Introduction
Dendritic cells (DCs) are an important distinct
subset of antigen-presenting cells (APCs) that have key roles in
the uptake, progression and presentation of foreign and
self-antigens, including tumor-associated antigens (TAAs) (1,2).
Cancer-associated DCs present antigens derived from dying cancer
cells to antigen-specific CD8+ cytotoxic T cells to
induce cell-mediated immune responses against cancer (1). In turn, these responses directly
induce cancer cell apoptosis via tumor necrosis factor-related
apoptosis-inducing ligand, Fas and tumor necrosis factor-α
receptors (3). Apoptosis triggered
by APCs enhances TAA acquisition because apoptotic cell death leads
to the release of TAAs from cancer cells. In addition, a group of
host proteins, termed alarmins, function to ‘warn’ the innate and
adaptive immune systems early and to interact with receptors that
activate APCs, such as DCs (4).
High mobility group box protein 1 (HMGB1) is one
such innate immune alarmin protein (5). It is a highly conserved non-histone
chromatin-associated protein that functions as an architectural
chromatin-binding protein and enables transcriptional protein
assemblies to gain access to specific DNA targets (6). In addition to the role of HMGB1 in
the nucleus as a chromatin stabilizing factor, it also functions as
an extracellular signaling molecule. It is expressed in the nuclei
of eukaryotic cells and is passively released by necrotic,
apoptotic or autophagic cells (6).
HMGB1 has roles in numerous biological processes, including
inflammation, cell migration, cell differentiation and cancer
metastasis (7,8). It has been previously reported that
HMGB1 is over-expressed in a variety of cancer cell types, and that
it has central roles in carcinogenesis and cancer progression,
invasion and metastasis by sustaining autophagy and limiting
apoptosis (9). Furthermore, in the
disordered microenvironment of cancer, HMGB1 recognition has a
paradoxical effect on cancer immunity, promoting angiogenesis while
inducing a protective anti-neoplastic T cell response (10).
Autophagy occurs at a low basal level in almost all
cells in response to disordered microenvironmental stimuli, such as
endoplasmic reticulum (ER) stress, mitochondrial toxins, hypoxia,
abnormal cell growth, and nutrient deprivation (11). As part of the normal catabolic
process required for homeostasis (12), autophagy has been implicated in
many physiological and pathophysiological conditions (13). In response to stress, the
upregulation of autophagy is a vital mechanism by which cells
remove damaged proteins and organelles (11,12).
Autophagy has been shown to be important in conferring resistance
to chemotherapy, radiation therapy and immunotherapy in cancer
(14). The paradoxical dual
effects of autophagy on carcinogenesis and cancer progression await
further verification. As a response to disordered
microenvironmental stimuli, autophagy can be activated by ER stress
(11). Autophagy is activated
sequentially following ER stress and saves cells undergoing ER
stress (15). The mechanism by
which ER stress activates autophagy is very complex. c-Jun
N-terminal kinase (JNK) is a crucial mediator of the ER
stress-induced autophagy pathway (16,17).
Multiple studies have reported that HMGB1 promotes autophagy by
interfering with the binding of B-cell lymphoma 2 (Bcl2) to Beclin
1 (18,19); however, the function of HMGB1
release from cancer cells in response to immune cell-mediated
stress remains unknown.
In the present study, it was demonstrated that HMGB1
is overexpressed in murine cancer cells and that it promotes the
resistance of CT26.WT murine colon cancer cells to apoptosis
triggered by murine bone marrow-derived DCs by promoting ER
stress-induced autophagy. Furthermore, the role of exogenous HMGB1
in the regulation of autophagy depends on the activation of JNK,
which links ER stress with autophagy. However, the suppression of
HMGB1 expression or the depletion of HMGB1 by an HMGB1-neutralizing
antibody in the culture medium increases the cytotoxicity of DCs to
CT26.WT cells; this effect indicates that HMGB1 is a potential
factor involved in cancer immune evasion.
Materials and methods
Mice and cell lines
Female BALB/c mice, 5–6 weeks old, were purchased
from the Jackson Laboratory (Bar Harbor, ME, USA) and were bred in
pathogen-free facilities in the Animal Biosafety Level-3 Lab of
Wuhan University (Wuhan, China). All live animal experiments were
approved by the Wuhan University Ethics Committee (Wuhan, China).
The CT26.WT (murine colon cancer cells) and L929 cell (mouse
immortal fibroblast cells) lines were purchased from the American
Type Culture Collection (Manassas, VA, USA) and were cultures in
RPMI-1640 medium (Invitrogen, Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 100 U/ml penicillin (Sigma
Aldrich; Merck KGaA, Darmstadt, Germany), 100 µg/ml streptomycin
(Sigma Aldrich; Merck KGaA) and 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) in petri dishes or Transwell dishes
(membrane pore size, 0.4 µm; Corning Incorporated, Corning, NY,
USA). The cells were maintained at 37°C in a humidified atmosphere
containing 5% CO2 and were passaged every 2 days; the
cell culture medium was changed once between the passages. The
viability of the cells was determined using a trypan blue exclusion
assay. For the cell viability and cytotoxicity assays, cells in the
exponential growth phase were trypsinized and resuspended in fresh
culture medium at a density of 5×103 cells/well in a
96-well plate for 24 h at 37°C. Following incubation, the medium
was replaced with a suspension of DCs (5×104
cells/well), which were derived from murine bone marrow cells. For
other experiments, the cells were trypsinized and resuspended in
fresh culture medium at a density of 105 cells/well in
the sub-layer of the Transwell or petri dishes for 24 h at 37°C.
Following incubation, the medium was changed, and the DC suspension
was added to the chamber.
Generation of bone marrow-derived
DCs
Bone marrow derived DCs were prepared according to
the protocol described by Lutz et al (20), with minor modifications. Briefly,
the bone marrow cells of BALB/c mice were isolated from the femurs
of adult mice and were cultured in RPMI-1640 medium supplemented
with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin, 100 µg/ml streptomycin, 5 µM β-mercaptoethanol, 2 mM
L-glutamine (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA),
20 ng/ml recombinant mouse granulocyte macrophage-colony
stimulating factor (GM-CSF; R&D Systems, Inc., Minneapolis, MN,
USA) and 10 ng/ml IL-4 (Cell Signaling Technology, Inc., Danvers,
MA, USA). On the third day of culture, all detached cells and
loosely adherent cells were removed by gentle swirling, and fresh
medium was added to the cells. On the 8th day, the non-adherent
cells were collected for experiments, and the purity of the bone
marrow-derived DCs was determined using flow cytometric analysis
(>85% CD11c+ cells). Cell viability was >98%, as
determined by trypan blue exclusion assay.
Annexin V assays
Annexin V assays were used to detect the CT26.WT
cells that had externalized phosphatidylserine on the outer layer
of the cell membrane, which is a marker of early apoptosis. The
CT25.WT cells were exposed to DCs at a ratio of 1:10 for 4 h. After
removal of the DCs, the CT26.WT cells (2×106 cells/ml)
were stained with Annexin V conjugated to fluorescein
isothiocyanate (FITC) and propidium iodide (PI) using the FITC
Annexin V/Dead Cell Apoptosis kit (eBioscience; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Briefly, cells were suspended in 500 µl Annexin V binding buffer
and incubated with 5 µl Annexin V and 5 µl PI for 5 min at room
temperature in the dark. Percentages of cells undergoing apoptosis
were determined by dual-color flow cytometric analysis. Immediately
after staining, the cells were analyzed on a flow cytometer using
488 nm as the excitation wavelength, a 525-nm band pass filter for
FITC and a 620-nm filter for PI detection. At least 20,000 cells
were acquired using a Beckman Coulter flow cytometer and data were
analyzed using Beckman Summit software version 6.1.0 (Beckman
Coulter, Inc., Brea, CA, USA).
Western blot analysis
The treated CT26.WT and L929 cells were harvested
and lysed in radioimmunoprecipitation assay lysis buffer (150 mM
NaCl, 1% SDS, 10 mg/ml leupeptin, 1 mM aprotinin, 50 mM Tris-HCl,
pH 7.4) containing the protease inhibitor phenylmethylsulfonyl
fluoride for 5 min on ice, prior to scraping and brief sonication.
The cell lysates were cleared by a 15-min centrifugation at 12,000
× g at 4°C; the protein concentration was determined using a
bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.) and the samples were then boiled for 10 min.
Equal amounts of extracted protein samples (10 µg) were loaded onto
10–12% gradient SDS-PAGE gels and run at 110 V at room temperature
in SDS-PAGE running buffer (Sigma Aldrich; Merck KGaA). Following
electrophoresis, the proteins were transferred to nitrocellulose
membranes by electroblotting at 30 V in Tris/glycine buffer with
20% methanol for 1.5 h at 4°C. The blots were blocked with 5%
bovine serum albumin (cat. no. ST023; Beyotime Institute of
Biotechnology, Haimen, China) in PBS washing buffer with 0.1%
Tween-20 (PBST) for 3 h at room temperature. After rinsing with
PBST (three times for 5 min), the membranes were incubated with
1:1,000 dilutions of the following primary antibodies at 4°C
overnight: HMGB1 (cat. no. 2639-1; Epitomics; Abcam, Cambridge,
UK), microtubule-associated protein 1 light chain 3 (LC3; cat. no.
4108; Cell Signaling Technology, Inc.), sequestosome 1 (P62; cat.
no. 3340-1; Epitomics; Abcam), glucose regulated protein 78 (GRP78;
cat. no. 3158-1; Epitomics; Abcam), JNK (cat. no. 3496-1;
Epitomics; Abcam), phospho (p)-JNK (cat. no. 3893-1; Epitomics;
Abcam), p-Bcl2 (cat. no. 8588-1; Epitomics; Abcam), Bcl2 (cat. no.
ab32124; Abcam), Beclin1 (cat. no. 2026-1; Epitomics; Abcam) or
β-actin (cat. no. ab8226; Abcam). The membranes were then incubated
with 1:1,000 dilutions of horseradish peroxidase-conjugated
secondary antibody (cat. no. ab191866; Abcam) for 1.5 h at room
temperature. The bound antibodies were visualized using the ECL
Supersignal West Pico Trial Kit (Pierce; Thermo Fisher Scientific,
Inc.). Densitometry analysis was performed using the ImageJ
software version 1.429 (National Institutes of Health, Bethesda,
MA, USA). The signal density was normalized to the expression of
b-actin, which was used as a loading control; at least three
separate experiments were analyzed.
Cytotoxicity assays
Lactate dehydrogenase (LDH) release was measured
using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit
(Promega Corporation, Madison, WI, USA) according to the
manufacturer's instructions. HMGB1 pretreated or untreated CT26.WT
cells were seeded at a density of 5,000 cells/well in 96-well
plates in 100 µl complete RPMI-1640 medium and were incubated
overnight. DCs were added to the CT26.WT cells at a ratio 1:10. The
DCs alone and the supernatant of the co-cultures mentioned above
were used as controls. After 4 h, the supernatants were collected,
and the resulting LDH release of the CT26.WT cells was
determined.
Cell viability assay
The CT26.WT cells were seeded in 96-well plates at a
density of 5,000 cells/well under each condition. The murine bone
marrow-derived DCs were then added to the wells at a ratio 1:10.
The plates were incubated at 37°C with 5% CO2 for 24 h
and were then subjected a Cell Counting Kit-8 (CCK-8) assay using
the CCK-8 assay kit (Beyotime Institute of Biotechnology) according
to the manufacturer's instructions.
Acridine orange (AO)/ethidium bromide
(EB) cell apoptosis assay
A total of 25 µl of CT26.WT cell suspension
(0.5–2.0×106 cells/ml) were incubated with 1 µl AO/EB
solution at 4°C for 1 min in darkness. Samples were evaluated
immediately by placing 10 µl cell suspension onto a microscope
slide, covering with a glass coverslip, and examined under a
fluorescence microscope (Olympus CX31; Olympus Corporation, Tokyo,
Japan) using a fluorescein filter at 510 nm and an ×40 objective.
At least 300 cells were observed.
Monodansylcadaverine (MDC)
staining
To evaluate autophagy, MDC powder was purchased from
Sigma Aldrich (Merck KGaA) and dissolved in dimethyl sulfoxide at
0.1 mol/l of the stock concentration; the working concentration was
50 µm/l. The CT26.WT cells (1×106 cells/ml) were
incubated with the MDC dye for 45 min in the dark at 4°C and then
washed with PBS thrice. Subsequently, they were visualized under a
fluorescence microscope (Olympus Corporation). The autophagosomes
emitted blue fluorescence and appeared in a punctate pattern in
CT26.WT cells.
Pharmacological and antibody
interventions
The following inhibitors were used in the present
study: ER stress inhibitor 4-phenylbutyrate (4-PBA; 10 mM; EMD
Millipore, Billerica, MA, USA) and JNK phosphorylation inhibitor
SP600125 (10 µM; Selleck Chemicals, Houston, TX, USA). The drugs
were dissolved in 10% FBS in RPMI-1640 medium and were added to the
CT26.WT cell cultures 30 min prior to initiating the DC exposure.
HMGB1 antibody (cat. no. 2639-1; Epitomics; Abcam) 1:50
(antibody/culture v/v) was applied to neutralize HMGB1 in cell
cultures, and rabbit IgG monoclonal antibody (cat. no. ab199376;
Abcam; 1:50) was used as its isotype control.
RNA transfection
Small interfering (si)RNA duplexes specifically
targeting HMGB1 and control siRNAs were obtained from Abgent
Biotech Co., Ltd. (Suzhou, China) and 100 nM siRNA was transfected
into 5×105 CT26.WT cells using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 24 h in 37°C
CO2 incubator, according to the manufacturer's protocol.
The following RNA oligonucleotides were used in the present study:
Control siRNA, 5′-GUUAUCGCAACGUGUCACGUA-3′; HMGB1 siRNA,
5′-GCAGCCCUAUGAGAAGAAATT-3′.
Statistical analysis
The results are presented as the mean ± standard
deviation. Experiments were repeated at least 3 times. Comparisons
between two groups were analyzed using Student's t-test, whereas
comparisons among multiple groups were analyzed using a one-way
analysis of variance followed by a post hoc Tukey test. P<0.05
was considered to indicate a statistically significant
difference.
Results
HMGB1 is associated with the viability
of CT26.WT cells exposed to DCs
To determine the role of HMGB1 in the colon cancer
cell response to DC exposure, HMGB1 expression in the murine colon
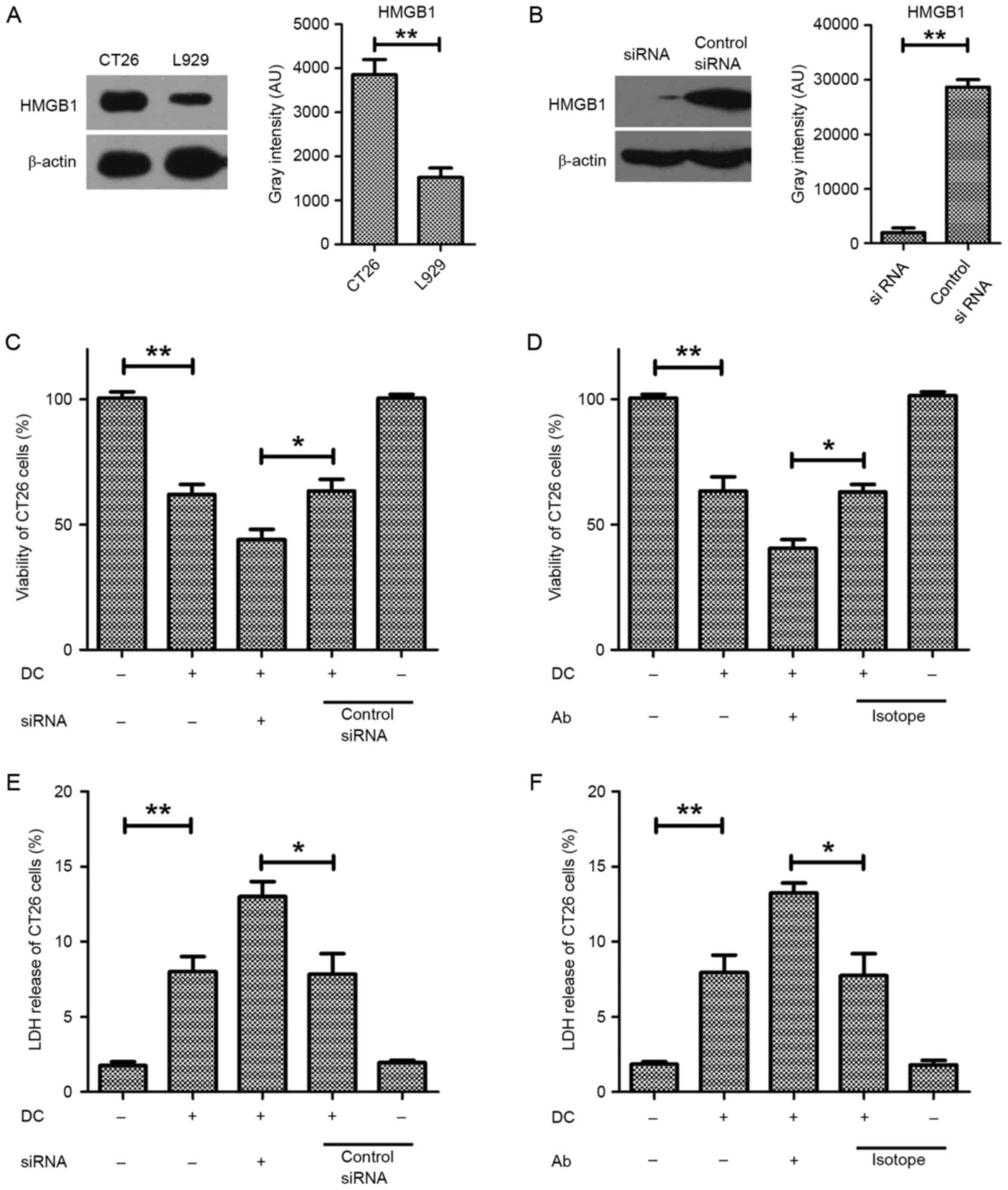
cancer cell line CT26.WT. As demonstrated in Fig. 1A, HMGB1 expression was markedly
higher in CT26.WT cells compared with the L929 immortalized murine
fibroblast cell line, as determined by immunoblot analysis,
indicating that HMGB1 expression may be upregulated in murine colon
cancer cells (Fig. 1A).
Subsequently, a specific siRNA targeting HMGB1 was used to knock
down its expression in CT26.WT cells (Fig. 1B). The viability of HMGB1-deficient
CT26.WT cells exposed to DCs was significantly lower than that of
control siRNA CT26.WT cells exposed to the DCs (Fig. 1C). Additionally, an
HMGB1-neutralizing antibody (1:50) was used to reduce the
concentration of HMGB1 in the supernatant, which resulted in
significantly decreased viability of the CT26.WT cells exposed to
the DCs compared with those exposed to DCs and an isotype antibody
control (Fig. 1D). These results
indicated that HMGB1 has an important role protecting the CT26.WT
cells from the effects of DCs. Additionally, the cytotoxic effect
of DCs on CT26.WT cells was significantly reduced following the
siRNA-mediated knockdown of HMGB1 or its depletion using a
neutralizing antibody, as measured by LDH release (Fig. 1E and F). These results indicate
that HMGB1 is highly important for maintaining the viability of
CT26.WT cells exposed to DCs.
HMGB1 increases the resistance of
CT26.WT cells to apoptosis after exposure to DCs
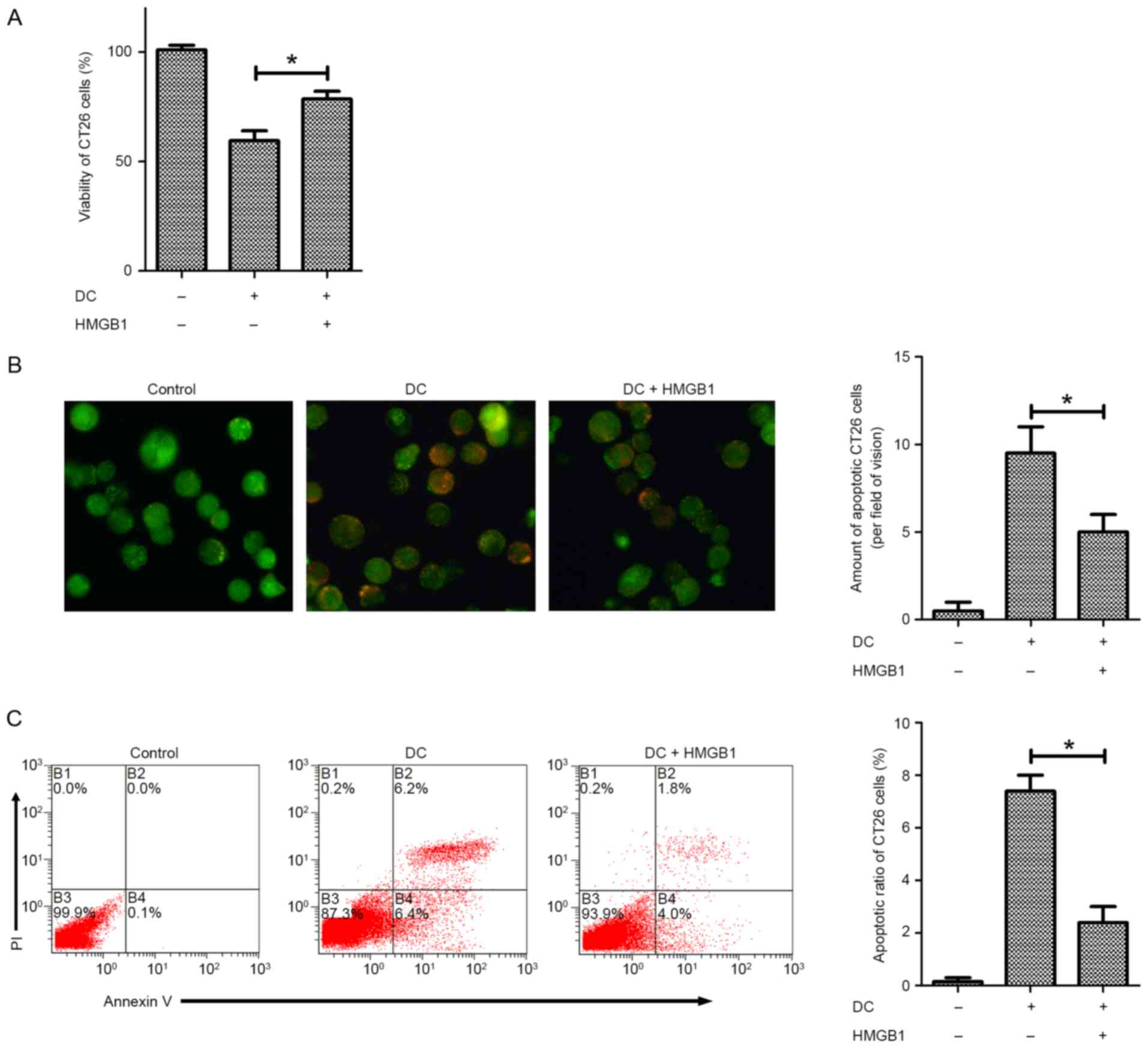
Further experiments were conducted to determine
whether HMGB1 is able to prevent apoptosis of CT26.WT cells exposed
to DCs. Following pretreatment with 10 ng/ml HMGB1, the viability
of DC-exposed CT26.WT cells was significantly increased compared
with that of cells that were not pretreated, as measured by CCK-8
assay (Fig. 2A). Furthermore, an
acridine orange/ethidium bromide cell apoptosis assay revealed that
pretreatment with HMGB1 significantly reduced the number of
non-viable, apoptotic CT26.WT cells after exposure to DCs and
postponed apoptosis (Fig. 2B). To
confirm these findings, an Annexin V assay was performed,
demonstrating similar results (Fig.
2C). These findings suggest that HMGB1 enhances the viability
of CT26.WT cells by preventing DC-induced apoptosis.
HMGB1 enhances the viability of
CT26.WT cells exposed to DCs through the upregulation of
autophagy
It has been suggested that two processes
simultaneously occur during programmed cell death: Autophagy and
apoptosis. Increasing evidence suggests that autophagy is an
important mechanism by which cells resist stress. The
aforementioned results suggest that HMGB1 promotes the resistance
of CT26.WT cells to stress induced by DCs, leading to speculation
that HMGB1 may influence autophagy in CT26.WT cells exposed to DCs.
To verify this hypothesis, the conversion of LC3 and the
degradation of P62 were measured by immunoblot analysis. Following
pretreatment with 10 ng/ml HMGB1, autophagic flux was significantly
increased in DC-exposed CT26.WT cells (Fig. 3A). This increased autophagic flux
was confirmed by MDC staining, which specifically labels autophagic
vacuoles. The mean fluorescence intensity of the DC-exposed CT26.WT
cells was obviously increased following pretreatment with HMGB1
compared with cells exposed to DCs only, as shown by fluorescence
microscopy (Fig. 3B).
Mechanistically, a significant increase in the expression of Beclin
1, a key molecule required for the initiation of autophagosomes,
was detected in the DC-exposed CT26.WT cells following pretreatment
with HMGB1 compared with those that did not receive pretreatment.
Increased phosphorylation of Bcl2 was also observed, suggesting the
disassociation of Beclin 1 and Bcl2 complexes. All of these data
suggest that HMGB1 induces autophagy in CT26.WT cells exposed to
DCs.
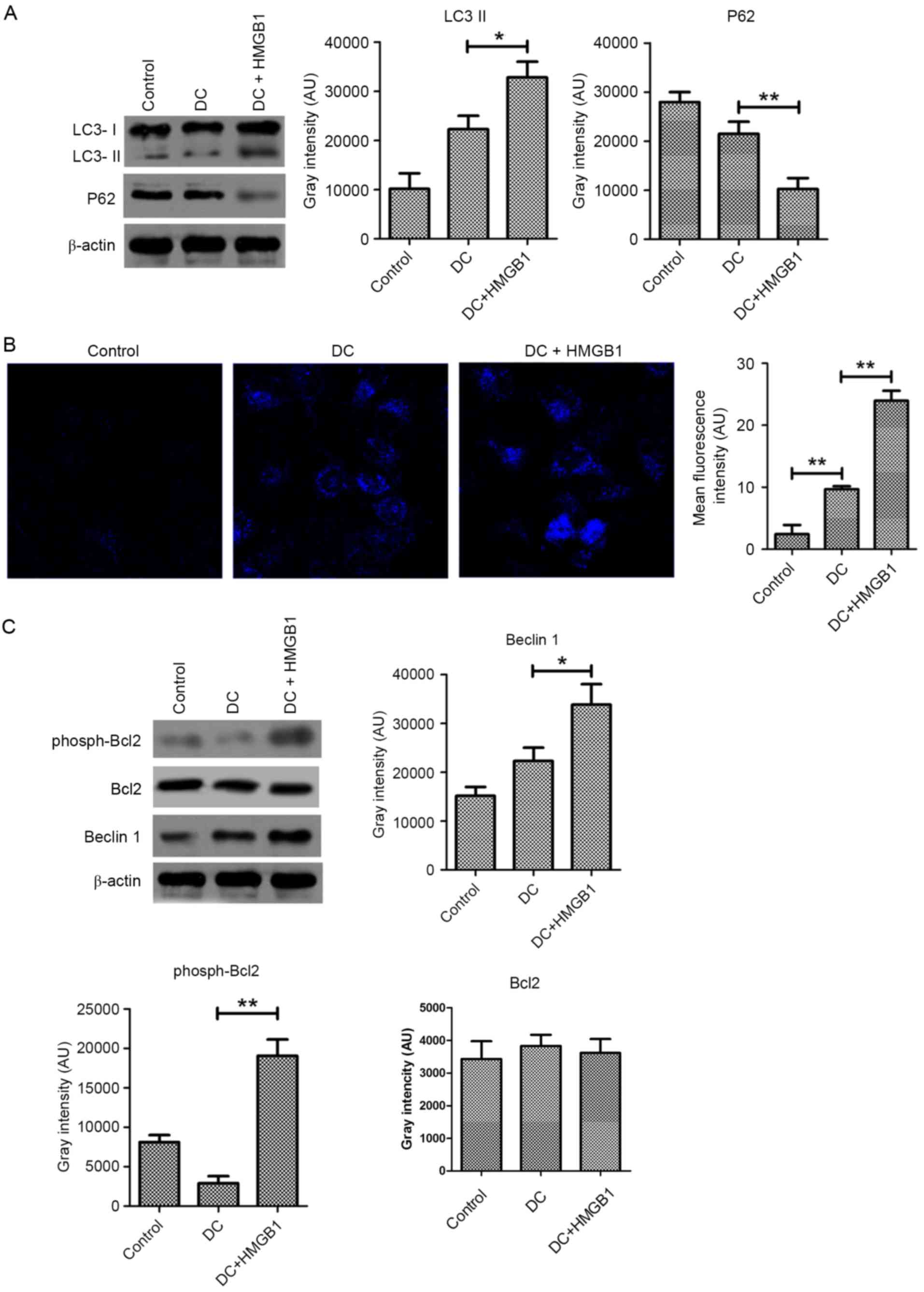 | Figure 3.HMGB1 upregulates autophagy in CT26.WT
cells exposed to DCs. (A), Pretreatment with 10 ng/ml HMGB1
increased the conversion of LC3 and the degradation of P62 in
CT26.WT cells exposed to DCs at a ratio of 1:10 for 24 h compared
with non-pretreated cells, as determined by immunoblot analysis.
(B) Monodansylcadaverine was used to label autophagic vesicles, and
the fluorescence intensity of the CT26.WT cells exposed to DCs at a
ratio of 1:10 for 24 h was determined by fluorescence microscopy.
Magnification, ×400. (C) The levels of Beclin 1 expression and
phosphorylated Bcl2 were determined by immunoblot analysis in 10
ng/ml HMGB1-pretreated CT26.WT cells exposed to DCs at a ratio of
1:10 for 24 h. The data represent the mean ± standard deviation
(n=4). *P<0.05, **P<0.01. LC3, microtubule-associated protein
1 light chain 3; P62, sequestosome 1; DC, dendritic cell; HMGB1,
high mobility group box protein 1; AU, arbitrary units; Bcl2,
B-cell lymphoma 2. |
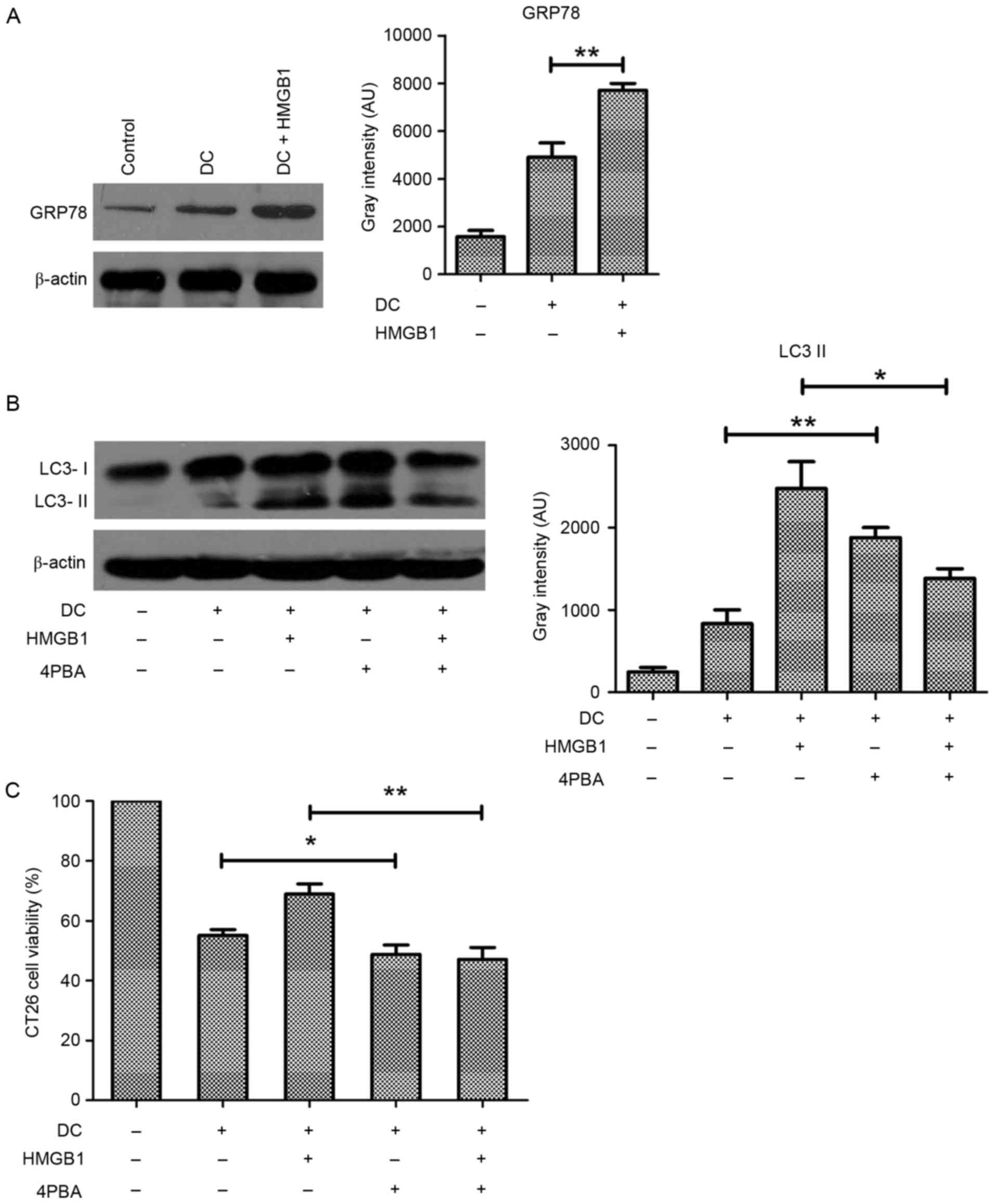
ER stress is involved in HMGB1-induced
autophagy in CT26.WT cells
Given that ER stress is an important disordered
microenvironmental stimulus for autophagy, it was aimed to
determine the role of ER stress in HMGB1-induced autophagy of
CT26.WT cells exposed to DCs. The expression of GRP78, a hallmark
of ER stress, was apparently increased in the HMGB1-treated,
DC-exposed CT26.WT cells (Fig.
4A), suggesting that HMGB1 promoted ER stress in these cells.
Whereas HMGB1 pretreatment resulted in a marked increase in the
conversion of LC3, treatment with 10 mM 4-PBA to block ER stress
reduced this conversion in the CT26.WT cells exposed to DCs
(Fig. 4B), suggesting that ER
stress was involved in HMGB1-induced autophagy in these cells.
Consistently, CCK-8 assay demonstrated that treatment with 4-PBA
resulted in a significant decrease in cell viability (Fig. 4C). These results suggest that ER
stress mediates HMGB1-induced protective autophagy in CT26.WT cells
exposed to DCs.
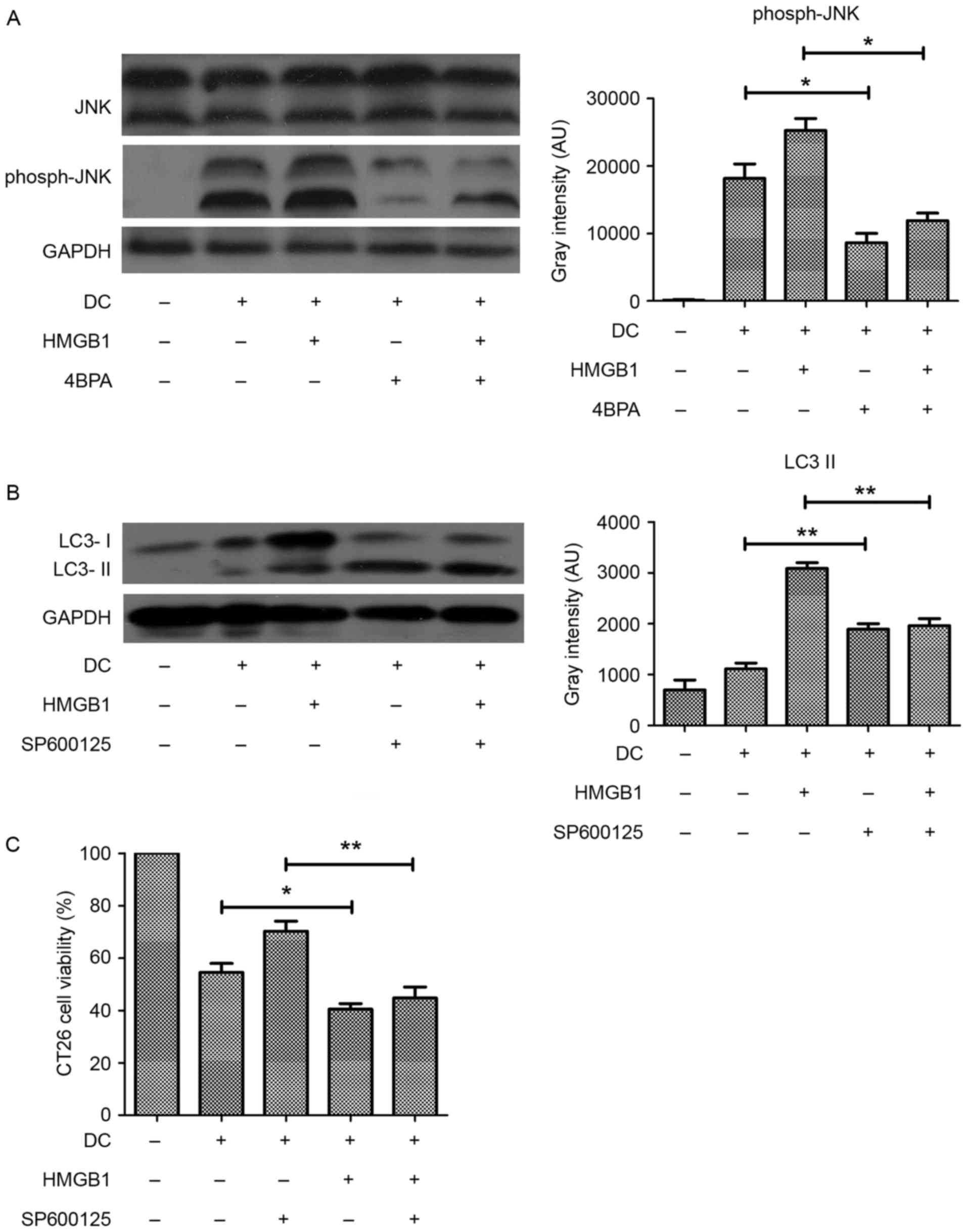
JNK phosphorylation links ER stress to
HMGB1-induced autophagy
Activated JNK phosphorylates Bcl2 to facilitate
disassociation of the Beclin 1-Bcl2 complex, which is essential for
the initiation of autophagosomes. The phosphorylation of Bcl2 was
increased in the HMGB1-pretreated CT26.WT cells, suggesting the
involvement of JNK kinase activity in HMGB1-induced autophagy. To
verify this, JNK phosphorylation in HMGB1-pretreated, DC-exposed
CT26.WT cells was evaluated and observed that JNK phosphorylation
was increased by DCs and further increased by HMGB1 pretreatment
(Fig. 5A). To clarify the
association between JNK activation, HMGB1-induced autophagy and ER
stress, 4-PBA was used to inhibit stress, and the resulting effect
on JNK phosphorylation was assessed. Treatment of DC-exposed
CT26.WT cells with 10 mM 4-PBA resulted in inhibition of JNK
phosphorylation (Fig. 5A). In
addition, the increased conversion of LC3 induced by HMGB1 was
inhibited by treatment with the JNK inhibitor SP600125, suggesting
that JNK activation may link ER stress with autophagy in the
CT26.WT cells exposed to DCs (Fig.
5B). In addition, treatment of HMGB1-pretreated CT26.WT cells
exposed to DCs with SP600125 also significantly decreased their
viability (Fig. 5C). These results
suggest that HMGB1-induced autophagy is activated by ER stress
through JNK phosphorylation in CT26.WT cells exposed to DCs.
Discussion
Immune evasion is an essential step in cancer
progression. One principle of immune evasion by cancer cells is
that these cells suppress the immune system, resulting in failure
of the immune system to detect them through TAAs, thereby enhancing
their survival. Following exposure of cancer cells to immunological
stress, they acquire phenotypes that allow them to escape from
immune surveillance (21). This
process involves dysfunction of the antigen presenting and
progressing abilities of APCs such as DCs. DCs, the most efficient
APCs, are critical for cancer antigen processing and presentation
and have the extraordinary ability to induce cancer-directed T cell
responses; therefore, these cells have the potential to be used in
biotherapy (22). However, there
are always obstacles to overcome. Some cancer cell phenotypes
inhibit the immune pathway, and one therapeutic strategy involves
impeding the acquisition of TAAs by APCs such as DCs (23,24).
Apoptosis results in the release of cancer cell contents, including
many TAAs. Therefore, if cancer cells elude DC-induced apoptosis,
they may partially contribute to immune system surveillance and
immune evasion. The results of this study have demonstrated that
HMGB1 is overexpressed in the colon cancer cell line CT26.WT and
that HMGB1 expression enables CT26.WT cells to avoid apoptotic cell
death triggered by bone marrow-derived dendritic cells. Our results
have also shown that HMGB1 treatment reduces the cytotoxicity of
DCs to CT26.WT cells, indicating that HMGB1 is closely associated
with cancer immune evasion.
HMGB1 is overexpressed in a variety of types of
cancer cells, and it can help cancer cells to cope with stresses
induced by chemotherapy (25) or
immune cytokines, resulting in their survival; thus, HMGB1 has been
demonstrated to be closely associated with chemotherapy resistance
and cancer evasion. A previous study demonstrated that the
cancer-associated expression of the receptor T-cell immunoglobulin
and mucin-domain containing-3 in DCs results in inhibition of the
anticancer efficacy of DNA vaccines and chemotherapy through its
binding to the damage-associated molecular pattern molecule HMGB1
(26). HMGB1 produced by cancer
cells has an inhibitory effect on DCs in mice and humans (27). Studies examining the mechanisms by
which HMGB1 contributes to carcinogenesis have been largely focused
on molecular biological and biochemical aspects and therapeutic
strategies based on the clinical targeting of HMGB1. The results of
this study revealed that HMGB1 is a damage-associated molecular
pattern molecule that rescues CT26.WT cells from stress caused by
DCs by promoting autophagy. siRNA-mediated depletion of HMGB1 or
antibody-mediated neutralization in the co-culture supernatant
resulted in decreased autophagy of CT26.WT cells exposed to DCs and
increased the vulnerability of these cells to the DCs. Thus, we
hypothesize that HMGB1 helps CT26.WT cells escape from apoptotic
cell death by promoting the autophagic pathway. Beclin 1 has a
central role in autophagy and is important for the localization of
autophagic proteins to pre-autophagosomes, depending on its
interaction with class 3 phosphoinositide 3 kinase (PI3K) (18). The current study also demonstrated
that HMGB1 is a direct regulator of autophagy in CT26.WT cells and
that it promotes LC3 conversion, P62 degradation, Beclin 1
upregulation and increased Bcl2 phosphorylation, which are key
molecular events in the formation of the PI3KIII/Beclin 1 complex.
Taken together, these results indicate that HMGB1-induced autophagy
is dependent upon class 3 PI3K/Beclin 1.
ER stress is a critical disordered
microenvironmental stimulus for autophagy. The results of the
current study demonstrated that GRP78 expression was increased
following pretreatment with HMGB1 in CT26.WT cells exposed to DCs
and that it was suppressed by treatment with the ER stress
inhibitor 4-PBA. HMGB1 treatment also increased the conversion of
LC3 I to LC3 II and the degradation of P62 in the CT26.WT cells
exposed to DCs, suggesting that ER stress is involved in
HMGB1-induced autophagy. The pathways that link ER stress with
autophagy are complex, and one important mechanism involves the
phosphorylation of JNK, which is important for cell survival
(27). As demonstrated in the
present study, JNK phosphorylation was increased following
treatment with HMGB1 in the CT26.WT cells exposed to DCs and that
it was suppressed by 4-PBA treatment. In addition, treatment of
these cells with the JNK inhibitor SP600125 inhibited the
conversion of LC3 and the degradation of P62.
In conclusion, HMGB1 functions as an autophagy
effector in CT26.WT cells exposed to DCs and promotes autophagy by
increasing ER stress through JNK phosphorylation and the PI3K class
3/Beclin 1 complex to promote resistance to DC-induced apoptotic
cell death. However, the specific region of HMGB1 that binds to
Beclin 1 and how this binding affects downstream genes in CT26.WT
cells are still largely unknown. Further studies are required to
address these questions. The results of the study suggest that
HMGB1 is released from CT26.WT cells during cancer immune evasion
and that it functions as a positive regulator of autophagy that
prevents DC-induced apoptosis. The findings also indicate that
HMGB1 activates class 3 PI3 K/Beclin 1, thereby regulating
autophagosome formation and the fusion of autophagosomes with
lysosomes. In light of promising autophagy inhibitors that increase
the susceptibility of cancer cells to conventional therapies, the
findings also suggest that HMGB1 is a crucial regulator of
autophagy in CT26.WT cells exposed to DCs. These findings may
facilitate the development of novel treatment strategies for colon
cancer.
Glossary
Abbreviations
Abbreviations:
|
HMGB1
|
high mobility group box 1
|
|
ER
|
endoplasmic reticulum
|
|
JNK
|
c-Jun N-terminal kinases
|
|
DC
|
dendritic cell
|
|
APC
|
antigen presenting cell
|
|
TAA
|
tumor-associated antigen
|
|
LDH
|
lactate dehydrogenase
|
|
GM-CSF
|
granulocyte macrophage-colony
stimulating factor
|
|
FBS
|
fetal bovine serum
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
GRP78
|
glucose regulated protein 78
|
|
PI3K
|
phosphoinositide 3-kinase
|
References
|
1
|
Engblom C, Pfirschke C and Pittet MJ: The
role of myeloid cells in cancer therapies. Nat Rev Cancer.
16:447–462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Banchereau J and Steinman RM: Dendritic
cells and the control of immunity. Nature. 392:245–252. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
James BR, Brincks EL, Kucaba TA, Boon L
and Griffith TS: Effective TRAIL-based immunotherapy requires both
plasmacytoid and CD8α dendritic cells. Cancer Immunol Immunother.
63:685–697. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oppenheim JJ and Yang D: Alarmins:
Chemotactic activators of immune responses. Curr Opin Immunol.
17:359–365. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chiba S, Baghdadi M, Akiba H, Yoshiyama H,
Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan
JD, et al: Tumor-infiltrating DCs suppress nucleic acid-mediated
innate immune responses through interactions between the receptor
TIM-3 and the alarmin HMGB1. Nat Immunol. 13:832–842. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scaffidi P, Misteli T and Bianchi ME:
Release of chromatin protein HMGB1 by necrotic cells triggers
inflammation. Nature. 418:191–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang D, Kang R, Zeh HJ III and Lotze MT:
High-mobility group box 1 and cancer. Biochim Biophys Acta.
1799:131–140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martinotti S, Patrone M and Ranzato E:
Emerging roles for HMGB1 protein in immunity, inflammation, and
cancer. Immunotargets Ther. 4:101–109. 2015.PubMed/NCBI
|
|
9
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III
and Lotze MT: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Campana L, Bosurgi L and Rovere-Querini P:
HMGB1: A two-headed signal regulating tumor progression and
immunity. Curr Opin Immunol. 20:518–523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hoyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li C, Capan E, Zhao Y, Zhao J, Stolz D,
Watkins SC, Jin S and Lu B: Autophagy is induced in CD4+ T cells
and important for the growth factor-withdrawal cell death. J
Immunol. 177:5163–5168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kang R, Livesey KM, Zeh HJ, Loze MT and
Tang D: HMGB1: A novel Beclin 1-binding protein active in
autophagy. Autophagy. 6:1209–1211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lutz MB, Kukutsch N, Ogilvie AL, Rössner
S, Koch F, Romani N and Schuler G: An advanced culture method for
generating large quantities of highly pure dendritic cells from
mouse bone marrow. J Immunol Methods. 223:77–92. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khong HT and Restifo NP: Natural selection
of tumor variants in the generation of ‘tumor escape’ phenotypes.
Nat Immunol. 3:999–1005. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Steinman RM and Banchereau J: Taking
dendritic cells into medicine. Nature. 449:419–426. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Banchereau J and Palucka AK: Dendritic
cells as therapeutic vaccines against cancer. Nat Rev Immunol.
5:296–306. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim R, Emi M and Tanabe K: Cancer
immunoediting from immune surveillance to immune escape.
Immunology. 121:1–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu
Y, Xie M, Yin X, Livesey KM, Lotze MT, et al: HMGB1-induced
autophagy promotes chemotherapy resistance in leukemia cells.
Leukemia. 25:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang D and Lotze MT: Tumor immunity times
out: TIM-3 and HMGB1. Nat Immunol. 13:808–810. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kusume A, Sasahira T, Luo Y, Isobe M,
Nakagawa N, Tatsumoto N, Fujii K, Ohmori H and Kuniyasu H:
Suppression of dendritic cells by HMGB1 is associated with lymph
node metastasis of human colon cancer. Pathobiology. 76:155–162.
2009. View Article : Google Scholar : PubMed/NCBI
|