7 and MCF-7/T breast cancer.
Previous studies observed that activation of c-Raf
signaling led to stabilization and accumulation of Aurora A mitotic
kinase in breast cancer cells, and deduced that c-Raf may regulate
the expression levels of Aurora A (24,37).
The current study found that T03 treatment led to inhibition of
c-Raf and decreased Aurora A in MCF-7 and MCF-7/T xenografts.
Previous studies have demonstrated that
over-expressed Aurora A inhibits apoptosis, promotes cell cycle
progression and metastasis, and mediates Taxol-resistance in breast
cancer and other types of cancer (38,39).
Inhibition of Aurora A by T03 may cause G2/M cell
accumulation, apoptosis and sensitivity to Taxol in breast cancer.
Based on the current results, the therapeutic efficacy of T03 on
breast cancer may be partially attributed to the inhibition of
c-Raf, PDGFRβ and the associated signaling pathways (40,41).
Although T03 presented similar anti-tumor activity
in Taxol-sensitive and -resistant breast cancer, and
Doxorubicin-resistant breast cancer as well, there are certain
efficacy differences, which merit further investigation. In
addition, based on our existing data, whether the anti-tumor
effects are transient or permanent could not be determined.
In conclusion, the current study demonstrated that
T03 induces cell cycle arrest and apoptosis, and inhibits cell
proliferation in MX-1, MCF-7, MX-1/T, and MCF-7/T breast cancer
in vitro and in vivo. These results demonstrate that
T03 inhibits breast cancer growth by downregulating PDGFRβ/Akt and
Ras/Raf/ERK signaling pathways, which are regulators of apoptosis,
proliferation and chemoresistance. These findings indicate that T03
may be a potential candidate for effective chemotherapy of breast
cancer, particularly for Taxol-resistant breast cancer.
Acknowledgements
The present study was financially supported by The
National Natural Science Foundation of China (grant no. 81102025)
and National S&T Major Special Project on ‘12·5 Major New Drug
Innovation’ (grant no. 2012ZX09103-101-019).
References
|
1
|
Reese JM, Suman VJ, Subramaniam M, Wu X,
Negron V, Gingery A, Pitel KS, Shah SS, Cunliffe HE, McCullough AE,
et al: ERβ1: Characterization, prognosis, and evaluation of
treatment strategies in ERα-positive and -negative breast cancer.
BMC Cancer. 14:7492014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nahta R, Hortobágyi GN and Esteva FJ:
Growth factor receptors in breast cancer: Potential for therapeutic
intervention. Oncologist. 8:5–17. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vlahovic G and Crawford J: Activation of
tyrosine kinases in cancer. Oncologist. 8:531–538. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Niehoff P, Mundhenke C, Kimmig B and Maas
N: Breast irradiation with brachytherapy: Approved techniques and
new concepts. Minerva Ginecol. 59:377–386. 2007.PubMed/NCBI
|
|
5
|
Hubbard SR: Juxtamembrane autoinhibition
in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 5:464–471.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shchemelinin I, Sefc L and Necas E:
Protein kinase inhibitors. Folia Biol (Praha). 52:137–148.
2006.PubMed/NCBI
|
|
7
|
Weigel MT, Meinhold-Heerlein I,
Bauerschlag DO, Schem C, Bauer M, Jonat W, Maass N and Mundhenke C:
Combination of imatinib and vinorelbine enhances cell growth
inhibition in breast cancer cells via PDGFR beta signalling. Cancer
Lett. 273:70–79. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weigel MT, Banerjee S, Arnedos M, Salter
J, A'Hern R, Dowsett M and Martin LA: Enhanced expression of the
PDGFR/Abl signaling pathway in aromatase inhibitor-resistant breast
cancer. Ann Oncol. 24:126–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin HJ, Hsieh FC, Song H and Lin J:
Elevated phosphorylation and activation of PDK-1/AKT pathway in
human breast cancer. Br J Cancer. 93:1372–1381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weigel MT, Dahmke L, Schem C, Bauerschlag
DO, Weber K, Niehoff P, Bauer M, Strauss A, Jonat W, Maass N and
Mundhenke C: In vitro effects of imatinib mesylate on
radiosensitivity and chemosensitivity of breast cancer cells. BMC
Cancer. 10:4122010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sridhar SS, Hedley D and Siu LL: Raf
kinase as a target for anticancer therapeutics. Mol Cancer Ther.
4:677–685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Z, Min W and Gou J: Knockdown of
cyclophilin A reverses paclitaxel resistance in human endometrial
cancer cells via suppression of MAPK kinase pathways. Cancer
Chemother Pharmacol. 72:1001–1011. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gong Y, He H, Liu H, Zhang C, Zhao W and
Shao RG: Phosphorylation of myofibrillogenesis regulator-1
activates the MAPK signaling pathway and induces proliferation and
migration in human breast cancer MCF7 cells. FEBS Lett.
588:2903–2910. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hashimoto K, Tsuda H, Koizumi F, Shimizu
C, Yonemori K, Ando M, Kodaira M, Yunokawa M, Fujiwara Y and Tamura
K: Activated PI3K/AKT and MAPK pathways are potential good
prognostic markers in node-positive, triple-negative breast cancer.
Ann Oncol. 25:1973–1979. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Heckler MM, Thakor H, Schafer CC and
Riggins RB: ERK/MAPK regulates ERRγ expression, transcriptional
activity and receptor-mediated tamoxifen resistance in ER+ breast
cancer. FEBS J. 281:2431–2442. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Im NK, Jang WJ, Jeong CH and Jeong GS:
Delphinidin suppresses PMA-induced MMP-9 expression by blocking the
NF-κB activation through MAPK signaling pathways in MCF-7 human
breast carcinoma cells. J Med Food. 17:855–861. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saini KS, Loi S, de Azambuja E,
Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE and
Piccart-Gebhart MJ: Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK
pathways in the treatment of breast cancer. Cancer Treat Rev.
39:935–946. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu J, Tan M, Huang WC, Li P, Guo H, Tseng
LM, Su XH, Yang WT, Treekitkarnmongkol W, Andreeff M, et al:
Mitotic deregulation by survivin in ErbB2-overexpressing breast
cancer cells contributes to Taxol resistance. Clin Cancer Res.
15:1326–3134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Y, Tang K, Zhang H, Zhang Y, Zhou W and
Chen X: Function of Aurora kinase A in Taxol-resistant breast
cancer and its correlation with P-gp. Mol Med Rep. 4:739–746.
2011.PubMed/NCBI
|
|
20
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43–9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Zhang ZF, Chen J, Huang D, Ding Y,
Tan MH, Qian CN, Resau JH, Kim H and Teh BT: VX680/MK-0457, a
potent and selective Aurora kinase inhibitor, targets both tumor
and endothelial cells in clear cell renal cell carcinoma. Am J
Transl Res. 2:296–308. 2010.PubMed/NCBI
|
|
22
|
Reynolds TH IV, Bodine SC and Lawrence JC
Jr: Control of Ser2448 phosphorylation in the mammalian target of
rapamycin by insulin and skeletal muscle load. J Biol Chem.
277:17657–17662. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Salmond RJ, Emery J, Okkenhaug K and
Zamoyska R: MAPK, phosphatidylinositol 3-kinase, and mammalian
target of rapamycin pathways converge at the level of ribosomal
protein S6 phosphorylation to control metabolic signaling in CD8 T
cells. J Immunol. 183:7388–7397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Biran A, Brownstein M, Haklai R and Kloog
Y: Downregulation of survivin and aurora A by histone deacetylase
and RAS inhibitors: A new drug combination for cancer therapy. Int
J Cancer. 128:691–701. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim BK, Lee JW, Park PJ, Shin YS, Lee WY,
Lee KA, Ye S, Hyun H, Kang KN, Yeo D, et al: The multiplex bead
array approach to identifying serum biomarkers associated with
breast cancer. Breast Cancer Res. 11:R222009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
de Jong JS, van Diest PJ, van der Valk P
and Baak JP: Expression of growth factors, growth-inhibiting
factors, and their receptors in invasive breast cancer. II:
Correlations with proliferation and angiogenesis. J Pathol.
184:53–57. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lev DC, Kim SJ, Onn A, Stone V, Nam DH,
Yazici S, Fidler IJ and Price JE: Inhibition of platelet-derived
growth factor receptor signaling restricts the growth of human
breast cancer in the bone of nude mice. Clin Cancer Res.
11:306–314. 2005.PubMed/NCBI
|
|
28
|
Klos KS, Wyszomierski SL, Sun M, Tan M,
Zhou X, Li P, Yang W, Yin G, Hittelman WN and Yu D: ErbB2 increases
vascular endothelial growth factor protein synthesis via activation
of mammalian target of rapamycin/p70S6K leading to increased
angiogenesis and spontaneous metastasis of human breast cancer
cells. Cancer Res. 66:2028–2037. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wen XF, Yang G, Mao W, Thornton A, Liu J,
Bast RC Jr and Le XF: HER2 signaling modulates the equilibrium
between pro- and antiangiogenic factors via distinct pathways:
Implications for HER2-targeted antibody therapy. Oncogene.
25:6986–6996. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seymour L and Bezwoda WR: Positive
immunostaining for platelet derived growth factor (PDGF) is an
adverse prognostic factor in patients with advanced breast cancer.
Breast Cancer Res Treat. 32:229–233. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Blagosklonny MV and Fojo T: Molecular
effects of paclitaxel: Myths and reality (a critical review). Int J
Cancer. 83:151–156. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Langley RR, Fan D, Tsan RZ, Rebhun R, He
J, Kim SJ and Fidler IJ: Activation of the platelet-derived growth
factor-receptor enhances survival of murine bone endothelial cells.
Cancer Res. 64:3727–3730. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sambade MJ, Camp JT, Kimple RJ, Sartor CI
and Shields JM: Mechanism of lapatinib-mediated radiosensitization
of breast cancer cells is primarily by inhibition of the
Raf>MEK>ERK mitogen-activated protein kinase cascade and
radiosensitization of lapatinib-resistant cells restored by direct
inhibition of MEK. Radiother Oncol 93: 639–644, 2009. Radiother
Oncol 93: 639–644, 2009. 93: 639–644, 2009:639-644, 2009–644, 2009.
2009.
|
|
34
|
Frogne T, Benjaminsen RV, Sonne-Hansen K,
Sorensen BS, Nexo E, Laenkholm AV, Rasmussen LM, Riese DJ II, de
Cremoux P, Stenvang J and Lykkesfeldt AE: Activation of ErbB3, EGFR
and Erk is essential for growth of human breast cancer cell lines
with acquired resistance to fulvestrant. Breast Cancer Res Treat.
114:263–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lian WJ, Liu G, Liu YJ, Zhao ZW, Yi T and
Zhou HY: Downregulation of BMP6 enhances cell proliferation and
chemoresistance via activation of the ERK signaling pathway in
breast cancer. Oncol Rep. 30:193–200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sokolosky M, Chappell WH, Stadelman K,
Abrams SL, Davis NM, Steelman LS and McCubrey JA: Inhibition of
GSK-3β activity can result in drug and hormonal resistance and
alter sensitivity to targeted therapy in MCF-7 breast cancer cells.
Cell Cycle. 13:820–833. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
D'Assoro AB, Liu T, Quatraro C, Amato A,
Opyrchal M, Leontovich A, Ikeda Y, Ohmine S, Lingle W, Suman V, et
al: The mitotic kinase Aurora-a promotes distant metastases by
inducing epithelial-to-mesenchymal transition in ERα(+) breast
cancer cells. Oncogene. 33:599–610. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Anand S, Penrhyn-Lowe S and Venkitaraman
AR: AURORA-A amplification overrides the mitotic spindle assembly
checkpoint, inducing resistance to Taxol. Cancer Cell. 3:51–62.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kwiatkowski N, Deng X, Wang J, Tan L,
Villa F, Santaguida S, Huang HC, Mitchison T, Musacchio A and Gray
N: Selective aurora kinase inhibitors identified using a
taxol-induced checkpoint sensitivity screen. ACS Chem Biol.
7:185–196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Horowitz JC, Lee DY, Waghray M, Keshamouni
VG, Thomas PE, Zhang H, Cui Z and Thannickal VJ: Activation of the
pro-survival phosphatidylinositol 3-kinase/AKT pathway by
transforming growth factor-beta1 in mesenchymal cells is mediated
by p38 MAPK-dependent induction of an autocrine growth factor. J
Biol Chem. 279:1359–1367. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Arany I, Faisal A, Nagamine Y and
Safirstein RL: p66shc inhibits pro-survival epidermal growth factor
receptor/ERK signaling during severe oxidative stress in mouse
renal proximal tubule cells. J Biol Chem. 283:6110–6117. 2008.
View Article : Google Scholar : PubMed/NCBI
|