Introduction
Liver failure is a serious and common clinical
syndrome induced in a number of different ways. With liver failure
there is severe dysfunction or decompensation in liver synthesis,
detoxification, excretion, and conversion. These are accompanied by
coagulation disorders, jaundice, hepatic encephalopathy, ascites,
and other clinical manifestations, that result in an extremely high
mortality (1). Acute and subacute
forms of liver failure (ALF/SLF) are the most critical.
Infantile liver failure syndrome-2 (ILFS2, OMIM
616483) is an autosomal recessive genetic disorder connected with
recurrent episodes of acute liver failure during intercurrent
febrile illness. Initial episodes occur in infancy or early
childhood, and with conservative treatment, full recovery is
achieved between episodes. Haack et al identified homozygous
or compound heterozygous mutations in the neuroblastoma amplified
sequence (NBAS) gene in five unrelated German patients with
ILFS2 (2). NBAS was
previously associated with short stature, optic nerve atrophy, and
Pelger-Huet anomaly (SOPH syndrome, MIM614800) in an isolated
Russian Yakut population, but without liver failure (3). Further studies have found that the
phenotypic spectrum of NBAS based diseases also involve brain,
connective tissue, and the immune system (4,5). The
NBAS protein is involved in Golgi-to-endoplasmic reticulum (ER)
retrograde transport (6) and is
considered to be a component of the SNAREs (Soluble
N-Ethylmaleimide-sensitive Factor (NSF) Attachment Protein
Receptors). Essentially every step of membrane transport is carried
out by a pair of different SNARE proteins (v-SNARE and t-SNARE).
The SNARE proteins mediate intracellular transport of vesicles,
such as ER to Golgi and Golgi to plasma membranes, and are
conserved from yeast to human. The NBAS protein interacts with
t-SNARE p31 directly and with other proteins, forming complex
syntaxin 18 (7). In the cells of
patients, a reduction in NBAS is accompanied by a decrease in p31,
supporting an important function for NBAS in the SNARE complex
(2). Although these intracellular
events are known, the specific mechanisms by which NBAS contributes
to liver disease and to fever, is not fully understood.
In this study, we performed targeted next-generation
sequencing (NGS) to identify mutations in 358 genes related to
diseases of growth and development. The purpose of this was to
devise a strategy useful for genetic diagnosis of patients with
abnormal growth and development. By use of this strategy, two novel
mutations in NBAS were identified in a 4-year-old Chinese
girl with ILFS2.
Materials and methods
Study participants
The research protocol was approved by the ethics
committee of the Hubei Provincial Hospital of Traditional Chinese
Medicine (Wuhan, China). The participant provided informed consent
for clinical and genetic studies. Patient associated with recurrent
respiratory infections, elevated transaminase, and severe
hypoglycemia within the last 2 years. Based on the Online Mendelian
Inheritance in Man website (OMIM), we chose specific genes linked
to teratogenic and fatal diseases that would most certainly be
involved in tests associated with family planning (available upon
request).
Enrichment and sequencing of disease
genes
Genomic DNA was extracted from blood samples using
the QIAamp DNA Blood Midi kit (Qiagen, Hilden, Germany) following
the manufacturer's standard procedure. The qualified genomic DNA
sample was randomly fragmented by Covaris (Covaris S2; Covaris,
Inc., Woburn, MA, USA) with the size of the library fragments
primarily distributed between 200 and 250 bp. Illumina HiSeq SBS
barcoded sequencing libraries were made based on the manufacturer's
protocols (Illumina, Inc., San Diego, CA, USA). We performed 4
cycles of polymerase chain reaction (PCR). The PCR products were
pooled and hybridized to the capture array for 72 h. The washing,
elution, and additional amplification steps were performed
according to NimbleGen protocols (F. Hoffmann-La Roche Ltd.,
Pleasanton, CA, USA) followed by 12 cycles of
capturedligation-mediated PCR (LM-PCR). An Agilent 2100 Bioanalyzer
(Agilent Technologies, Waldbronn, Germany) and ABI stepOne (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) were
used to estimate the magnitude of enrichment and library insert
size for the final products. The libraries were sequenced
byHiSeq2000 (8).
NGS data analysis
After base calling by Illumina Pipeline, the primary
data were obtained by fastq. Clean data for further analysis were
obtained by filtering adapter and low quality reads (9). To identify single nucleotide variants
(SNVs) and indels, we aligned the 100 bp clean reads against the
National Center for Biotechnology Information (NCBI) reference
human genome sequence using Burrows-Wheeler Aligner software
(10). Indels and SNVs were
identified using the GATK (Genome Analysis Toolkit) (11). All SNVs were annotated using the
dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), HapMap (http://hapmap.ncbi.nlm.nih.gov/), HGMD
(http://www.hgmd.org/), and the 1000 Genome
(http://www.1000genomes.org/),
respectively.
Sanger sequencing
Polymerase chain reaction (PCR) amplification was
executed using primers specific for mutations of NBAS, which
NBAS-c.209 forward, 5′-GGAAATACTTGAAACTTTTGAATAACAC-3′, and
reverse, 5′-ACCTAAATGTTTGAAATGTCTCATACTG-3′; NBAS-c.3596 forward,
5′-TTATGTACTGTGGATGTAACTGTGGG-3′, and reverse,
5′-TGTTGGCTTTATTTTTTCTTTTGG-3′. PCR conditions were: i) 98°C for 30
sec; ii) 98°C for 10 sec, 55°C for 30 sec, 72°C for 30 sec, 25
cycles; ii) 72°C for 10 min; and iv) hold at 4°C. The amplified
products were sequenced using an ABI 3730 DNA Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.) by capillary
electrophoresis and analyzed using BioEdit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html).
NM_015909.3 was chosen as the reference sequence.
Calculation of the stability of
predicted mutations
Potential deleterious effects of identified sequence
variants were assessed by various algorithms; PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2/) (12), SIFT (http://sift.jcvi.org/) (13), and Mutation Taster (http://mutationtaster.org/) (14). In theory, mutations usually alter
the structural stability of the protein and thus affect its
functional activity. Human Splicing Finder (http://umd.be/HSF3/) was used to analyze pre-mRNA
splicing (15). In order to check
the energy stability of the mutants, the Web server I-Mutant 3.0
(http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi)
was used (16). The I-Mutant 3.0
suite is based on a support vector machine (SVM) algorithm that
calculates protein stability related to a single mutation in units
of free energy (ΔΔG).
Sequence conservative analysis and
computer modeling
Conservation across species indicates that a
sequence has been maintained by evolution despite speciation. In
this study, conservation analysis showed the degree of conservation
across species at each residue of a given protein. CLUSTALW
(http://www.genome.jp/tools/clustalw/)
was used to compare homologous protein sequences among nine
representative species; Homo sapiens (human), Pan
troglodytes (chimpanzee), Macaca mulatta (Rhesus
monkey), Canis lupusfamiliaris (dog), Bos taurus
(cattle), Mus musculus (house mouse), Gallus gallus
(chicken), Xenopus tropicalis (tropical clawed frog), and
Danio rerio (zebrafish). The alignment result was exported
by EsPript (http://espript.ibcp.fr/ESPript/ESPript/). Homology
modeling and refining were conducted by the YASARA program using
default parameters (http://www.yasara.org/). The model was visualized with
the Pymol Molecular Graphics system (http://pymol.org/).
Results
Clinical status
The patient was a 4-year-old female with repeated
upper respiratory tract infections that were associated with fever.
There were no obvious physical abnormalities upon examination.
Routine blood analysis showed a slightly elevated white blood cell
count and a slightly elevated level of C-reactive protein (CRP).
Liver function was abnormal: Alanine transaminase (ALT) 4,790 U/l,
aspartate transaminase (AST) 7,690 U/l, alkaline phosphatase: 253
U/l; total bile acid: 267.5 µmol/l, blood glucose: 7.80 mmol/l,
lactate dehydrogenase: 3,230 U/l; α-hydroxybutyrate dehydrogenase:
1,541 U/l, creatine kinase (CK) isoenzyme: 30.9 U/l, and CKMB/CK:
0.36. After anti-infection and liver-protection treatment (10%
glucose 100 ml, reduced glutathione 0.45 i.v., 10% glucose 200 ml,
and compound glycyrrhizin glucoside 40 mg, i.v. drip), symptoms
mitigated and liver function normalized. After hospital discharge,
the patient had repeated upper respiratory tract infections
associated with fever and was readmitted to hospital for treatment.
AST and ALT were abnormal for unknown reasons, even though there
was no history of hepatitis.
High-throughput sequencing, mapping,
and coverage
In this study, we designed a unique high-density
array to capture all exons and 200 bp adjacent intron sequences of
358 genes related to growth and development diseases. The enriched
DNA was sequenced at single-base resolution using the Illumina
HiSeq2000 Analyzer. Of the reads, 53.66% mapped to target regions
and 52.64% mapped to flanking 200 bp regions. Coverage of target
regions was 99.51% and the average sequencing depth for target
regions was 108.1x. The fraction of target regions covered was at
least 4x, 10x, 20× and were respectively, 99.25, 98.34, and
96.01%.
Determination clinical significance of
genetic variants
All variants meeting the filtering criteria,
described below, are listed in Table
I. First, the allele frequency of variants should be less than
0.05 in the dbSNP, HapMap, 1,000 human genome dataset. Second,
variants were considered as likely disease mutations, if sequence
variation was previously reported and was a recognized cause of the
disorder in dbSNP and HGMD. Third, variants were retained as
disease mutations if they were predicted to result in a premature
stop codon or loss of a substantial portion of the protein. Fourth,
the variation was new, was forecasted to be harmful, and its allele
frequency was less than 0.05. If these four criteria were met then
the mutation was considered a disease mutation.
 | Table I.Possible pathogenic mutations. |
Table I.
Possible pathogenic mutations.
| Gene | Transcript | Variation | Hom/Het | rs-ID | Fr.1 | Fr.2 | Fr.3 |
|---|
| NBAS | NM_015909.3 |
c.6220G>A;p.A2074T | Het | rs6710817 | 0.1821 | 0.234 | 0.1841 |
| NBAS | NM_015909.3 |
c.3596G>A;p.C1199Y | Het | – | – | – | 0 |
| NBAS | NM_015909.3 | c.209+1G>A | Het | – | – | – | 0 |
| NBAS | NM_015909.3 |
c.1964A>G;p.K655R | Het | rs4668909 | 0.497 | 0.343 | 0.3397 |
| NBAS | NM_015909.3 |
c.727A>G;p.I243V | Het | rs13029846 | 0.4999 | 0.333 | 0.3535 |
In this study, the following NBAS SNVs were
detected; c.6220G>A (p.A2074T), c.3596G>A (p.C1199Y),
c.209+1G>A, c.1964A>G (P.K655R), and c.727A>G (p.I243V).
Among them, although SIFT and PolyPhen-2 predicted that
c.6220G>A was, respectively, harmful and potentially harmful,
the frequency of the SNV in the three databases was 0.1821, 0.2434,
and 0.1841 and was not studied further. In other words, this SNV
appears to be common. In addition, c.1964A>G in the three
databases was respectively, 0.497, 0.343, and 0.3397. The frequency
of c.727A>G in the three databases was respectively, 0.4999,
0.333, and 0.3535. From this frequency information these two SNVs
were also common in the population. Two uncommon SNVs were
c.3596G>A and c.209 + 1G>A, which had no demonstrable allele
frequency in the three databases. For c.3596G>A, SIFT's
prediction was harmful and the I-Mutant 3.0 ΔΔG value was predicted
to be −0.22 kcal/mol and SVM3 predicted a Large Decrease (Table II). This information identified
these two sites as potentially pathogenic.
| Table II.In silico single nucleotide
variant predictions. |
Table II.
In silico single nucleotide
variant predictions.
| Variation | SIFT prediction | PolyPhen-2 | Mutation tester | Human splicing
finder | I-Mutant 3.0 |
|---|
|
c.6220G>A;p.A2074T | Damaging | Possibly
damaging | Disease causing |
| ΔΔG value
prediction | −0.56 Kcal/mol |
|
|
|
|
|
| SVM3 prediction
effect | Large decrease |
|
c.3596G>A;p.C1199Y | Damaging | Probably
damaging | Disease causing |
| ΔΔG value
prediction | −0.22 Kcal/mol |
|
|
|
|
|
| SVM3 prediction
effect | Large decrease |
| c.209+1G>A | – | – |
| Broken WT |
|
|
|
| – | – |
| Donor site |
|
|
|
c.1964A>G;p.K655R | Damaging | Benign | Polymorphism |
| ΔΔG value
prediction | −0.22 Kcal/mol |
|
|
|
|
|
| SVM3 prediction
effect | Neutral |
|
c.727A>G;p.I243V | Damaging | Benign | Disease causing |
| ΔΔG value
prediction | −0.66 Kcal/mol |
|
|
|
|
|
| SVM3 prediction
effect | Large decrease |
Mutations analysis
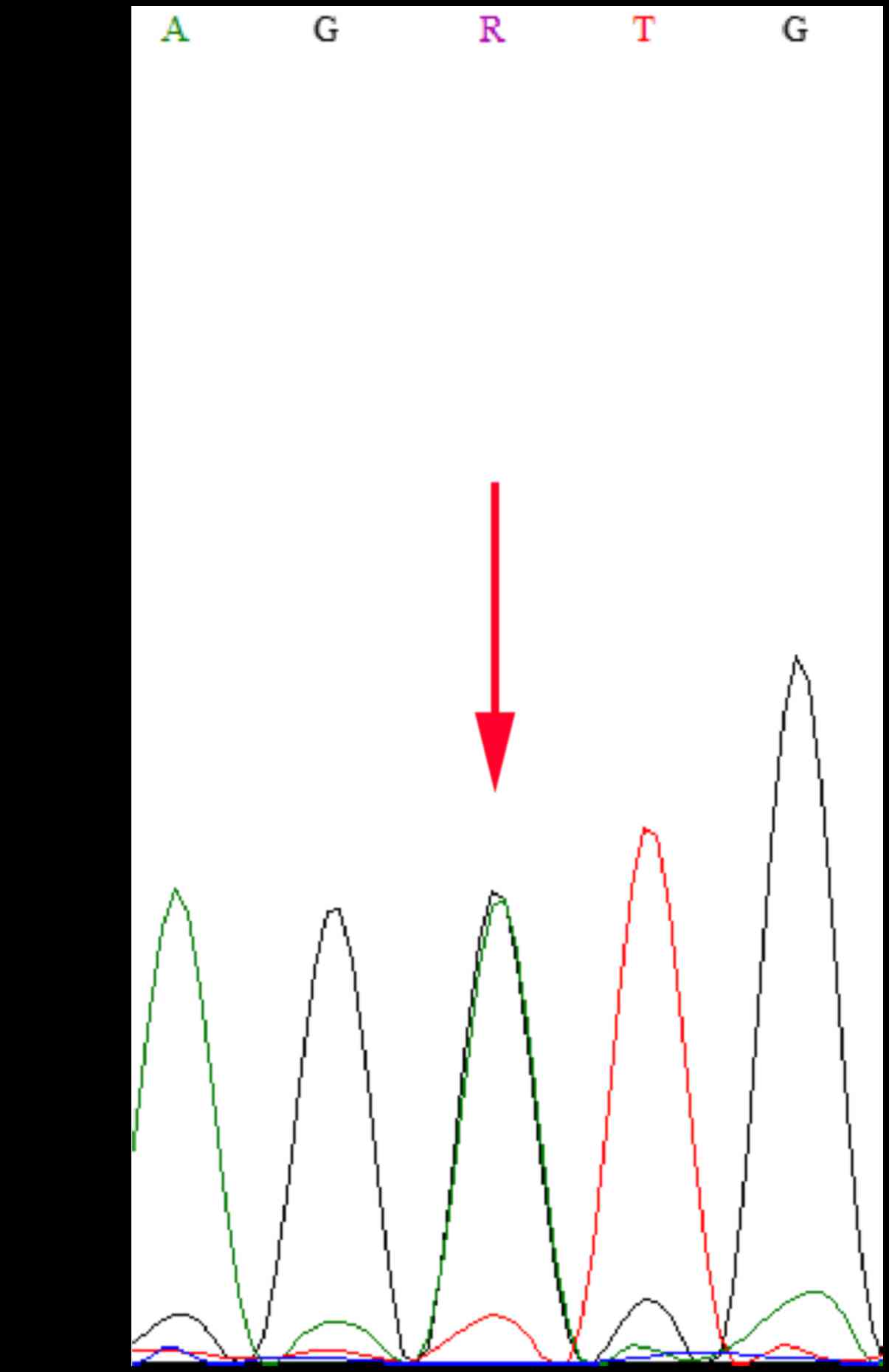
Mutations c.3596G>A and c.209+1G>A were
further confirmed by directional Sanger sequencing (Fig. 1). We analyzed the mutation,
c.209+1G>A, using the Human Splicing Finder program (http://umd.be/HSF3/index.html). Mutations found in
this way alter the wild-type donor site, most likely affecting
splicing. This heterozygous mutation was located at the splice
donor site of intron 3, forming a new cleavage donor site that
would result in the formation of frame shift mutations in the
protein product and ultimately premature coding (Table II). Such a truncated protein
product would lose its normal function, resulting in disease
(17). The amino acid
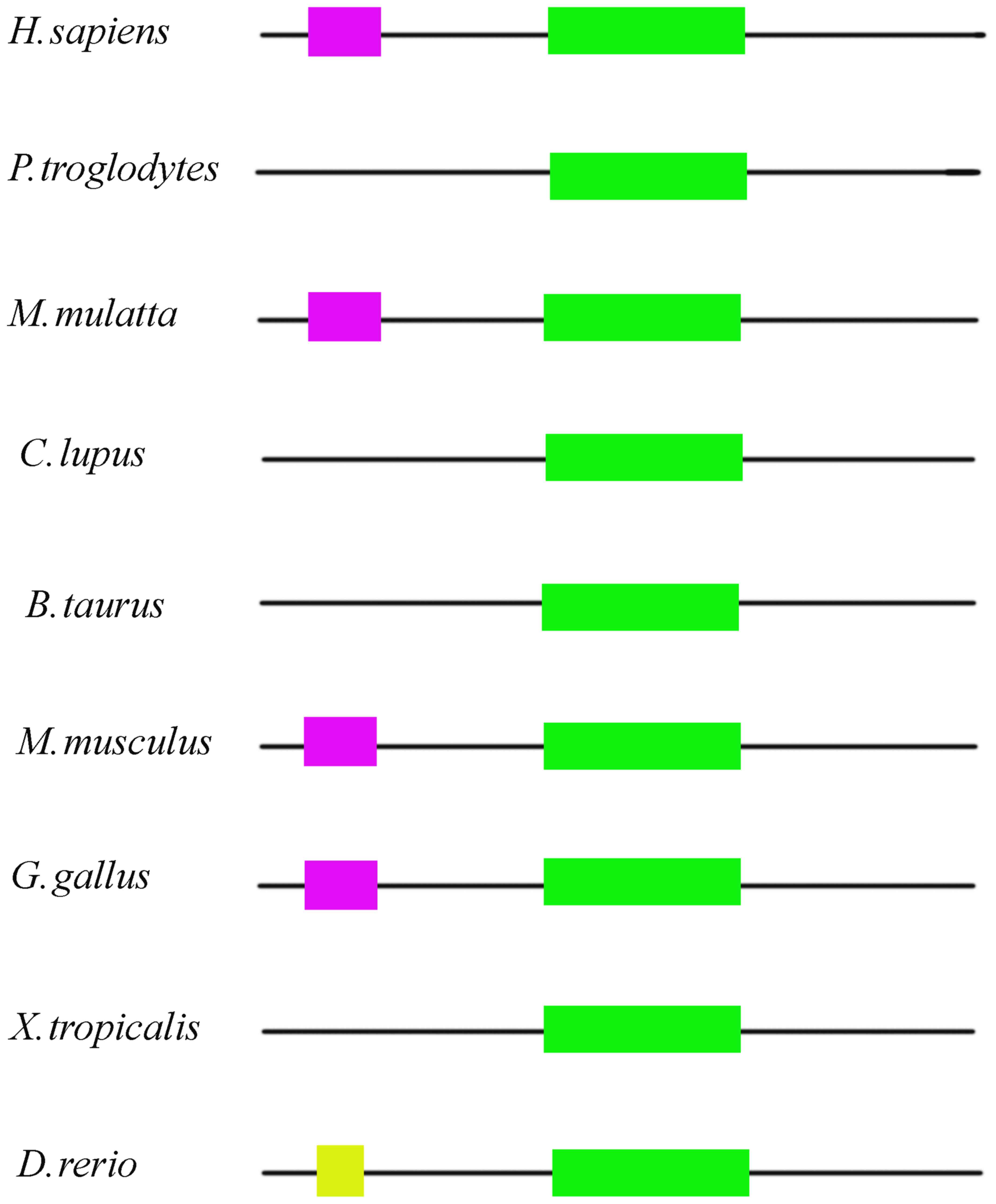
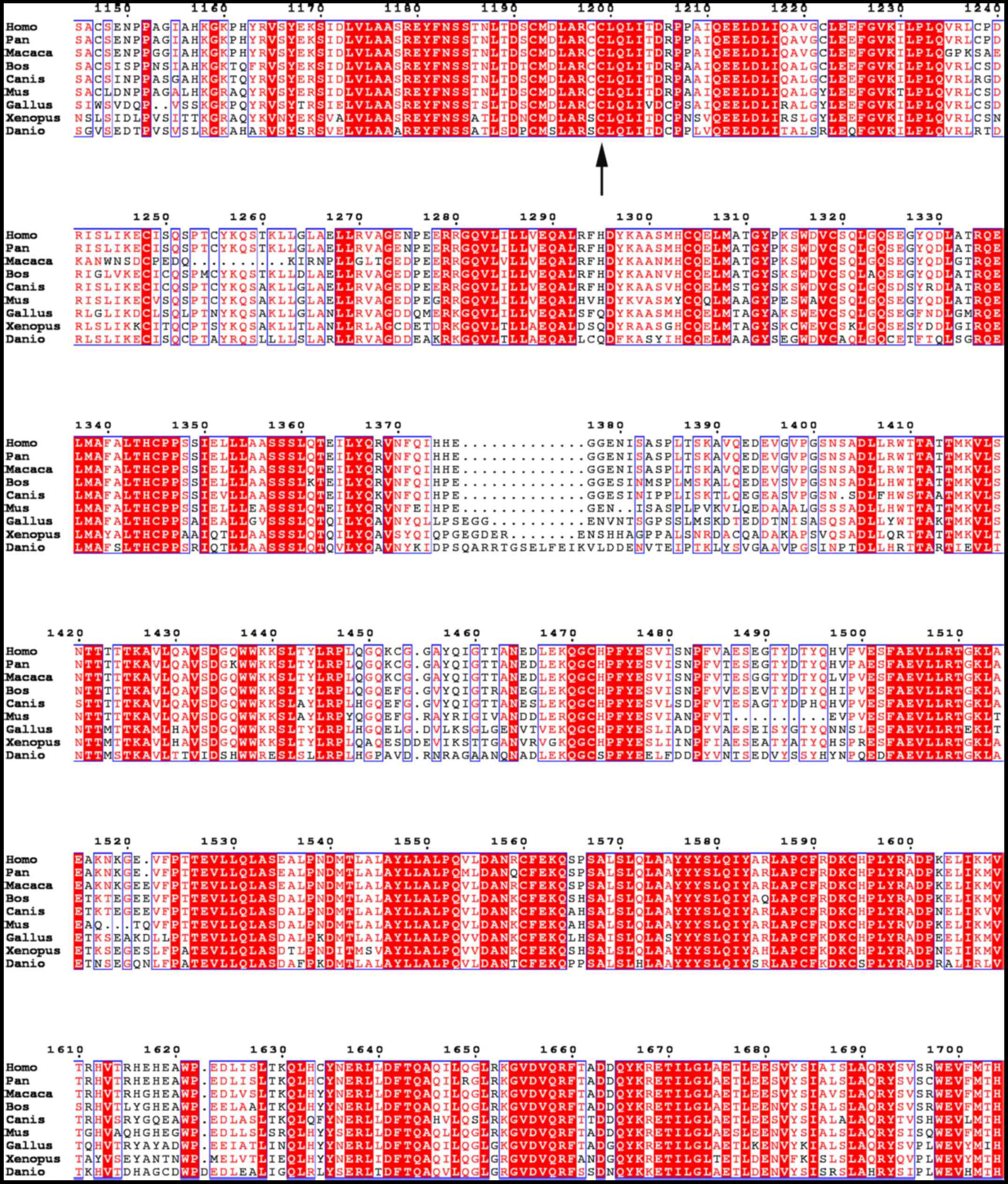
conservativeness of the c.3596G>A mutation was analyzed.
NBAS was found to be conserved in human, chimpanzee, Rhesus
monkey, dog, cattle, house mouse, chicken, tropical clawed frog,
and zebrafish (Fig. 2). The SEC39
region of the protein was found to be highly conserved. The SEC39
region is a necessary part of Golgi-ER retrograde transport, and
the mutation was located in this evolutionary conserved region
(Fig. 3).
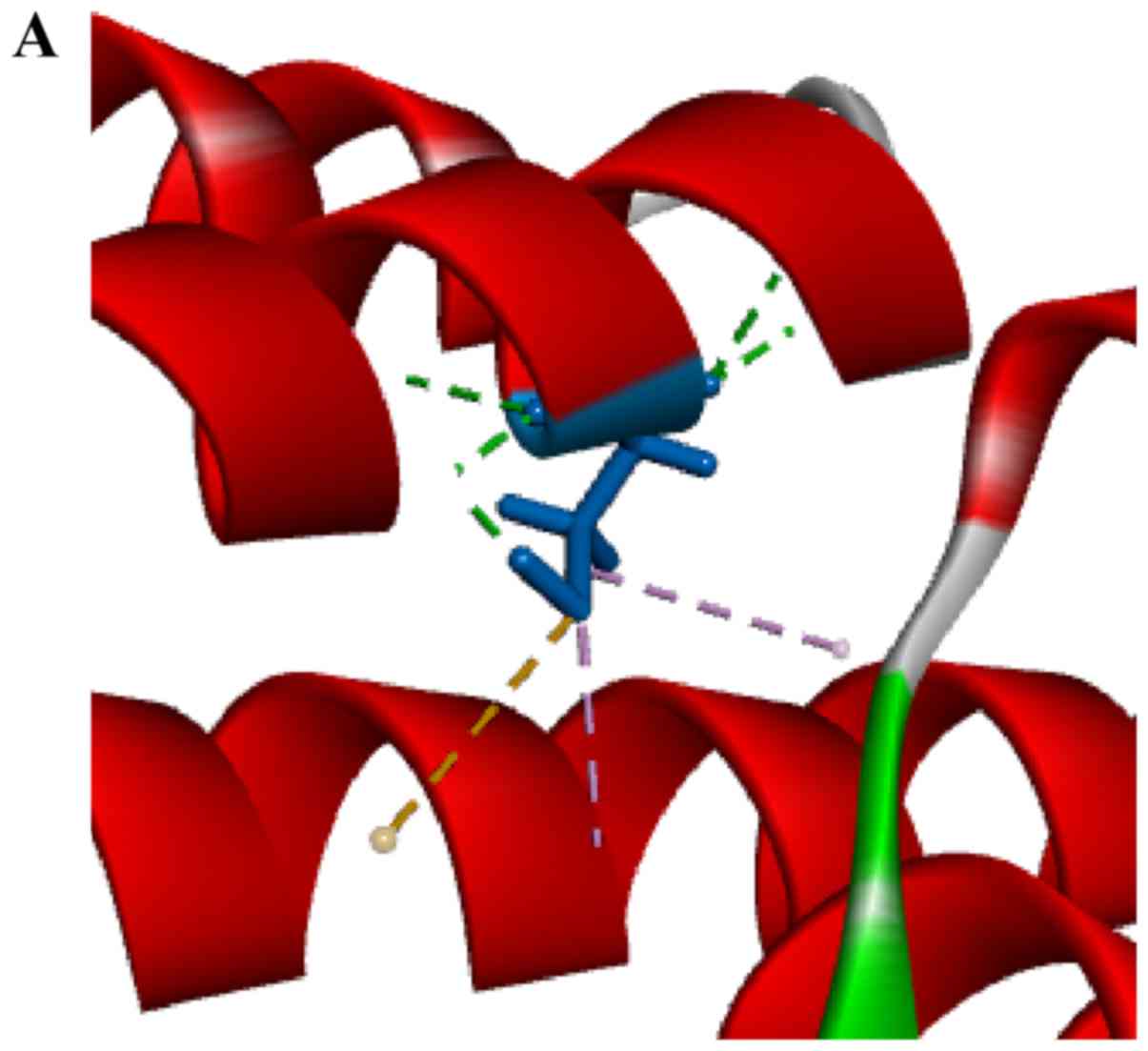
Computer modeling
In order to understand the effect of the
c.3596G>A mutation on protein structure, a homology simulation
for the SEC39 region was performed. The homology simulation was
based on protein transport protein SEC39 (3k8p). Modeling and
optimization was performed using YASARA software. Based on the
protein model obtained by homology modeling, both cysteine and
tyrosine were uncharged polar R-based amino acids, and on the
whole, the model did not change (Fig.
4A). The wild-type model appears more reasonable (Fig. 4B) in that the mutation model has a
large amount of unreasonable steric hindrance. This conclusion
implies that this mutation may lead to structural changes
throughout the SEC39 region or the entire protein.
Discussion
Human genetic diseases typically occur in about 8%
of the population (18). Over the
past decade, advances in technology have enabled the detection of
all sequence changes in a single genome in a cost-effective manner.
High throughput sequencing combined with capture techniques are
increasingly used for routine diagnosis of Mendelian disease.
Infantile liver failure is a rare but life-threatening disease.
Mutations in NBAS are known causes of acute liver failure in
children with fever. However, the physiological role of NBAS is not
particularly clear.
In this study, we have identified two deleterious
and rare variants by target NGS sequencing of 358 growth and
development associated genes in a 4-year-old girl with ILFS2. Based
on the child's medical history, clinical manifestations, laboratory
examination, the young age of the patient, and an unknown etiology,
hereditary liver disease was considered. However, the results here
in show that by second-generation and Sanger sequencing NBAS
mutations are the likely cause of the child's medical condition and
that a diagnosis of ILFS2 is appropriate. One missense mutation
c.3596G>A (p.C1199Y) and one splice site mutation c.209 +
1G>A were detected in the coding region of NBAS (NM_015909.3),
both of which were heterozygotic. Missense mutations can lead to
amino acid changes and may affect protein function while splice
site mutations can cause intronic shear errors, which impact mRNA
expression. The NBAS protein interacts with ZW10 and RINT1 at amino
acid position 1,036 to 2,371, which supports the classification of
the missense variant c.3596G>A as pathogenic (7,19).
This finding increases the number of NBAS
mutations associated with the disease and will facilitate further
studies of the syndrome. These results provide further insight into
the molecular pathogenesis of the disease and identifies
relationships among gene mutations and clinical manifestations of
this syndrome.
References
|
1
|
Lee WM: Recent developments in acute liver
failure. Best Pract Res Clin Gastroenterol. 26:3–16. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haack TB, Staufner C, Kopke MG, Straub BK,
Kölker S, Thiel C, Freisinger P, Baric I, McKiernan PJ, Dikow N, et
al: Biallelic mutations in NBAS cause recurrent acute liver failure
with onset in infancy. Am J Hum Genet. 97:163–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maksimova N, Hara K, Nikolaeva I,
Chun-Feng T, Usui T, Takagi M, Nishihira Y, Miyashita A, Fujiwara
H, Oyama T, et al: Neuroblastoma amplified sequence gene is
associated with a novel short stature syndrome characterised by
optic nerve atrophy and Pelger-Huet anomaly. J Med Genet.
47:538–548. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Segarra NG, Ballhausen D, Crawford H,
Perreau M, Campos-Xavier B, van Spaendonck-Zwarts K, Vermeer C,
Russo M, Zambelli PY, Stevenson B, et al: NBAS mutations cause a
multisystem disorder involving bone, connective tissue, liver,
immune system, and retina. Am J Med Genet A. 167A:1–2912.
2015.PubMed/NCBI
|
|
5
|
Capo-Chichi JM, Mehawej C, Delague V,
Caillaud C, Khneisser I, Hamdan FF, Michaud JL, Kibar Z and
Mégarbané A: Neuroblastoma amplified sequence (NBAS) mutation in
recurrent acute liver failure: Confirmatory report in a sibship
with very early onset, osteoporosis and developmental delay. Eur J
Med Genet. 58:637–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Staufner C, Haack TB, Köpke MG, Straub BK,
Kölker S, Thiel C, Freisinger P, Baric I, McKiernan PJ, Dikow N, et
al: Recurrent acute liver failure due to NBAS deficiency:
Phenotypic spectrum, disease mechanisms and therapeutic concepts. J
Inherit Metab Dis. 39:3–16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aoki T, Ichimura S, Itoh A, Kuramoto M,
Shinkawa T, Isobe T and Tagaya M: Identification of the
neuroblastoma-amplified gene product as a component of the syntaxin
18 complex implicated in Golgi-to-endoplasmic reticulum retrograde
transport. Mol Biol Cell. 20:2639–2649. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang Y, Mao B, Wang L, Mao L, Zhou A, Cao
J, Hu J, Zhou Y, Pan Y, Wei X, et al: Targeted next generation
sequencing reveals a novel intragenic deletion of the LAMA2 gene in
a patient with congenital muscular dystrophy. Mol Med Rep.
11:3687–3693. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T,
Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence
variants in genetic disease-causing genes using targeted
next-generation sequencing. PloS One. 6:e295002011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–33. 2013.
|
|
12
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Desmet FO, Hamroun D, Lalande M,
Collod-Beroud G, Claustres M and Beroud C: Human splicing finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Capriotti E, Fariselli P, Rossi I and
Casadio R: A three-state prediction of single point mutations on
protein stability changes. BMC Bioinformatics. 9 Suppl 2:S62008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the american college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baird PA, Anderson TW, Newcombe HB and
Lowry RB: Genetic disorders in children and young adults: A
population study. Am J Hum Genet. 42:677–693. 1988.PubMed/NCBI
|
|
19
|
Civril F, Wehenkel A, Giorgi FM,
Santaguida S, Di Fonzo A, Grigorean G, Ciccarelli FD and Musacchio
A: Structural analysis of the RZZ complex reveals common ancestry
with multisubunit vesicle tethering machinery. Structure.
18:616–626. 2010. View Article : Google Scholar : PubMed/NCBI
|