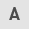Necrostatin-1 attenuates sepsis-associated acute kidney injury by promoting autophagosome elimination in renal tubular epithelial cells
- Authors:
- Published online on: December 7, 2017 https://doi.org/10.3892/mmr.2017.8214
- Pages: 3194-3199
Abstract
Introduction
Acute kidney injury (AKI) is generally characterized by a rapid decline in renal function due to a variety of causes (1). AKI is a common illness with an incidence rate of 2–12% among general inpatients (2,3), and an incidence of up to 30% among critically ill patients (4). AKI not only affects patient survival, but also increases the incidence of long-term cardiovascular events and the likelihood of developing end-stage renal diseases (3,5). Sepsis is an important cause of AKI, and 29.6–32.4% of AKI cases are associated with sepsis (6,7). Recently, the incidence rate of sepsis-associated AKI (SA-AKI) has increased, and the mortality rate remains high (8,9); thus, the prevention and control of SA-AKI is a major challenge for nephrology and critical care medicine.
Previous studies have identified necroptosis as a regulated form of necrosis that participates in AKI caused by multiple factors (10–12). However, necrostatin-1 (Nec-1) has been reported to reduce the necroptosis of renal tubular epithelial cells by inhibiting the kinase activity of receptor-interacting protein-1 (RIP-1), thereby protecting against ischemia-reperfusion-induced or cisplatin-induced AKI (10,11). However, the pathophysiology of SA-AKI differs from that of these previous AKI models, as no significant necrosis of renal tubular epithelial cells is observed in SA-AKI (13). This indicates that necroptosis may not contribute to the development of SA-AKI, and thus the protective effective of Nec-1 in SA-AKI remains unclear.
Autophagy is a highly conserved physiological mechanism that enables cells to maintain their normal activities (14). In cases of nutrient starvation, cells transport cytoplasmic substances from autophagosomes into lysosomes in order to synthesize essential energy compounds and nutrients, while in cases of intracellular damage, injured mitochondria and endoplasmic reticulum can be transported into lysosomes in order to remove the damaged organelles (14). Previous studies have reported that the promotion of autophagy may reduce renal damage caused by SA-AKI (15–17). Additionally, we previously reported that Nec-1 reduced necroptosis in renal tubular epithelial cells and also affected the levels of the autophagy marker microtubule associated protein 1 light chain 3 (LC3)-II (18). In the present study, a model of SA-AKI was established in mice to observe the influence of Nec-1 on the autophagy of renal epithelial cells, with the aim of evaluating the protective effect of Nec-1 against kidney damage, and of determining whether Nec-1 can attenuate SA-AKI by affecting autophagy.
Materials and methods
Experimental animals and grouping
A total of 12 C57BL/6 mice (weight 18–22 g, aged 8–10 weeks) were purchased from the Experimental Animal Center of Southern Medical University (Guangzhou, China), and were housed in the Experimental Animal Center of Sun Yat-sen University, Zhongshan School of Medicine (Guangzhou, China). The mice were maintained under a 12-h light-dark cycle in an atmosphere of 0.03% CO2 at 24°C and received food and water ad libitum prior to use in experiments. All animal experiments were approved by the Research Ethics Committee at Guangdong General Hospital (Guangzhou, China). The 12 mice were randomly assigned into the following three groups (n=4 per group): i) Control group, in which mice were intraperitoneally injected with saline; ii) a sepsis group, in which mice were intraperitoneally injected with lipopolysaccharide (LPS; 10 mg/kg); and iii) a sepsis + Nec-1 pretreatment group, in which mice were intraperitoneally injected with Nec-1 (1.65 mg/kg) and then with LPS (10 mg/kg) 15 min later. All mice were observed for 18 h after LPS/saline treatment, after which the mice were euthanized. Blood was then isolated by inferior vena cava puncture and the kidneys were harvested.
Detection of renal function
Renal function was determined by assaying the serum for blood urea nitrogen (BUN) and serum creatinine (SCr) using a QuantiChrom™ Urea Assay kit and QuantiChrom™ Creatinine Assay kit (BioAssay Systems, Hayward, CA, USA), respectively.
Western blot analysis
Renal tissues from the mice in the different experimental groups were lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China). The total protein concentration of the resultant supernatant was determined with a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Samples containing equal protein content (30 µg) were subjected to SDS-PAGE using 8–15% gels according to the molecular weight of different proteins, and the separated proteins were transferred to polyvinylidene difluoride membranes. The membranes were blocked with 3% bovine serum albumin (Beyotime Institute of Biotechnology) in TBS-Tween for 1 h at room temperature and then incubated overnight at 4°C with anti-β-actin antibody (1:1,000; cat. no. 8457; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-LC3A/B antibody (1:1,000; cat. no. 4108; Cell Signaling Technology, Inc.) and anti-p62 antibody (1:1,000; cat. no. 5114; Cell Signaling Technology, Inc.). After washing in TBS, the blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody (cat. no. 7074; Cell Signaling Technology, Inc.) for 1 h at room temperature. Following a final wash with TBS, the specifically bound secondary antibodies were visualized with enhanced Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific, Inc.). Signals were quantified using a chemiluminescence detector and the accompanying densitometric software (Tanon Science and Technology Co., Ltd., Shanghai, China).
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the isolated kidney tissues using TRIzol reagent according to the manufacturer's instructions (Thermo Fisher Scientific, Inc.). Total RNA (500 ng) was subjected to reverse transcription using a PrimeScript RT Reagent kit (Perfect Real Time; Takara Biotechnology Co., Ltd., Dalian, China). The resulting cDNA was used for qPCR analysis using SYBR Premix Ex Taq (Perfect Real Time; Takara Biotechnology Co., Ltd.). Following 1 min denaturation at 95°C; amplification was performed for 40 cycles, including 95°C for 5 sec and 60°C for 25 sec. The levels of target mRNA were determined using the comparative cycle threshold method of relative quantitation. GAPDH was used as an internal control (19). The primer sequences used are presented in Table I.
Periodic acid-Schiff (PAS) staining of renal tissues
Renal tissues were fixed with 10% formalin at room temperature for 2 h. Formalin-fixed paraffin-embedded kidney tissues were sectioned at a thickness of 4 µm. The sections from all specimens were batch-stained with 1% periodic acid for 15 min and Schiff reagent for 30 min at room temperature.
Electron microscopy
Renal tissues were fixed at 4°C in a fixing solution containing 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) overnight. Samples were rinsed in 0.1 M cacodylate buffer, post-fixed in 1% osmium tetroxide for 2 h, and then washed in 0.1 M cacodylate buffer. The samples were dehydrated in a graded series of ethanol (50, 70, 80, 90 and 100%) and 100% propylene oxide, and then embedded in resin (Epon 812) for 12 h. Ultra-thin sections (60–70 nm) were counterstained with 2% uranyl acetate for 5 min and lead citrate for 10 min. A transmission electron microscope (JEM-100CX; JEOL Ltd., Tokyo, Japan) was used to observe and capture images of the kidney tissue ultrastructure.
Statistical analysis
All values are expressed as the mean ± standard deviation. Multiple comparisons among the groups were conducted by one way analysis of variance followed by a least significant difference multiple comparison test. All statistical analyses were performed using SPSS software (v21.0; IBM Corp., Armonk, NY, USA), and P<0.05 was considered to indicate a statistically significant difference.
Results
Nec-1 attenuates SA-AKI
After 18 h of intervention with LPS, when compared with the control group, LPS-treated mice had significantly higher levels of SCr (LPS group 30.08±4.18 µmol/l vs. control group 9.80±1.84 µmol/l; P<0.001) and BUN (LPS group 32.54±5.46 mmol/l vs. control group 13.87±4.83 mmol/l; P<0.001; Fig. 1). This indicated that the mice had developed AKI following the LPS injection. Compared with the LPS group, mice pretreated with Nec-1 had significantly lower levels of SCr (LPS + Nec-1 group 11.50±1.67 µmol/l vs. LPS group 30.08±4.18 µmol/l; P<0.001) and BUN (LPS + Nec-1 group 14.15±4.14 mmol/l vs. LPS group 32.54±5.46 mmol/l; P<0.001), and no significant differences in SCr and BUN levels were identified between the LPS + Nec-1 and control groups (Fig. 1), indicating that Nec-1 may serve a role in protecting renal function from the effects of sepsis in mice.
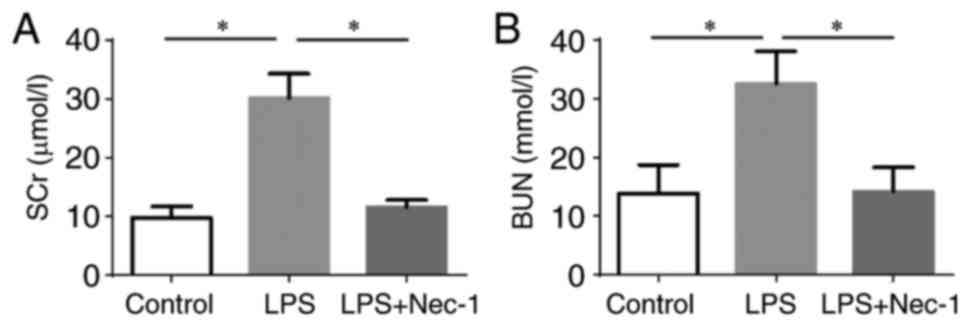
Nec-1 does not attenuate SA-AKI by reducing cell necrosis
Although the renal tissues of mice injected with LPS exhibited local vacuolar degeneration, renal tubular epithelial cell necrosis and apoptosis were not obvious upon assessment of tissue morphology (Fig. 2). This result suggested that the protective effect of Nec-1 against AKI was not achieved through the suppression of renal tubular epithelial cell necrosis.
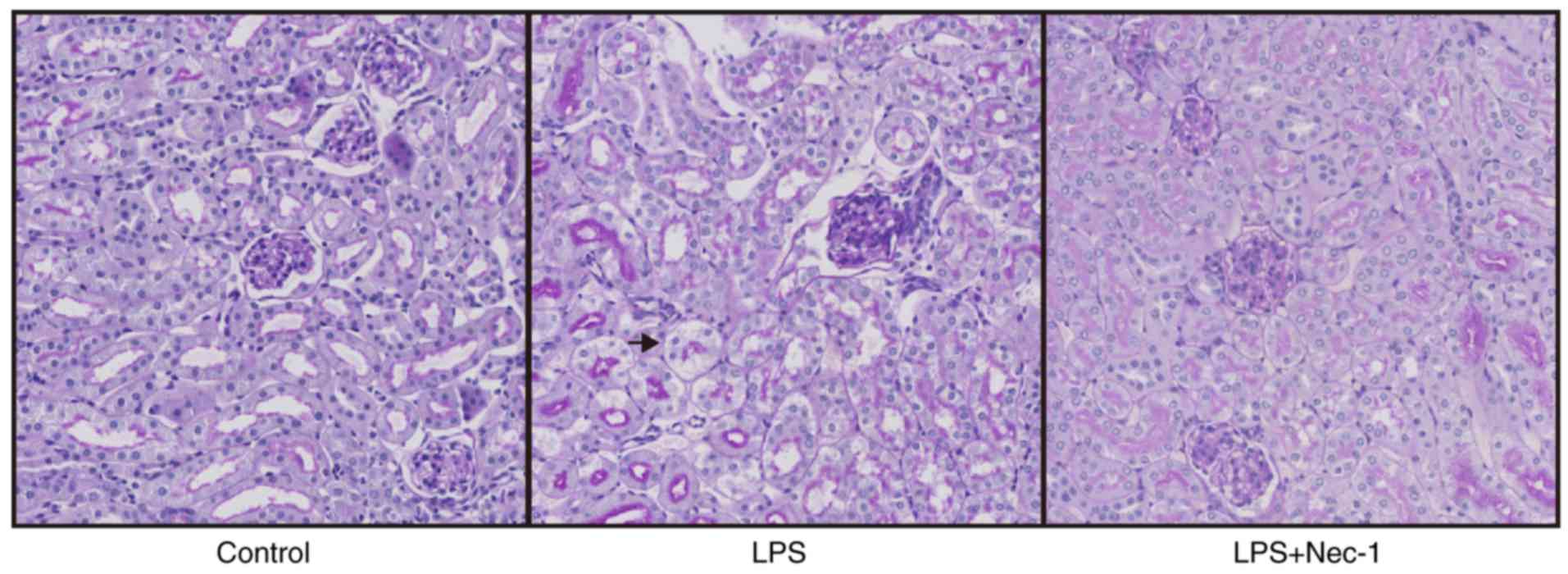
Disrupted autophagosome elimination occurs during SA-AKI
As a marker protein of autophagosomes, LC3-II can be used to identify changes in cell autophagy; when the number of autophagosomes increases, the level of LC3-II rises. Additionally, when fusion events between autophagosomes and lysosomes are disrupted, the clearance of autophagosomes cannot be completed, and thus the level of LC3-II increases (14). As a substrate of autophagy, the level of p62 increases markedly when autophagy is reduced (14). Thus, we performed RT-qPCR and western blotting to detect the mRNA and protein levels of LC3 and p62 in renal tissues (Fig. 3). The results showed that in mice with SA-AKI, the protein levels of LC3-II and p62 were significantly higher than those in control mice (Fig. 3A-D); however, the LC3-II and p62 mRNA levels did not differ significantly from those in the controls (Fig. 3E and F). These results indicated that the increases in LC3-II and p62 may not be caused by increased synthesis, but rather by disrupted autophagosome elimination. Thus, we speculate that disrupted autophagosome elimination might be a manifestation of SA-AKI.
Electron microscopy was subsequently performed to determine whether autophagosome elimination occurred in renal tubular epithelial cells during SA-AKI. Autophagosomes are vacuolar structures bordered by double layer membranes that can sequester cellular contents. Autophagosomes fuse with lysosomes to form autolysosomes, upon which the inner membrane of the autophagosome and the cytoplasm-derived materials contained in the autophagosome are degraded. Thus, autolysosomes may be identified by electron microscopy as vacuolar structures with monolayer membranes containing degraded cytoplasmic components and organelles (20). In the present study, it was observed by electron microscopy that no apparent autophagosomes were formed in the renal epithelial cells of the control group, whereas many autophagosomes, but not autolysosomes, were observed in the renal epithelial cells of mice with SA-AKI (Fig. 4). This is consistent with the expected features of disrupted autophagosome elimination.
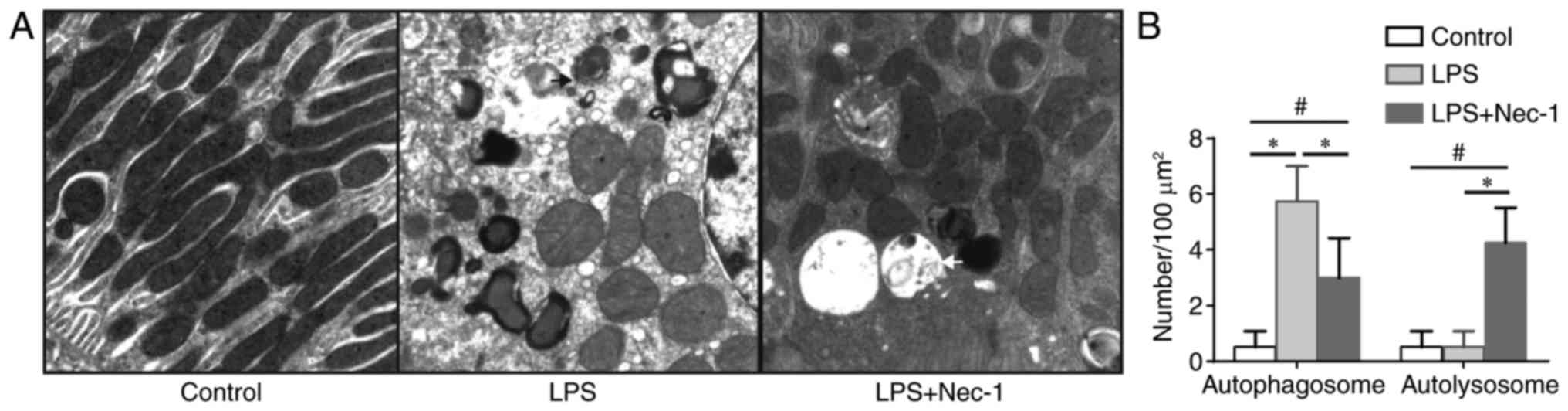
Nec-1 promotes autophagosome elimination during SA-AKI
In mice pretreated with Nec-1, the protein levels of both LC3-II and p62 were significantly decreased when compared with those in mice injected with LPS only (Fig. 3A-D); however, the mRNA levels of LC3-II and p62 did not differ significantly between the two groups (Fig. 3E and F). Additionally, electron microscopy identified both autophagosomes and autolysosomes in the renal epithelial cells of Nec-1-pretreated mice (Fig. 4), indicating that Nec-1 may promote autophagosome elimination during SA-AKI.
Discussion
Sepsis is a major cause of AKI and, due to a lack of effective prevention and treatment methods, the incidence and mortality rates of SA-AKI remain high at present (8,9). Thus, it is necessary to investigate the mechanisms underlying the development and progression of SA-AKI, and to identify effective prevention and treatment measures.
Necroptosis, a regulated form of cell necrosis, is involved in ischemia-reperfusion-induced or cisplatin-induced AKI (10,11). Two serine/threonine kinases, RIP-1 and RIP-3, mediate necroptosis through their kinase activity (21,22). Nec-1 reduces necroptosis by inhibiting the kinase activity of RIP-1 (21,23). Previous studies have revealed that Nec-1 may be capable of alleviating ischemia-reperfusion- and cisplatin-induced AKI by decreasing the necrosis of renal tubular epithelial cells (10–12). However, due to its differing etiology, SA-AKI does not show significant necrosis of renal epithelial cells, and thus the protective effect of Nec-1 in SA-AKI remains undetermined.
Our study found that, following Nec-1 pretreatment, mice injected with LPS had significantly lower levels of SCr and BUN, indicating that Nec-1 alleviated SA-AKI. Meanwhile, no obvious necrosis was observed in the renal epithelial cells of SA-AKI mice, which was consistent with previous results (13). This indicated that Nec-1 did not attenuate SA-AKI by inhibiting necroptosis, and thus the mechanism by which Nec-1 functions to protect SA-AKI was further investigated.
At present, the role of apoptosis in septic AKI remains controversial. Some studies have reported that apoptosis is involved in SA-AKI (24,25), while other studies have drawn the opposite conclusion (26,27). In our study, morphological assessment by renal tissue staining identified no significant renal tubular epithelial cell apoptosis. It has previously been documented that the kinase activity of RIP-1 is not required for apoptosis (28), and there is no evidence showing that Nec-1, as an RIP-1 kinase inhibitor, can alleviate apoptosis. Therefore, we concluded that the protective effect of Nec-1 against SA-AKI may not have resulted from the suppression of apoptosis.
Although RIPs are involved in the regulation of nuclear factor-κB activation and inflammatory cytokine production, the kinase activities of RIP-1 and RIP-3 are not required for these effects (29,30). Hence, we speculated that the RIP-1 kinase inhibitor Nec-1 did not affect inflammatory cell responses induced by LPS.
In our previous study in a necroptosis model in renal tubular epithelial cells, an increased level of the autophagy marker LC3-II was identified, and Nec-1 treatment not only reduced renal tubular epithelial cell necroptosis, but also decreased the level of LC3-II (18). Thus, we speculate that Nec-1 may protect against SA-AKI by affecting autophagy. Autophagy is a physiological reaction that occurs when cells are starved, stressed or injured. Autophagy enables starved cells to degrade and recycle cytoplasmic components as a survival mechanism, and removes damaged organelles in cases of intracellular injury (14). The process of autophagy includes autophagosome formation, autolysosome formation by autophagosome and lysosome fusion, and degradation of the autolysosome contents (14). Disruption in any one step can result in autophagy disorder. When the number of autophagosomes increases, the level of LC3-II increases. Additionally, when autolysosome formation or lysosome function are disrupted, the level of LC3-II increases due to reduced autophagosome elimination. Furthermore, it has been reported that the level of P62, which serves as a substrate for autophagy, was significantly increased with a decreased rate of autophagy (14). Previous studies have indicated that the promotion of renal tubular epithelial cell autophagy may alleviate SA-AKI (15–17), and that inhibition of either tubular epithelial cell autophagy initiation or autophagosome elimination may aggravate SA-AKI (31–33). The results of the present study showed that both LC3-II and p62 increased during SA-AKI. However, when autophagy is increased, the corresponding increase in LC3-II should be accompanied by a decrease in p62. Thus, we speculate that during SA-AKI, the increase in LC3-II should not be attributed to increased autophagy, but to disrupted autophagosome elimination. By electron microscopy, it was observed that although the number of autophagosomes were notably increased during SA-AKI, no obvious autolysosomes were present, which was in line with the features of disrupted autophagosome elimination, and verified that disrupted autophagosome elimination was present during SA-AKI. Pretreatment with Nec-1 attenuated the LPS-induced increases in LC3-II and p62, and the formation of autolysosomes was also observed by electron microscopy. This result indicated that Nec-1 does not enhance autophagy initiation but improves autophagosome elimination. We therefore propose that Nec-1 exerts its protective effect in SA-AKI by promoting autophagosome elimination in renal tubular epithelial cells.
A previous study reported that basal autophagy was negatively regulated by RIP-1 (34). RIP-1 activates extracellular signal-regulated kinase, which negatively regulates Transcription factor EB (TFEB) through phosphorylation of serine 142. The phosphorylation of TFEB has been shown to negatively regulate its transcriptional enhancement of autophagy and lysosomal genes. Therefore, we speculate that the stimulatory effect of Nec-1 on autophagosome elimination may be associated with increased TFEB phosphorylation. The mechanism by which Nec-1 promotes autophagosome elimination during SA-AKI will be investigated in future studies.
In conclusion, the current study confirmed that Nec-1 improved renal function in mice with sepsis. The protective effect of Nec-1 in SA-AKI might not involve the suppression of necroptosis, but rather the promotion of autophagosome elimination in renal tubular epithelial cells.
Acknowledgements
The present study was supported by the National Natural Science Foundation (grant nos. 81570609 and 81200544), the Natural Science Foundation of Guangdong Province (grant no. 2014A030313545), and the National Clinical Key Specialty Construction Preparatory Projects.
References
|
Bellomo R, Kellum JA and Ronco C: Acute kidney injury. Lancet. 380:756–766. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Yang L, Xing G, Wang L, Wu Y, Li S, Xu G, He Q, Chen J, Chen M, Liu X, et al: Acute kidney injury in China: A cross-sectional survey. Lancet. 386:1465–1471. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Xu X, Nie S, Liu Z, Chen C, Xu G, Zha Y, Qian J, Liu B, Han S, Xu A, et al: Epidemiology and clinical correlates of AKI in Chinese Hospitalized adults. Clin J Am Soc Nephro. 10:1510–1518. 2015. View Article : Google Scholar | |
|
Zhou J, Qian C, Zhao M, Yu X, Kang Y, Ma X, Ai Y, Xu Y, Liu D, An Y, et al: Epidemiology and outcome of severe sepsis and septic shock in intensive care units in mainland China. PLoS One. 9:e1071812014. View Article : Google Scholar : PubMed/NCBI | |
|
Chertow GM, Burdick E, Honour M, Bonventre JV and Bates DW: Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 16:3365–3370. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Bagshaw SM, George C and Bellomo R; ANZICS Database Management Committee, : Early acute kidney injury and sepsis: A multicentre evaluation. Crit Care. 12:R472008. View Article : Google Scholar : PubMed/NCBI | |
|
Thomas M, Sitch A and Dowswell G: The initial development and assessment of an automatic alert warning of acute kidney injury. Nephrol Dial Transplant. 26:2161–2168. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Perner A, Haase N, Guttormsen AB, Tenhunen J, Klemenzson G, Åneman A, Madsen KR, Møller MH, Elkjær JM, Poulsen LM, et al: Hydroxyethyl starch 130/0.42 versus Ringer's acetate in severe sepsis. N Engl J Med. 367:124–134. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Kellum JA, Chawla LS, Keener C, Singbartl K, Palevsky PM, Pike FL, Yealy DM, Huang DT and Angus DC; ProCESS and ProGReSS-AKI Investigators, : The effects of alternative resuscitation strategies on acute kidney injury in patients with septic shock. Am J Respir Crit Care Med. 193:281–287. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U and Krautwald S: Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 81:751–761. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, et al: A role for tubular necroptosis in cisplatin-induced AKI. J Am Soc Nephrol. 26:2647–2658. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Linkermann A, Heller JO, Prókai A, Weinberg JM, De Zen F, Himmerkus N, Szabó AJ, Bräsen JH, Kunzendorf U and Krautwald S: The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J Am Soc Nephrol. 24:1545–1557. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Takasu O, Gaut JP, Watanabe E, To K, Fagley RE, Sato B, Jarman S, Efimov IR, Janks DL, Srivastava A, et al: Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Resp Crit Care Med. 187:509–517. 2013. View Article : Google Scholar : PubMed/NCBI | |
|
Green DR and Levine B: To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 157:65–75. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Kimura A, Ishida Y, Inagaki M, Nakamura Y, Sanke T, Mukaida N and Kondo T: Interferon-γ is protective in cisplatin-induced renal injury by enhancing autophagic flux. Kidney Int. 82:1093–1104. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Howell GM, Gomez H, Collage RD, Loughran P, Zhang X, Escobar DA, Billiar TR, Zuckerbraun BS and Rosengart MR: Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS One. 8:e695202013. View Article : Google Scholar : PubMed/NCBI | |
|
Wang LT, Chen BL, Wu CT, Huang KH, Chiang CK and Hwa LS: Protective role of AMP-activated protein kinase-evoked autophagy on an in vitro model of ischemia/reperfusion-induced renal tubular cell injury. PLoS One. 8:e798142013. View Article : Google Scholar : PubMed/NCBI | |
|
Liang X, Chen Y, Zhang L, Jiang F, Wang W, Ye Z, Liu S, Yu C and Shi W: Necroptosis, a novel form of caspase-independent cell death, contributes to renal epithelial cell damage in an ATP-depleted renal ischemia model. Mol Med Rep. 10:719–724. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Berg TO, Fengsrud M, Stromhaug PE, Berg T and Seglen PO: Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 273:21883–21892. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al: Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI | |
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L and Wang X: Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009. View Article : Google Scholar : PubMed/NCBI | |
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J: Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 1:112–119. 2005. View Article : Google Scholar : PubMed/NCBI | |
|
Takahashi K, Mizukami H, Kamata K, Inaba W, Kato N, Hibi C and Yagihashi S: Amelioration of acute kidney injury in lipopolysaccharide-induced systemic inflammatory response syndrome by an aldose reductase inhibitor, fidarestat. PLoS One. 7:e301342012. View Article : Google Scholar : PubMed/NCBI | |
|
Guo R, Wang Y, Minto AW, Quigg RJ and Cunningham PN: Acute renal failure in endotoxemia is dependent on caspase activation. J Am Soc Nephrol. 15:3093–3102. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Langenberg C, Gobe G, Hood S, May CN and Bellomo R: Renal histopathology during experimental septic acute kidney injury and recovery. Crit Care Med. 42:e58–e67. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, et al: PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. 121:4003–4014. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Luo J, Tao Y, Liang X, Chen Y, Zhang L, Jiang F, Liu S, Ye Z, Li Z and Shi W: Flow control effect of necrostatin-1 on cell death of the NRK-52E renal tubular epithelial cell line. Mol Med Rep. 16:57–62. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Liu ZG, Hsu H, Goeddel DV and Karin M: Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 87:565–576. 1996. View Article : Google Scholar : PubMed/NCBI | |
|
Moriwaki K, Bertin J, Gough PJ and Chan FK: A RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing. J Immunol. 194:1938–1944. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, et al: Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 22:902–913. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM and Hsu C: The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 37:289–296. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, et al: Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 180:517–525. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Yonekawa T, Gamez G, Kim J, Tan AC, Thorburn J, Gump J, Thorburn A and Morgan MJ: RIP1 negatively regulates basal autophagic flux through TFEB to control sensitivity to apoptosis. EMBO Rep. 16:700–708. 2015. View Article : Google Scholar : PubMed/NCBI |