Introduction
The routine karyotype analysis is the gold standard
method currently used for the detection of fetal chromosomal number
and structural abnormalities; however, this method has certain
limitations, such as limited resolution, which may result in
chromosomal structural abnormalities <5Mb not being diagnosed
(1–4). The fluorescence in situ
hybridization (FISH) technique sets the probe according to the
target chromosomal fragment and detects whether the number of
chromosomes or the particular gene(s) is/are abnormal or not. FISH
has proven invaluable as an ancillary technique for the
identification of clinically significant chromosomal aberrations,
as FISH can be performed on metaphase or non-dividing interphase
cells, and can detect genomic abnormalities with a resolution from
200 to 1,000 kb, depending on the probe size. In genetic diagnosis,
FISH enables the rapid detection of relatively low-level mosaicism
in cells and FISH may facilitate the characterization of complex
chromosome rearrangements identified by G-banded analysis. Despite
numerous advantages, FISH does not provide genome-wide analyses and
must be targeted to selected genomic regions believed to be of
interest in a given case. When the chromosomal type(s) is/are
unclear, the FISH results are often difficult to analyze.
Furthermore, FISH only detects the chromosomal structural
abnormality in the local regions covered by the detection probes.
FISH may yield false negative results in cases where genomic
imbalances are smaller than the size of a FISH probe or chromosomal
rearrangements are complex, or false positive results when two
fluorescent signals co-localize due to viewing a three-dimensional
nucleus in two dimensions. The advent of the whole genome
microarray technique, including comparative genomic hybridization
(CGH) and single nucleotide polymorphism (SNP), has enabled the
detection of submicroscopic copy number variations of clinical
significance, e.g., small genomic deletions and duplications, known
as copy-number variants, that are not routinely observed in
karyotype analysis (5,6). SNP arrays can also detect genomic
regions with loss of heterozygosity (LOH) (6–8).
Microarray analysis eliminates the need for dividing cells and can
be performed on direct (uncultured) specimens to provide a more
accurate assessment of abnormalities. However, the whole genome
microarray technique cannot recognize the different types of
chromosomal structural abnormalities, including chromosomal
inversion, translocation or insertion. Furthermore, the precise
physical location of genomic gains cannot be determined by
microarray analysis and requires G-banding or FISH for further
characterization. Therefore, prenatal diagnosis should be based on
the characteristics of different cases and use comprehensive
diagnostic methods in order to obtain more accurate diagnostic
results. The present study reports a case of complex inversion,
translocation and deletion, as diagnosed by such integrated
prenatal diagnostic techniques as amniotic fluid-karyotype
analysis, FISH, and whole genome microarray. In addition, the
etiology and clinical manifestations were investigated.
Materials and methods
General information
The female patient (age, 27 years), experienced
regular menstruation (4 days/30 days), gravida 1 para 0, and had
decorated her house prior to pregnancy and during early pregnancy,
thus she may have been exposed to hazardous chemicals in her home
environment. The maternal serum screening at 16 weeks and 5 days of
gestation showed the risk of Down's syndrome value as 1:261. The
noninvasive prenatal testing of the maternal peripheral blood at 18
weeks and 4 days of gestation showed aneusomic fetal chromosomes,
suggesting suspicious abnormalities of the fetal sex chromosomes.
The patient attended the Prenatal Diagnosis Center of the People's
Hospital of Peking University (Beijing, China) at 21 weeks and 1
day of gestation for amniocentesis. After the patient provided
written informed consent, 20 ml amniotic fluid was sampled under
ultrasound guidance for routine amniotic fluid cell culture,
karyotype analysis and the corresponding FISH assays. The results
indicated that the fetal sex chromosome exhibited inversion and
translocation, but the existence of fragment deletion or
duplication could not be ruled out, so further assays were
recommended. The current study was conducted in accordance with the
declaration of Helsinki and with approval from the Ethics Committee
of Peking University. Amniotic fluid (10 ml) was sampled again at
25 weeks of gestation for the whole genome microarray assay. The
results confirmed the existence of the inversion and deletion in
one X chromosome and partial fragment translocation in the Y
chromosome. The patient and her family requested to terminate the
pregnancy, so an induction delivery was performed in the at 27
weeks and 4 days of gestation. The couple was non-consanguineous
and had no family history of genetic disease. There was no history
of contact with hazardous substances, although the couple had
performed home renovations prior to and in the early stages of
pregnancy. The karyotype analysis of the peripheral blood of this
couple identified the karyotypes of the husband and wife as 46,XY
and 46,XX, respectively.
Amniotic fluid cell culture and
karyotype analysis
Amniotic fluid (20 ml) was obtained (with the first
1–2 ml amniotic fluid discarded) and injected into two disposable
sterile centrifuge tubes for the centrifugation at 190 × g for 10
min; the supernatant was subsequently discarded, and 2.5–3.0 ml
cell suspension was inoculated into 5 ml of each of Gibco
Amniomax-II (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
BIO-AMF-2 (Biological Industries, Kibbutz Beit-Haemek, Israel)
amniotic fluid media, under sterile conditions, for 6~7-day static
culture at 37°C and 5% CO2. The cell growth was observed
every day after the medium was changed. When the amniotic fluid
cells adhered to the wall and grew vigorously, and the cells in
metakinesis exhibited multiple clones under an inverted microscope,
the amniotic fluid chromosomes in each culture bottle were
collected separately and the slides produced as follows: Colcemid
solution (Thermo Fisher Scientific, Inc.) was added to the culture
bottle and incubated for a further 2 h at 37°C. The medium was
removed from the culture bottle and saved in a prelabeled
centrifuge tube. Trypsin-EDTA (1 ml) was added to the bottle and
the cells washed by tilting the bottle from side to side. The
solution was removed from the bottle and 1.5 ml of fresh
trypsin-EDTA solution added and the cells bathed thoroughly with
the solution by tilting the bottle. The cells were incubated at
37°C for 5 min and then added to the contents of the centrifuge
tube and mixed prior to centrifugation at 190 × g for 10 min. The
supernatant was removed and 5 ml of potassium chloride hypotonic
solution 0.075 mol/l added to the cell, resuspended and incubated
for 10–15 min in a water bath at 37°C. Freshly made fixative was
added to each tube and mixed gently by inverting the tubes twice
then centrifuged at 190 × g for 10 min and the supernatant
discarded and the fixative step repeated an additional three times.
Then the cells were suspended in a small volume of fixative to give
a slightly opaque suspension and 3 to 4 drops were placed evenly on
a cold wet slide an allow to dry. G-band staining (plus C-band
staining when necessary) was used to prepare the chromosome
specimens, and, in accordance with the International System for
Human Cytogenomic Nomenclature (2013) (9), each specimen was analyzed 30
well-dispersed moderately-long metakinesis phases under a light
microscope. When the chimera or abnormal karyotype was identified,
the analysis was performed for a total of 100 mitotic phases.
FISH analysis
In order to determine the complex chromosome
rearrangements and chromosome breakpoints, FISH analysis was
performed on the fetus using the locus probes of XYpter/XYqter,
DXZ1, STS, RP11-64L19 and SRY (Vysis; Abbott Molecular, Abbott
Park, IL, USA), according to the manufacturer's instructions. Human
chromosomes were stained by 4–6-diamidino-2-phenylindole (DAPI) in
the dark for 10–15 min at room temperature and exhibited bright
fluorescence at secondary constriction regions of chromosomes 1, 9
and 16, the proximal short arm of 15, and the distal long arm of
Y.
Affymetrix CytoScan 750K array
Genomic DNA was extracted from 10 ml amniotic fluid
using a commercially available Genomic DNA Extraction kit (QIAamp
DNA Blood Mini kit; Qiagen GmBH, Hilden, Germany) according to the
manufacturer's instructions. The standard experimental procedure
incorporated the following: Digestion, ligation, polymerase chain
reaction (PCR), PCR purification, fragmentation, labeling,
hybridization, washing, staining and scanning. A microarray
(Affymetrix CytoScan 750K Array) was used to detect copy number
variants (losses or gains of chromosome material). This platform
includes 25-mer oligonucleotide probes covering the entire human
genome with an overall mean probe spacing of 4 kb. Following
hybridization, the laser scanner (GeneChip® 3000 Scanner
with 7G upgrade) was used for scanning the arrays, and the images
were extracted and analyzed using Affymetrix GeneChip Command
Console software (version 4.0) and Chromosome analysis software
(Chromosome Analysis Suite version 2.1) (both from Affymetrix;
Thermo Fisher Scientific, Inc.), respectively.
The Affymetrix CytoScan 750K Array includes 550,000
non-polymorphic markers and 200,000 gene-centric single-nucleotide
polymorphisms (SNPs), which enables cytogeneticists to detect and
analyze relevant chromosomal aberrations with confidence. This
solution provides high-resolution coverage of cancer and
constitutional genes of interest, along with high-density SNP
coverage for loss of heterozygosity and uniparental disomy
detection.
Results
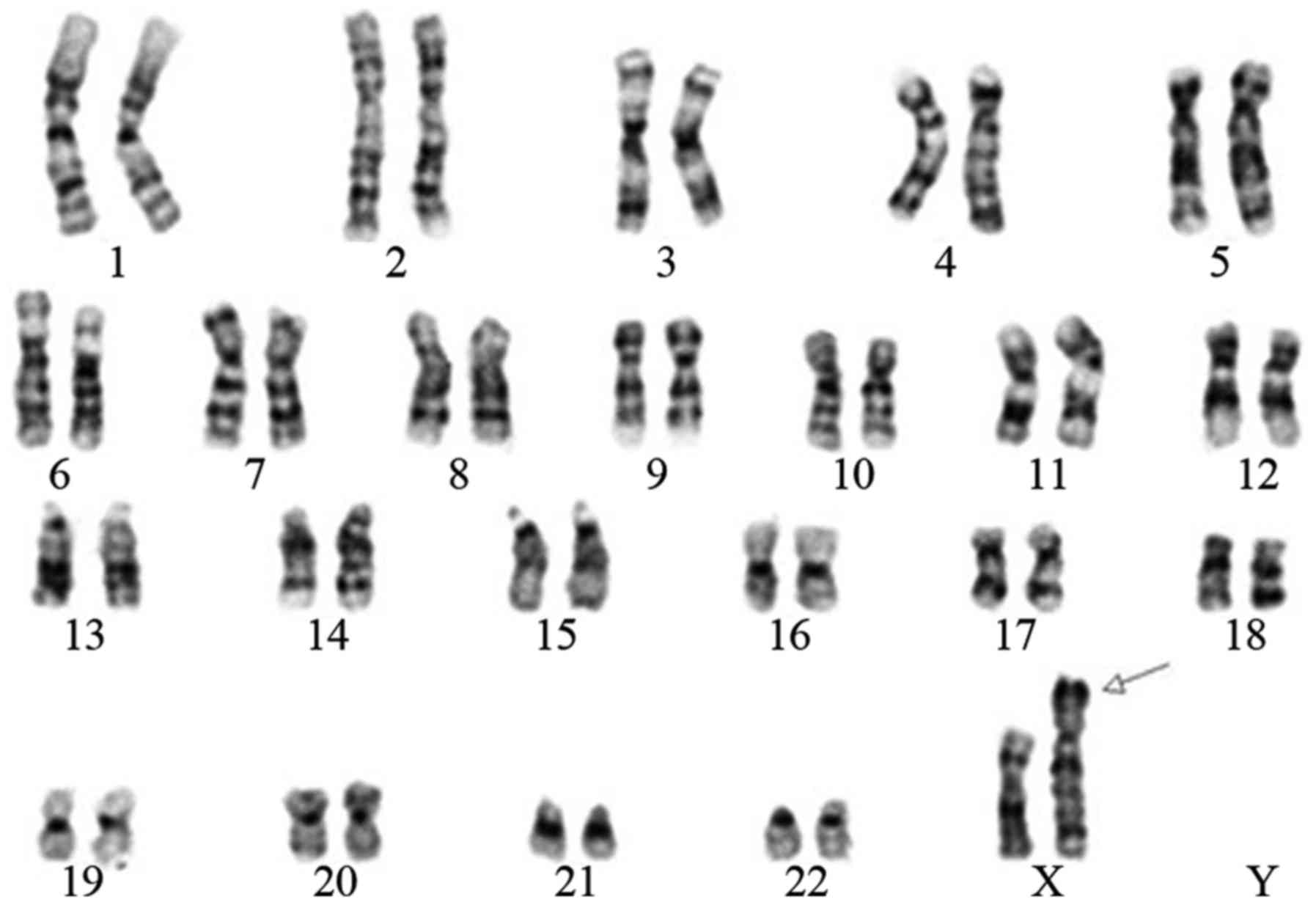
Karyotype analysis
The routine G-band staining analysis indicated that
the fetus had 46 chromosomes; the sex chromosomes were two X
chromosomes, of which one was normal while the other was abnormal.
This abnormal X chromosome may have been due to a pericentric
inversion occurring between Xp22.3 and Xq28. In addition, the short
arm of the suspicious X chromosome exhibited fragment translocation
of the Y chromosome (Fig. 1).
FISH
The DAPI staining results indicate that the short
arm of the abnormal X chromosome exhibited blue fluorescence, which
corresponded with the staining characteristics of the long arm of
the Y chromosome; therefore, it confirmed the results of the
karyotype analysis that this abnormal X chromosome exhibited the
translocated long arm segment of the Y chromosome (Fig. 2A). The results of FISH using the
XYpter (green)/XYqter (red) and STS (red) probe showed that the red
fluorescent signal of Xqter, which should appear at the end of the
chromosomal long arm, appeared at the end of the short arm of the
abnormal X chromosome, but the positive signals of Xpter and STS at
the end of the X-chromosomal short arm appeared at the end of the
long arm of the abnormal X chromosome. The results confirmed that
the abnormal X chromosome exhibited pericentric inversion. Further
FISH using the RP11-64L19 (red) probe (located in Xq28)
demonstrated that the positive signal appeared at the end of the
long arm of the abnormal X chromosome, indicating that the
breakpoint of the long arm inversion should be located before this
site. Therefore, the X chromosome exhibited pericentric inversion
between Xp22.3 and Xq28. Additional FISH using SRY (red) and DXZ1
(green) probes on the abnormal X chromosome confirmed that this
abnormal X chromosome was SRY-negative, indicating that the
translocated Y chromosome was only in the partial long arm of the Y
chromosome rather than the segment that contains the
sex-determining region Y (testis-determining factor) gene in the
short arm. The positive green fluorescence of DXZ1 indicated that
the centromere was from the X chromosome, namely the abnormal
chromosome was the X chromosome, namely ish der(X) inv(X)(p22.3q28)
t(X;Y) (q28;q11.23) (SRY-, DXZ1+, XYpter+, XYqter+, STS+,
RP11-64L19+); therefore, the fetal karyotype was 46, X, ish der(X)
inv(X) (p22.3q28)t(X;Y) (q28;q11.23) (XYqter+, SRY-, DXZ1+,
RP11-64L19+, STS+, XYpter+) (Fig.
2B-E).
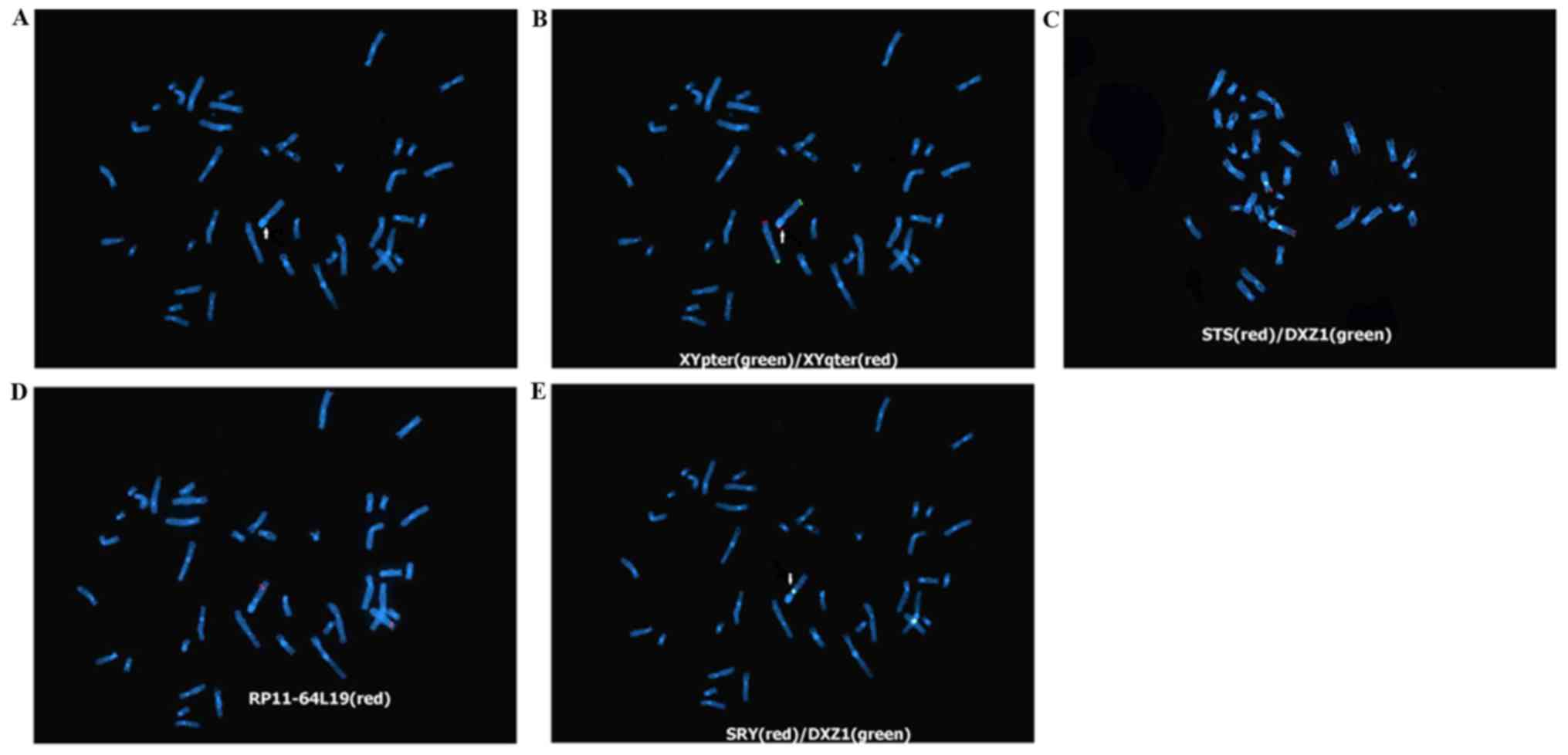 | Figure 2.DAPI staining results (magnification,
×1,000). (A) Fetal chromosomal DAPI staining. The arrow indicates
the short arm of the derivative chromosome X with blue
fluorescence, confirming it exhibited the translocated long arm
fragment of the Y chromosome. (B) FISH assay using XYpter
(green)/XYqter (red) probes. The arrow indicates the red
fluorescent signal (XYqter) appeared at the end of the
X-chromosomal short arm, while the green fluorescent signal
(XYpter) appeared at the end of the long arm, confirming
pericentric inversion in this X chromosome. (C) FISH assay using
DXZ1 (green) and STS (red) probes. The positive signal of the STS
probe appeared at the end of the abnormal X-chromosomal long arm,
reconfirming that this abnormal X chromosome exhibited pericentric
inversion. The positive green fluorescent signal of the DXZ1probe
confirmed that this centromere was on the X chromosome. (D) FISH
assay using a RP11-64L19 (red) probe. The positive signal of the
RP11-64L19 (red) probe appeared at the end of the abnormal
X-chromosomal long arm, indicating that the breakpoint of the long
arm inversion should be located before this site. (E) FISH assay
using the SRY (red)/DXZ1 (green) and STS (red) probes. The red
fluorescence signal of the male testis-determining factor was
negative SRY (−). The arrow indicates the positive green
fluorescent signal of the DXZ1probe, namely the derivative
chromosome was an X chromosome. DAPI, 4–6-diamidino-2-phenylindole;
FISH, fluorescence in situ hybridization. |
SNP microarray assay
The array analysis performed on the DNA extracted
from amniotic fluid revealed a 1.241-Mb deletion involving the
chromosome region Xq28 and a 12.329-Mb gain involving chromosome
region Yq11.21q11.23, or arr[hg19] Xq28(153,689,031-154,930,045)
×1, Yq11.21q11.23 (13,979,645-27,799,653) ×1 (Fig. 3). The results of the SNP microarray
assay confirmed that the long arm of the newly derivative X
chromosome contained a deletion of one 1.241-Mb fragment within
q28, which contained several Online Mendelian Inheritance in Man
(OMIM) genes, including as coagulation factor VIII (F8),
glucose-6-phosphate dehydrogenase (G-6-PD), inhibitor of nuclear
factor-κB kinase subunit γ (IKBKG), trimethyllysine hydroxylase ε
(TMLHE), Ras-related protein Rab-39B (RAB39B) and chloride
intracellular channel 2 (CLIC2). In addition, this derivative X
chromosome contained the 12.329-Mb fragment from the Yq11.21-q11.23
interval of the Y chromosome rather than the SRY gene. As the
patient does not have SRY, he does not secrete testosterone and
does not show male characteristics.
Prenatal diagnosis
Using karyotype analysis, FISH and SNP microarray
assay, the number of this fetal chromosomes was determined as 46,
and the two sex chromosomes were X, of which one X chromosome was
normal while the other one was unbalanced derivative X chromosome
exhibiting complex inversion, translocation and deletion, notably
exhibiting a pericentric inversion between Xp22.3 and Xq28 (one
1.241-Mb deletion in Xq28, including the OMIM genes, F8, G-6-PD,
IKBKG, TMLHE, RAB39B and CLIC2). The derivative X chromosome also
exhibited fragment translocation between Yq11.21 and q11.23 (not
containing the SRY gene). Therefore, this fetus demonstrated
inversion, translocation, and deletion (deletion/duplication)
syndrome; therefore, it exhibited X-linked recessive type A
hemophilia and X-linked incomplete dominant favism simultaneously.
Furthermore, as a result of a lack of the RAB39B and CLIC2 genes,
this fetus may have experienced various degrees of cognitive
impairment and developmental abnormalities in future. The novel
derivative X chromosome contained a fragment in Yq11.21q11.23,
although it did not contain the SRY gene; therefore, its social
gender would have been female. However, whether this fragment may
have impacted the growth and development of this fetus after birth
remains unknown.
Pregnancy outcome and autopsy
results
The two parents had normal karyotypes, but the fetal
chromosomal abnormality was a novel chromosome derivation. As this
derivative chromosome exhibited complex inversion, translocation
and deletion, the genetic counseling expert informed the patient
and her family of the corresponding clinical phenotypes after
birth, and the certificate of prenatal diagnosis was issued in
accordance with the Ethics Committee of the People's Hospital of
Peking University. The patient and her family decided to terminate
the pregnancy, and labor was induced at the 27 weeks and 4 days of
gestation. The autopsy and pathological results identified that it
was a female fetus, exhibiting a vulva, vagina, uterus and ovaries,
and the appearance of the fetus was not abnormal. The ovarian
pathological results included ovarian tissue cells and no
testicular tissue components were observed.
Discussion
Different genetic testing methods have their
advantages, disadvantages and applications (1,2,6);
therefore, prenatal diagnosis should be tailored to the
characteristics of different cases and use comprehensive diagnostic
methods to determine an accurate diagnosis. When the routine
karyotyping analysis indicates novel structural aberration in
suspicious chromosomes, site-specific probes should be designed to
test the corresponding genes at the breakpoints of the structurally
abnormal chromosome so as to confirm or rule out the chromosomal
structural abnormalities. When the suspicious chromosome exhibits
micro-fragment deletion or duplication, the gene microarray
technique could be used to detect copy-number variants (10–12).
Therefore, in the present study, routine karyotype
analysis of the case revealed that one X chromosome was a de
novo derivative, which was abnormal and exhibited pericentric
inversion and long arm translocation of the Y chromosome. The DAPI
staining confirmed that this abnormal X chromosome contained a
fragment from the Y chromosome. The FISH assay using the SRY/DXZ1
probe identified a negative SRY signal, confirming that the
abnormal chromosome-translocated Y chromosome only had the long arm
fragment instead of the testis-determining factor region in the
short arm. The positive DXZ1 signal confirmed the centromere was
from the X chromosome, notably this derivative chromosome was an X
chromosome. The FISH assay using the XYpter (green)/XYqter (red)
and STS probes confirmed the presence of the pericentric inversion
in this abnormal X chromosome, and the FISH test using the
RP11-64L19 probe (located in Xq28) confirmed the breakpoint of the
chromosomal inversion was located before the Xq28 locus; thus, the
inversion occurred between Xp22.3 and Xq28. Whole gene microarray
detection of the fetus confirmed the deletion of one 1.241-Mb
fragment in q28 of the long arm of the X chromosome, as well as the
fragment translocation between Yq11.21 and q11.23.
During the diagnostic process of the current case,
G-banding karyotype analysis and DAPI staining only determined that
this abnormal chromosome was derivative from the translocation of
the X and Y chromosomes; if the FISH had not been performed,
pericentric inversion and breakpoints in the abnormal chromosome
would not have been observed. Furthermore, without performing the
SNP microarray, neither the deletion at the distal end of Xq28, nor
the fragment size containing the Y chromosome would not have been
determined. Thus, only by comprehensively applying the various
detection methods of karyotyping analysis, FISH or SNP were the
nature, origin and manifestations of this chromosomal-derivative
abnormalities established. This enabled the successful diagnosis of
this case of unbalanced sex chromosomal inversion, translocation
and deletion.
When one chromosome breaks during the ‘hit’ event,
translocation, rearrangement and reconnection of different
chromosomes occurs, thus forming a novel derivative chromosome
during the repair process (13,14).
If the derivative chromosome exhibits fragment deletion or gene
damage at the breakpoints, it's termed unbalanced translocation
and/or inversion. The patients with unbalanced translocation and/or
inversion would often exhibit the corresponding genetic effects due
to gene loss or damage. As the formation of complex chromosomal
translocation and/or inversion results from extrinsic factors
acting on chromosomal exogenous DNA molecules, causing breakage,
translocation, and reconnection of the chromosomes at the molecular
level, men and women at childbearing ages should take measures to
protect their reproductive organs when exposed to large doses of
ionizing radiation or rays (15–17).
The couple in the present study decorated their house before and
during the early stages of pregnancy; therefore, contact with
hazardous materials prior to pregnancy may have resulted in a ‘hit’
event in the parental germ cells (haploid chromosome) prior to
fertilization or even after the father's sperm or the mother's egg
were formed, thus the chromosome breakage, repair and reconnection
occurred. The fetal derivative X chromosome only contained the
partial long arm of the Y chromosome, while it did not contain the
short arm that had the testis-determining genes; therefore, without
the roles of male hormone testosterone, its genitalia would not
differentiate into male, and its social gender would have been
female. However, whether the translocated Y chromosome fragment
would affect the growth and development of the fetus in future
remains unknown. In addition, the long arm of the derivative X
chromosome missed a 1.241-Mb fragment containing F8, G-6-PD, IKBKG,
TMLHE, RAB39B, and CLIC2 (18–24).
The F8 gene encodes the clotting factor VIII, therefore a
homozygous mutation or deletion of this gene would cause X-linked
recessive type A hemophilia; thus, the patient would be a type A
hemophilia gene carrier. The mutation or deletion of the G-6-PD
gene results in the deficiency of G-6-PD, thus exhibiting X-chain
incomplete dominant ‘favism’. It has been reported that mutations
of the RAB39B and CLIC2 genes were associated with X-linked mental
retardation diseases, manifesting as mental retardation,
developmental delay, congenital heart disease or epilepsy (25). It was also reported that the
females carrying the CLIC2 mutation may exhibit mild cognitive
impairment (26). Mutation of the
DKC1 gene has been associated with the X-linked dyskeratosis
congenita (27), manifesting as
skin and mucosal abnormalities, progressive myelodysplasia or organ
abnormalities. In addition, it was reported that the female
DKC1-mutation carriers would exhibit increased risks of the
above-mentioned diseases with variable phenotypic expression.
Therefore, it could be hypothesized that certain X-linked recessive
genetic diseases may exhibit specific clinical phenotypes in female
carriers, which may be due to the random X chromosome inactivation
or haploinsufficiency. Thus, it could not be ruled out that the
fetus in the present case may have been at risk of different
degrees of cognitive impairment, as well as the above-mentioned
diseases. This was a case of unbalanced chromosomal inversion,
translocation and deletion. Due to the gene deletion or
displacement effects at the breakpoints of the X chromosome,
clinical manifestations, such as corresponding gonad and
reproductive dysfunction, could not be ruled out. The parents had
normal chromosomal karyotypes, and the abnormal chromosome of the
fetus was a de novo derivative, therefore its recurrence
risk would not be high; however, routine prenatal diagnosis,
ultrasound or SNP microarray should be considered according to the
regular examinations during the pregnancy period.
In conclusion, the karyotype analysis, FISH, and
whole genome microarray were performed for the prenatal diagnosis
of a high-risk pregnant woman, and the fetus was diagnosed with sex
chromosomal inversion, translocation, and deletion, so the
corresponding clinical phenotypes associated with this derivative
unbalanced chromosome (of chromosomal deletion/duplication) may
have presented after birth. The current study provided adequate
genetic counseling to the patient and her family, and the family
decided to terminate the pregnancy as the fetus would have been
born with birth defects. The present study may provide guidance for
future pregnancy and a healthy birth.
References
|
1
|
Cancer Genome Atlas Research Network, ;
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A,
Hoadley K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic
landscapes of adult de novo acute myeloid leukemia. N Engl J Med.
368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garcia-Manero G: Myelodysplastic
syndromes: 2014 update on diagnosis, risk-stratification, and
management. Am J Hematol. 89:97–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kantarjian HM, Larson RA, Cortés JE,
Deering KL and Mauro MJ: Current practices in the management of
chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 13:48–54.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stilgenbauer S, Schnaiter A, Paschka P,
Zenz T, Rossi M, Döhner K, Bühler A, Böttcher S, Ritgen M, Kneba M,
et al: Gene mutations and treatment outcomes in chronic lymphocytic
leukemia: Results from the CLL8 trial. Blood. 123:3247–3254. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gibson SE, Luo J, Sathanoori M, Liao J,
Surti U and Swerdlow SH: Whole-genome single nucleotide
polymorphism array analysis is complementary to classical
cytogenetic analysis in the evaluation of lymphoid proliferations.
Am J Clin Pathol. 141:247–255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu X, Johnson EB, Leverton L, Arthur A,
Watson Q, Chang FL, Raca G and Laffin JJ: The advantage of using
SNP array in clinical testing for hematological malignancies-a
comparative study of three genetic testing methods. Cancer Genet.
206:317–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dougherty MJ, Wilmoth DM, Tooke LS, Shaikh
TH, Gai X, Hakonarson H and Biegel JA: Implementation of high
resolution single nucleotide polymorphism array analysis as a
clinical test for patients with hematologic malignancies. Cancer
Genet. 204:26–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okada M, Suto Y, Hirai M, Shiseki M, Usami
A, Okajima K, Teramura M, Mori N and Motoji T: Microarray CGH
analyses of chromosomal 20q deletions in patients with
hematopoietic malignancies. Cancer Genet. 205:18–24. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shaffer LG, McGowan-Jordan J and Schmid M:
An International System for Human Cytogenomic Nomenclature.
Cytogenetic and Genome Research, Basel. 2013.
|
|
10
|
Mullighan CG: The molecular genetic makeup
of acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ
Program. 2012:389–396. 2012.PubMed/NCBI
|
|
11
|
Kolquist KA, Schultz RA, Furrow A, Brown
TC, Han JY, Campbell LJ, Wall M, Slovak ML, Shaffer LG and Ballif
BC: Microarray-based comparative genomic hybridization of cancer
targets reveals novel, recurrent genetic aberrations in the
myelodysplastic syndromes. Cancer Genet. 204:603–628. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heinrichs S, Li C and Look AT: SNP array
analysis in hematologic malignancies: Avoiding false discoveries.
Blood. 115:4157–4161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee MY, Seo CS, Kim JY and Shin HK:
Genotoxicity evaluation of Guibi-Tang extract using an in vitro
bacterial reverse mutation assay, chromosome aberration assay, and
in vivo micronucleus test. BMC Complement Altern Med. 14:2152014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jenderny J: Chromosome aberrations in a
large series of spontaneous miscarriages in the German population
and review of the literature. Mol Cytogenet. 7:382014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tawn EJ, Curwen GB, Jonas P, Riddell AE
and Hodgson L: Chromosome aberrations determined by sFISH and
G-banding in lymphocytes from workers with internal deposits of
plutonium. Int J Radiat Biol. 92:312–320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fucić A, Zeljezić D, Kasuba V, Kopjar N,
Rozgaj R, Lasan R, Mijić A, Hitrec V and Lucas JN: Stable and
unstable chromosome aberrations measured after occupational
exposure to ionizing radiation and ultrasound. Croat Med J.
48:371–377. 2007.PubMed/NCBI
|
|
17
|
Themis M, Garimberti E, Hill MA and
Anderson RM: Reduced chromosome aberration complexity in normal
human bronchial epithelial cells exposed to low-LET γ-rays and
high-LET α-particles. Int J Radiat Biol. 89:934–943. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
http://omim.org/http://genome.ucsc.edu/https://decipher.sanger.ac.uk/https://decipher.sanger.ac.uk/
|
|
19
|
Antonarakis SE, Kazazian HH and Tuddenham
EG: Molecular etiology of factor VIII deficiency in hemophilia A.
Hum Mutat. 5:1–22. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tuddenham EG, Cooper DN, Gitschier J,
Higuchi M, Hoyer LW, Yoshioka A, Peake IR, Schwaab R, Olek K,
Kazazian HH, et al: Haemophilia A: Database of nucleotide
substitutions, deletions, insertions and rearrangements of the
factor VIII gene. Nucleic Acids Res. 19:4821–4833. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaplan M, Renbaum P, Levy-Lahad E,
Hammerman C, Lahad A and Beutler E: Gilbert syndrome and
glucose-6-phosphate dehydrogenase deficiency: A dose-dependent
genetic interaction crucial to neonatal hyperbilirubinemia. Proc
Nat Acad Sci USA. 94:pp. 12128–12132. 1997; View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Bruggen R, Bautista JM, Petropoulou T,
de Boer M, van Zwieten R, Gómez-Gallego F, Belohradsky BH, Hartwig
NG, Stevens D, Mason PJ and Roos D: Deletion of leucine 61 in
glucose-6-phosphate dehydrogenase leads to chronic nonspherocytic
anemia, granulocyte dysfunction, and increased susceptibility to
infections. Blood. 100:1026–1030. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Giannandrea M, Bianchi V, Mignogna ML,
Sirri A, Carrabino S, D'Elia E, Vecellio M, Russo S, Cogliati F,
Larizza L, et al: Mutations in the small GTPase gene RAB39B are
responsible for X-linked mental retardation associated with autism,
epilepsy, and macrocephaly. Am J Hum Genet. 86:185–195. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wilson GR, Sim JC, McLean C, Giannandrea
M, Galea CA, Riseley JR, Stephenson SE, Fitzpatrick E, Haas SA,
Pope K, et al: Mutations in RAB39B cause X-linked intellectual
disability and early-onset Parkinson disease with α-synuclein
pathology. Am J Hum Genet. 95:729–735. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leonard H and Wen X: The epidemiology of
mental retardation: Challenges and opportunities in the new
millennium. Ment Retard Dev Disabil Res Rev. 8:117–134. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takano K, Liu D, Tarpey P, Gallant E, Lam
A, Witham S, Alexov E, Chaubey A, Stevenson RE, Schwartz CE, et al:
An X-linked channelopathy with cariomegaly due to a CLIC2 mutation
enhancing ryanodine receptor channel activity. Hum Mol Genet.
21:4497–4507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alder JK, Parry EM, Yegnasubramanian S,
Wagner CL, Lieblich LM, Auerbach R, Auerbach AD, Wheelan SJ and
Armanios M: Telomere phenotypes in females with heterozygous
mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum Mutat.
34:1481–1485. 2013. View Article : Google Scholar : PubMed/NCBI
|