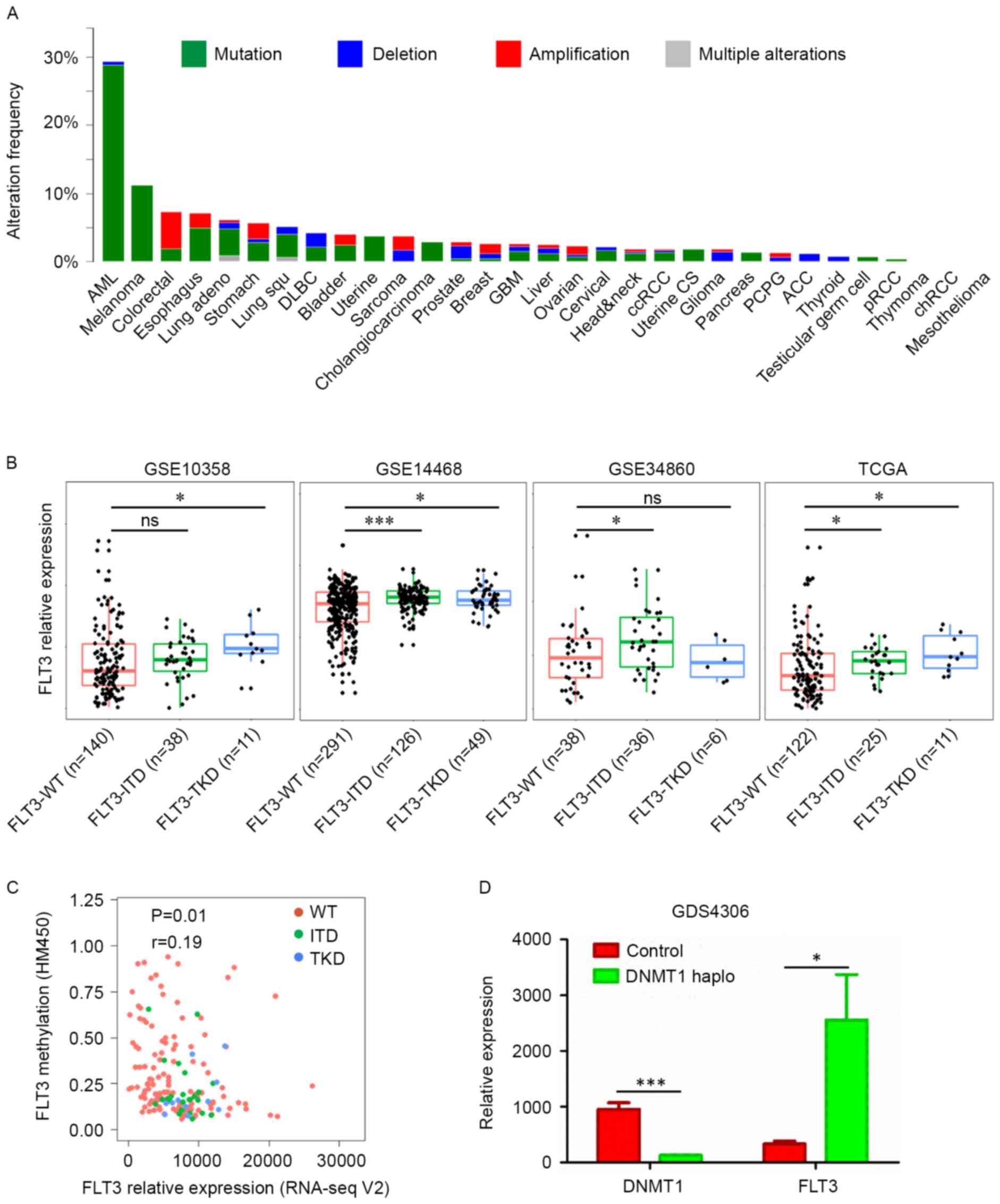
The GDS4306 dataset was used to evaluate the effect
of a DNA methyltransferase 1 (DNMT1) haploinsufficiency on
the expression of FLT3. To compare the correlation between
FLT3 methylation and expression, data were downloaded from
cBioportal to perform linear regression. The FLT3 mutated
samples and wild-type samples were analyzed separately.
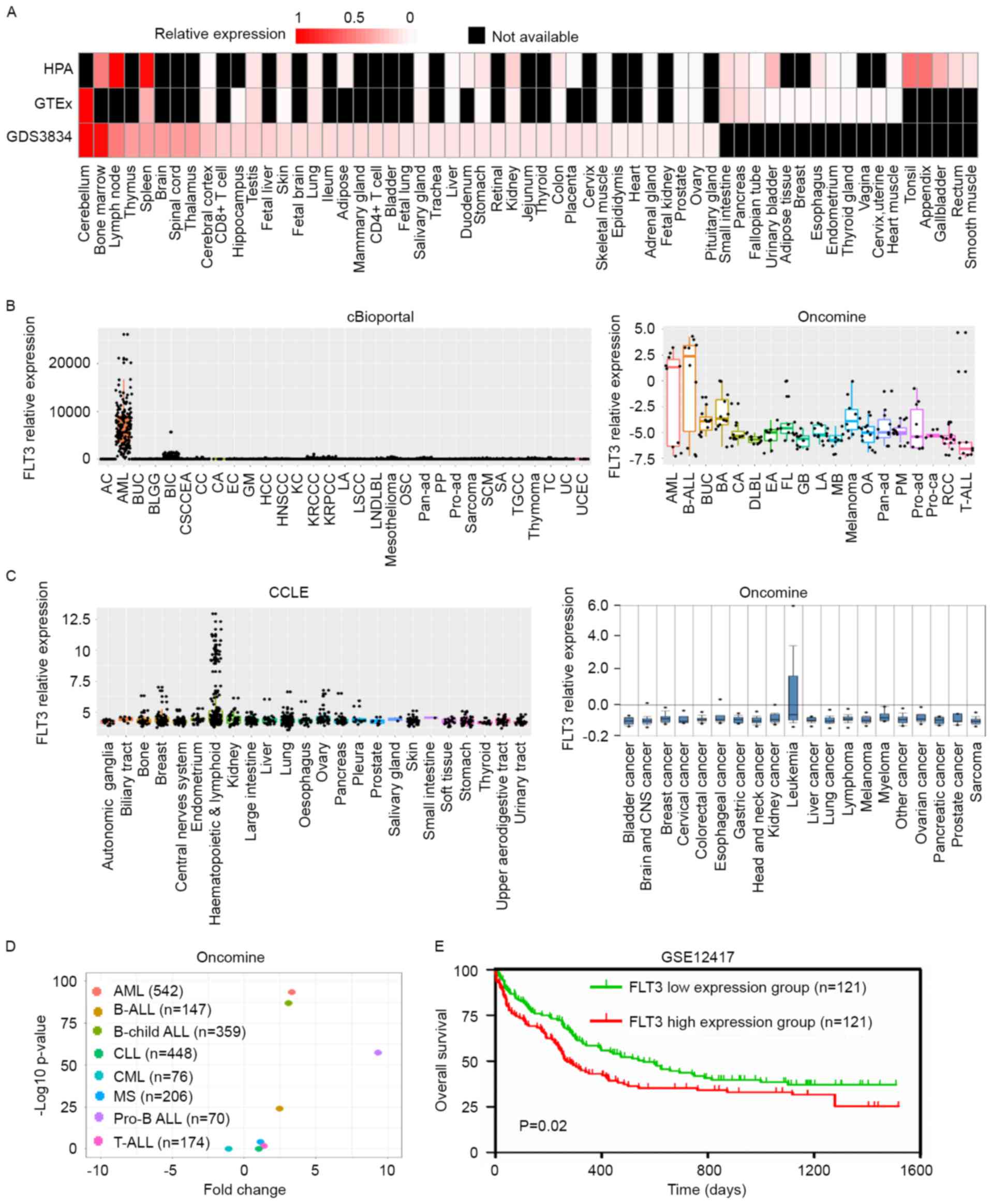
The GSE12417 GEO dataset, which contains data on
survival rates and survival status, was selected to draw the
overall survival curve, and the median expression of FLT3
was used as the cut-off, according to a previous report (33), to classify patients into a high
expression group and low expression group. The top 50% of patients
were classified as the FLT3 high expression group and the
lowest 50% patients were defined as the FLT3 low expression
group, according to the expression of FLT3 from high to low.
The statistical difference between two curves was calculated using
a log-rank test.
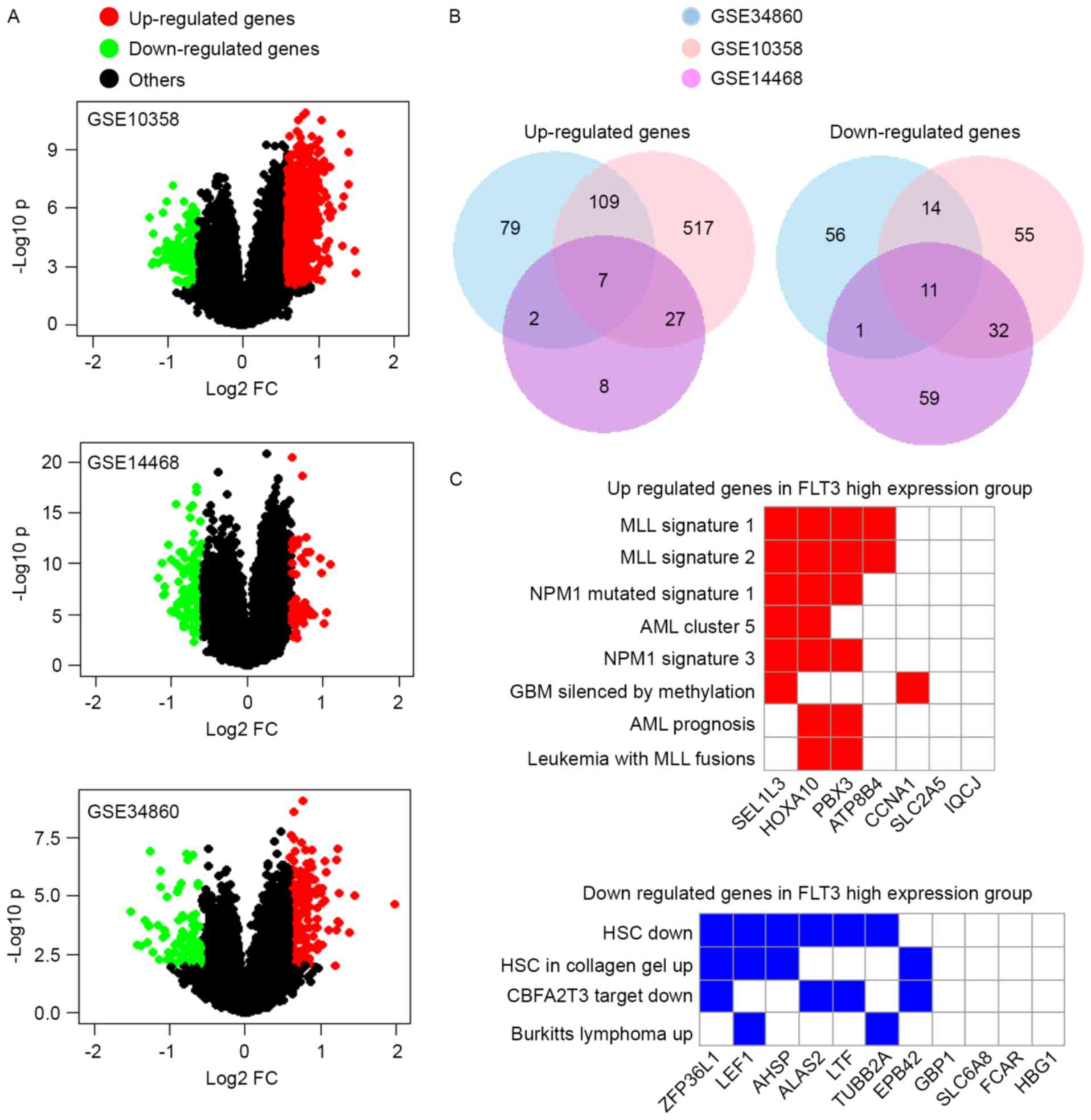
Three GEO datasets of AML (GSE10358, GSE14468 and
GSE34860) were selected to analyze the DEGs between the FLT3
high expression group and FLT3 low expression group.
Initially, the raw data in the CEL file were downloaded from the
GEO database (34), and the robust
multiarray average algorithm in the affy R-3.3.1 package was used
to perform background correction, normalization and expression
calculation (35–37). The Limma package in R (38) was used to identify the DEGs at the
probe level between these two groups. P<0.05 and |log2 fold
change (FC)|>0.585 were used as the cut-off criteria. Finally,
these probes were annotated into gene names.
Student's t-test was used to calculate statistically
significant differences between quantitative variables. The
log-rank test was used to compare the overall survival curve.
P<0.05 was considered to indicate a statistically significant
difference. GraphPad Prism 5.01 was used for statistical analysis
(GraphPad Software, Inc., La Jolla, CA, USA).
FLT3-related pathways were activated in AML by ITD
mutations or the overexpression of FLT3. Several interacting
proteins are also reported to interact with FLT3 to negatively or
positively regulate FLT3 pathways. Spleen tyrosine kinase (SYK) and
the mucin 1-C-terminal subunit (MUC1-C) oncoprotein are reported to
directly bind to and activate FLT3-related pathways (50,51),
whereas suppressor of cytokine signaling 2 (SOCS2) and src-like
adaptor protein 2 (SLAP2) interact with FLT3 protein to inhibit its
signaling (52,53). In addition, the transcription of
FLT3 can be regulated in AML. Certain AML-related
transcription factors, including CCAAT/enhancer binding protein α
and the proto-oncogene MYB, can bind to the FLT3 promoter to
activate the transcription of FLT3 (54). In the present study, another two
mechanism were found to increase the expression of FLT3 in
AML. ITD and TKD mutations, and the methylation of FLT3
increased its expression in AML (Fig.
2B and C). In addition, DNMT1 was identified as is a potential
regulator of FLT3 (Fig.
2D).
This study was supported by a grant from the
National Natural Science Foundation of China (grant no.
8130044).
|
1
|
Volpe G, Clarke M, Garcìa P, Walton DS,
Vegiopoulos A, Del Pozzo W, O'Neill LP, Frampton J and Dumon S:
Regulation of the Flt3 gene in haematopoietic stem and early
progenitor cells. PLoS One. 10:e01382572015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ebihara Y, Wada M, Ueda T, Xu MJ, Manabe
A, Tanaka R, Ito M, Mugishima H, Asano S, Nakahata T and Tsuji K:
Reconstitution of human haematopoiesis in non-obese diabetic/severe
combined immunodeficient mice by clonal cells expanded from single
CD34+CD38- cells expressing Flk2/Flt3. Br J Haematol. 119:525–534.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nakao M, Yokota S, Iwai T, Kaneko H,
Horiike S, Kashima K, Sonoda Y, Fujimoto T and Misawa S: Internal
tandem duplication of the flt3 gene found in acute myeloid
leukemia. Leukemia. 10:1911–1918. 1996.PubMed/NCBI
|
|
4
|
Kiyoi H, Towatari M, Yokota S, Hamaguchi
M, Ohno R, Saito H and Naoe T: Internal tandem duplication of the
FLT3 gene is a novel modality of elongation mutation which causes
constitutive activation of the product. Leukemia. 12:1333–1337.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kiyoi H, Naoe T, Yokota S, Nakao M, Minami
S, Kuriyama K, Takeshita A, Saito K, Hasegawa S, Shimodaira S, et
al: Internal tandem duplication of FLT3 associated with
leukocytosis in acute promyelocytic leukemia. Leukemia Study Group
of the Ministry of Health and Welfare (Kohseisho). Leukemia.
11:1447–1452. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stirewalt DL, Kopecky KJ, Meshinchi S,
Appelbaum FR, Slovak ML, Willman CL and Radich JP: FLT3, RAS, and
TP53 mutations in elderly patients with acute myeloid leukemia.
Blood. 97:3589–3595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meshinchi S, Woods WG, Stirewalt DL,
Sweetser DA, Buckley JD, Tjoa TK, Bernstein ID and Radich JP:
Prevalence and prognostic significance of Flt3 internal tandem
duplication in pediatric acute myeloid leukemia. Blood. 97:89–94.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schnittger S, Schoch C, Dugas M, Kern W,
Staib P, Wuchter C, Löffler H, Sauerland CM, Serve H, Büchner T, et
al: Analysis of FLT3 length mutations in 1003 patients with acute
myeloid leukemia: Correlation to cytogenetics, FAB subtype, and
prognosis in the AMLCG study and usefulness as a marker for the
detection of minimal residual disease. Blood. 100:59–66. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thiede C, Steudel C, Mohr B, Schaich M,
Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M,
Neubauer A, et al: Analysis of FLT3-activating mutations in 979
patients with acute myelogenous leukemia: Association with FAB
subtypes and identification of subgroups with poor prognosis.
Blood. 99:4326–4335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abu-Duhier FM, Goodeve AC, Wilson GA, Gari
MA, Peake IR, Rees DC, Vandenberghe EA, Winship PR and Reilly JT:
FLT3 internal tandem duplication mutations in adult acute myeloid
leukaemia define a high-risk group. Br J Haematol. 111:190–195.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kottaridis PD, Gale RE, Frew ME, Harrison
G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett
AK, et al: The presence of a FLT3 internal tandem duplication in
patients with acute myeloid leukemia (AML) adds important
prognostic information to cytogenetic risk group and response to
the first cycle of chemotherapy: Analysis of 854 patients from the
United Kingdom Medical Research Council AML 10 and 12 trials.
Blood. 98:1752–1759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu F, Taki T, Yang HW, Hanada R, Hongo T,
Ohnishi H, Kobayashi M, Bessho F, Yanagisawa M and Hayashi Y:
Tandem duplication of the FLT3 gene is found in acute lymphoblastic
leukaemia as well as acute myeloid leukaemia but not in
myelodysplastic syndrome or juvenile chronic myelogenous leukaemia
in children. Br J Haematol. 105:155–162. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kiyoi H, Naoe T, Nakano Y, Yokota S,
Minami S, Miyawaki S, Asou N, Kuriyama K, Jinnai I, Shimazaki C, et
al: Prognostic implication of FLT3 and N-RAS gene mutations in
acute myeloid leukemia. Blood. 93:3074–3080. 1999.PubMed/NCBI
|
|
14
|
Horiike S, Yokota S, Nakao M, Iwai T,
Sasai Y, Kaneko H, Taniwaki M, Kashima K, Fujii H, Abe T and Misawa
S: Tandem duplications of the FLT3 receptor gene are associated
with leukemic transformation of myelodysplasia. Leukemia.
11:1442–1446. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yokota S, Kiyoi H, Nakao M, Iwai T, Misawa
S, Okuda T, Sonoda Y, Abe T, Kahsima K, Matsuo Y and Naoe T:
Internal tandem duplication of the FLT3 gene is preferentially seen
in acute myeloid leukemia and myelodysplastic syndrome among
various hematological malignancies. A study on a large series of
patients and cell lines. Leukemia. 11:1605–1609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R,
Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C,
et al: Activating mutation of D835 within the activation loop of
FLT3 in human hematologic malignancies. Blood. 97:2434–2439. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abu-Duhier FM, Goodeve AC, Wilson GA, Care
RS, Peake IR and Reilly JT: Identification of novel FLT-3 Asp835
mutations in adult acute myeloid leukaemia. Br J Haematol.
113:983–988. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayakawa F, Towatari M, Kiyoi H, Tanimoto
M, Kitamura T, Saito H and Naoe T: Tandem-duplicated Flt3
constitutively activates STAT5 and MAP kinase and introduces
autonomous cell growth in IL-3-dependent cell lines. Oncogene.
19:624–631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kiyoi H, Ohno R, Ueda R, Saito H and Naoe
T: Mechanism of constitutive activation of FLT3 with internal
tandem duplication in the juxtamembrane domain. Oncogene.
21:2555–2563. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizuki M, Fenski R, Halfter H, Matsumura
I, Schmidt R, Müller C, Grüning W, Kratz-Albers K, Serve S, Steur
C, et al: Flt3 mutations from patients with acute myeloid leukemia
induce transformation of 32D cells mediated by the Ras and STAT5
pathways. Blood. 96:3907–3914. 2000.PubMed/NCBI
|
|
21
|
Spiekermann K, Bagrintseva K, Schoch C,
Haferlach T, Hiddemann W and Schnittger S: A new and recurrent
activating length mutation in exon 20 of the FLT3 gene in acute
myeloid leukemia. Blood. 100:3423–3425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Williams AB, Nguyen B, Li L, Brown P,
Levis M, Leahy D and Small D: Mutations of FLT3/ITD confer
resistance to multiple tyrosine kinase inhibitors. Leukemia.
27:48–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim Y, Lee GD, Park J, Yoon JH, Kim HJ,
Min WS and Kim M: Quantitative fragment analysis of FLT3-ITD
efficiently identifying poor prognostic group with high mutant
allele burden or long ITD length. Blood Cancer J. 5:e3362015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stirewalt DL, Kopecky KJ, Meshinchi S,
Engel JH, Pogosova-Agadjanyan EL, Linsley J, Slovak ML, Willman CL
and Radich JP: Size of FLT3 internal tandem duplication has
prognostic significance in patients with acute myeloid leukemia.
Blood. 107:3724–3726. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Griffith M, Griffith OL, Krysiak K,
Skidmore ZL, Christopher MJ, Klco JM, Ramu A, Lamprecht TL, Wagner
AH, Campbell KM, et al: Comprehensive genomic analysis reveals FLT3
activation and a therapeutic strategy for a patient with relapsed
adult B-lymphoblastic leukemia. Exp Hematol. 44:603–613. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lilakos K, Viniou NA, Mavrogianni D,
Vassilakopoulos TP, Dimopoulou MN, Plata E, Angelopoulou MK,
Variami E, Stavrogianni N, Liapi D, et al: FLT3 overexpression in
acute promyelocytic leukemia patients without detectable FLT3-ITD
or codon 835–836 mutations: A pilot study. Anticancer Res.
26:1201–1207. 2006.PubMed/NCBI
|
|
27
|
Staffas A, Arabanian LS, Wei SY, Jansson
A, Ståhlman S, Johansson P, Fogelstrand L, Cammenga J, Kuchenbauer
F and Palmqvist L: Upregulation of Flt3 is a passive event in
Hoxa9/Meis1-induced acute myeloid leukemia in mice. Oncogene.
36:1516–1524. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Spiekermann K, Bagrintseva K, Schwab R,
Schmieja K and Hiddemann W: Overexpression and constitutive
activation of FLT3 induces STAT5 activation in primary acute
myeloid leukemia blast cells. Clin Cancer Res. 9:2140–2150.
2003.PubMed/NCBI
|
|
29
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barretina J, Caponigro G, Stransky N,
Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV,
Sonkin D, et al: The cancer cell line encyclopedia enables
predictive modelling of anticancer drug sensitivity. Nature.
483:603–607. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Uhlén M, Fagerberg L, Hallström BM,
Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C,
Sjöstedt E, Asplund A, et al: Proteomics. Tissue-based map of the
human proteome. Science. 347:12604192015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu BH, Chen H, Cai CM, Fang JZ, Wu CC,
Huang LY, Wang L and Han ZG: Epigenetic silencing of JMJD5 promotes
the proliferation of hepatocellular carcinoma cells by
down-regulating the transcription of CDKN1A. Oncotarget.
7:6847–6863. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Troyanskaya O, Cantor M, Sherlock G, Brown
P, Hastie T, Tibshirani R, Botstein D and Altman RB: Missing value
estimation methods for DNA microarrays. Bioinformatics. 17:520–525.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fujita A, Sato JR, Rodrigues Lde O,
Ferreira CE and Sogayar MC: Evaluating different methods of
microarray data normalization. BMC Bioinformatics. 7:4692006.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:pp. 15545–15550. 2005;
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1alpha-responsive genes
involved in oxidative phosphorylation are coordinately
downregulated in human diabetes. Nat Genet. 34:267–273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liao C, Wang XY, Wei HQ, Li SQ, Merghoub
T, Pandolfi PP and Wolgemuth DJ: Altered myelopoiesis and the
development of acute myeloid leukemia in transgenic mice
overexpressing cyclin A1. Proc Natl Acad Sci USA. 98:pp. 6853–6858.
2001; View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang H, Lindsey S, Konieczna I, Bei L,
Horvath E, Huang W, Saberwal G and Eklund EA: Constitutively active
SHP2 cooperates with HoxA10 overexpression to induce acute myeloid
leukemia. J Biol Chem. 284:2549–2567. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garcia-Cuellar MP, Steger J, Füller E,
Hetzner K and Slany RK: Pbx3 and Meis1 cooperate through multiple
mechanisms to support Hox-induced murine leukemia. Haematologica.
100:905–913. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen WL, Wang YY, Zhao A, Xia L, Xie G, Su
M, Zhao L, Liu J, Qu C, Wei R, et al: Enhanced fructose utilization
mediated by SLC2A5 is a unique metabolic feature of acute myeloid
leukemia with therapeutic potential. Cancer Cell. 30:779–791. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hodson DJ, Janas ML, Galloway A, Bell SE,
Andrews S, Li CM, Pannell R, Siebel CW, MacDonald HR, De
Keersmaecker K, et al: Deletion of the RNA-binding proteins ZFP36L1
and ZFP36L2 leads to perturbed thymic development and T
lymphoblastic leukemia. Nat Immunol. 11:717–724. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ozeki K, Kiyoi H, Hirose Y, Iwai M,
Ninomiya M, Kodera Y, Miyawaki S, Kuriyama K, Shimazaki C, Akiyama
H, et al: Biologic and clinical significance of the FLT3 transcript
level in acute myeloid leukemia. Blood. 103:1901–1908. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Döhner H, Estey EH, Amadori S, Appelbaum
FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson
RA, et al: Diagnosis and management of acute myeloid leukemia in
adults: Recommendations from an international expert panel, on
behalf of the European Leukemia Net. Blood. 115:453–474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Estey EH: Acute myeloid leukemia: 2012
update on diagnosis, risk stratification, and management. Am J
Hematol. 87:89–99. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Patel JP, Gönen M, Figueroa ME, Fernandez
H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S,
Aminova O, et al: Prognostic relevance of integrated genetic
profiling in acute myeloid leukemia. N Engl J Med. 366:1079–1089.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Puissant A, Fenouille N, Alexe G, Pikman
Y, Bassil CF, Mehta S, Du J, Kazi JU, Luciano F, Rönnstrand L, et
al: SYK is a critical regulator of FLT3 in acute myeloid leukemia.
Cancer Cell. 25:226–242. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu S, Yin L, Stroopinsky D, Rajabi H,
Puissant A, Stegmaier K, Avigan D, Kharbanda S, Kufe D and Stone R:
MUC1-C oncoprotein promotes FLT3 receptor activation in acute
myeloid leukemia cells. Blood. 123:734–742. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Moharram SA, Chougule RA, Su X, Li T, Sun
J, Zhao H, Rönnstrand L and Kazi JU: Src-like adaptor protein 2
(SLAP2) binds to and inhibits FLT3 signaling. Oncotarget.
7:57770–57782. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kazi JU and Rönnstrand L: Suppressor of
cytokine signaling 2 (SOCS2) associates with FLT3 and negatively
regulates downstream signaling. Mol Oncol. 7:693–703. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Volpe G, Walton DS, Del Pozzo W, Garcia P,
Dassé E, O'Neill LP, Griffiths M, Frampton J and Dumon S: C/EBPα
and MYB regulate FLT3 expression in AML. Leukemia. 27:1487–1496.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Holm C, Ora I, Brunhoff C, Anagnostaki L,
Landberg G and Persson JL: Cyclin A1 expression and associations
with disease characteristics in childhood acute lymphoblastic
leukemia. Leuk Res. 30:254–261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Palmqvist L, Argiropoulos B, Pineault N,
Abramovich C, Sly LM, Krystal G, Wan A and Humphries RK: The Flt3
receptor tyrosine kinase collaborates with NUP98-HOX fusions in
acute myeloid leukemia. Blood. 108:1030–1036. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li Z, Zhang Z, Li Y, Arnovitz S, Chen P,
Huang H, Jiang X, Hong GM, Kunjamma RB, Ren H, et al: PBX3 is an
important cofactor of HOXA9 in leukemogenesis. Blood.
121:1422–1431. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Li Z, Chen P, Su R, Hu C, Li Y, Elkahloun
AG, Zuo Z, Gurbuxani S, Arnovitz S, Weng H, et al: PBX3 and MEIS1
cooperate in hematopoietic cells to drive acute myeloid leukemias
characterized by a core transcriptome of the MLL-rearranged
disease. Cancer Res. 76:619–629. 2016. View Article : Google Scholar : PubMed/NCBI
|