Introduction
Chronic pancreatitis (CP) is a painful inflammatory
disease, characterized by progressive destruction of the pancreatic
gland and severe abdominal pain (1). Approximately 85–90% CP patients
suffer from abdominal pain (2).
Pain in CP has been associated with malnutrition, narcotic
addiction, and physical and emotional disability, which leads to
major socioeconomic problems (3).
However, the currently available therapies for CP pain remain
inadequate and the underlying mechanisms remain to be completely
elucidated. Previous reports have demonstrated that CP-induced pain
exhibits numerous characteristics similar to neuropathic pain,
especially the alterations located in the central nervous system
(CNS) (4). Epigenetic modulations
of gene expression have been indicated to be involved in the
development of chronic pain (5).
Epigenetic alterations are required for long-lasting neuronal
plasticity that is essential for the development of chronic pain
state modifications (6,7). Epigenetic modifications regulate the
compaction of chromatin and include a variety of facets;
significant epigenetic control is achieved via histone acetylation.
Histone acetylation modification is dynamic and reversible, and is
regulated by histone acetyltransferases (HATs) and histone
deacetylases (HDACs) (8).
Typically, HATs acetylate the histones to produce an open chromatin
conformation, thus favoring gene expression, while HDACs
deacetylate the DNA and result in a closed chromatin conformation
and ultimately gene repression (9). Studies on HDACs inhibitors have
demonstrated obvious analgesic effect on nociceptive responses of
rodents, either delivered systemically or intrathecally (10–13).
However, little is known on how this mechanism operates and which
target genes are involved (14).
Furthermore, whether selective interruption of HDAC activity on CP
could alleviate allodynia remains to be investigated.
In the nervous system, activation of µ-opiod
receptor (MOR) leads to neuronal inhibition and causes an
endogenous analgesia effect (15).
The expression level of MOR in the mouse brain correlates with
alterations in histone modifications (16). However, limited studies have
analyzed the epigenetic processes that contribute to gene
repression and activation in pain states. Furthermore, a study
reported that MOR is mainly negatively regulated by the c-Jun
NH2-terminal kinase (JNK) signaling pathway (17). Our previous study demonstrated that
HDAC2 activity was significantly upregulated in the thoracic spinal
cord, based on a rat CP model induced by intrapancreatic infusion
of trinitrobenzene sulfonic acid (TNBS) (18), indicating that epigenetic
regulation mechanisms are involved in chronic pain induced by
CP.
The present study aimed to investigate whether
upregulation of HDAC2 affects MOR expression, and thus has an
impact on CP allodynia. It was hypothesized that the elevated HDAC2
expression suppressed MOR activation via JNK signaling pathways,
and aggravated CP pain. To test this hypothesis, AR-42 was used as
a selective HDAC2 inhibitor (19),
and the underlying mechanisms of CP pain were investigated.
Materials and methods
Animals
The present study was approved by the Animal Use and
Care Committee for Research and Education of the Fourth Military
Medical University (Xi'an, China), following the ethical guidelines
on investigating experimental pain in conscious animals. 54 young
adult male Sprague-Dawley rats (age, 10–12 weeks; weight, 180–220
g) were purchased from the Laboratory Animals Center (Fourth
Military Medical University, Xi'an, China) and caged in a
temperature-controlled environment at 22–25°C and 55±5% relative
humidity with a 12-h light/dark cycle. Free access to water and
food was available until 12 h before pancreatitis induction.
Minimum animals were used for consistent effects.
Induction of pancreatitis and pain
behavioral test
All rats were randomly divided into three main
groups: TNBS (n=30), sham (n=18) and controls (n=6). Rats in the
TNBS and sham group were further divided for drug injection:
TNBS-HDAC inhibitor (i)/saline and sham-HDACi/saline groups
(n=6/group). In order to study the time course of HDAC2 and MOR
changes, 6 rats in the TNBS group were sacrificed at 1, 3 and 5
weeks each following TNBS infusion, in addition to 6 rats in the
control and 6 rats in the sham group. All rats were sacrificed
following anesthetization with pentobarbital (cat. no. 1507,002;
Sigma-Aldrich; Merck KGaA; Darmstadt, Germany; 60 mg/kg,
intraperitoneal injection) for further experiments 5 weeks after
surgery. The protocol has been published in our previous study
(18) with certain improvement for
minimizing surgical injury. All the procedures in sham group were
identical to the TNBS group, except the same volume of saline
infusion. Mechanical allodynia was measured with von Frey filaments
(Stoelting Co., Wood Dale, IL, USA) and performed fully randomized
and blinded. The protocol was performed according to our previous
study (18).
Intrathecal operation and drug
administration
Intrathecal drug administration procedure was
performed as previous described (18). The HDAC2-selective inhibitor AR-42
(AdooQ Bioscience, Irvine, CA, Canada) was used for the testing
(19). The treatment group
received an intrathecal injection of AR-42 (30 nmol) at each time
point (0, 1, 3 and 5 weeks), while the same volume of saline was
administered to the control group. Drug administration was
performed under anesthetizing with isoflurane using a Vaporizer
(Harvard Apparatus, Holliston, MA, USA), through an intrathecal
catheter controlled by a micro-injection pump.
Immunofluorescent staining
analysis
Rats were perfused through the ascending aorta with
100 ml 0.9% saline after deeply anesthetizing with pentobarbital
(60 mg/kg, intraperitoneally), following administration of 500 ml
0.1 M phosphate buffer (PB, pH 7.4) containing 4% paraformaldehyde
and 2% picric acid. The thoracic spinal segments T10-T12 were
harvested and post-fixed with the same fixative for 4 h, then
cryosectioned at 4°C for 24 h in 0.1 M PB containing 30% sucrose.
Samples were transversely cut (25-µm thick) in a cryostat, then
washed in PBS (0.01 M, pH 7.3) for 10 min three times. Sections
were then blocked with 2% goat serum (EMD Millipore, Billerica, MA,
USA) in 0.01 M PBS containing 0.3% Triton X-100 for 1 h at room
temperature. These sections were incubated at 4°C overnight with
the following primary antibodies: rabbit anti-HDAC2 (cat. no.
sc-437285; 1:1,000; Santa-Cruz Biotechnology, Inc., Dallas, TX,
USA) guinea pig anti-MOR (cat. no. AB5509; 1:200; EMD Millipore),
mouse anti-neuronal-specific nuclear protein (NeuN; cat. no.
MAB377; 1:500; EMD Millipore), mouse anti-glial fibrillary acidic
protein (GFAP; cat. no. NE1015; 1:500; EMD Millipore) and mouse
anti-integrin α-M (CD11b) antibody (OX-42; cat. no. CBL1512; 1:500;
EMD Millipore). The sections were then washed three times with PBS
(10 min each) and then incubated for 2 h at room temperature with
the corresponding secondary antibody: Alexa 488-conjugated donkey
anti-rabbit IgG (cat. no. R37118; 1:800; Molecular Probes; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) and Alexa 594-conjugated
donkey anti-guinea pig IgG (cat. no. A-11076; 1:200; Molecular
Probes; Thermo Fisher Scientific, Inc.). Images were obtained under
a confocal laser microscope (FV1000; Olympus Corporation, Tokyo,
Japan), and digital pictures were captured with the Fluoview 1000
imaging system (Olympus Corporation). A total of 12 nonadjacent
sections were randomly selected for scanning. The z-separation was
4.6 µm under a ×20 objective magnification, and was 1.0 µm under a
×60 objective magnification. For semiquantification, the
fluorescent brightness value of immunoreactivities was detected on
the same areas of the thoracic spinal dorsal horn using software
(Fluoview; version 1.5.0.14; Olympus Corporation, Tokyo, Japan)
under an Olympus IX-70 confocal microscope (Olympus
Corporation).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Rat thoracic spinal cord samples were harvested as
described above. RNA extraction was performed using TRIzol (Gibco;
Thermo Fisher Scientific, Inc.), and oligo(dT) primer and
SuperScript II reverse transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.) were used in cDNA RT. The mRNA expression levels
of the pro-inflammatory cytokines interleukin (IL)-6, IL-1β and
tumor necrosis factor (TNF)-α were assessed by RT-qPCR. The
protocol was performed according to a previous report (18): 3 min at 9°C, followed by 45 cycles
of 10 sec at 95°C for denaturation, and 45 sec at 60°C for
annealing and extension. All experiments were repeated twice, and
PCR reactions were triplicate in each test. Target cDNA quantities
were evaluated from the quantitation cycle number (Cq) (20) using Sequence Detection System
software (version 2.4.1; Applied Biosystems; Thermo Fisher
Scientific, Inc.). Primer sequences are provided in Table I. GAPDH served as an endogenous
internal standard control.
 | Table I.Primers sequence for the rat genes
characterized in this experiment. |
Table I.
Primers sequence for the rat genes
characterized in this experiment.
| Gene | Sequence |
|---|
| TNF-α | F:
5′-TGATCGGTCCCAACAAGGA-3′ |
|
| R:
5′-TGCTTGGTGGTTTGCTACGA-3′ |
| IL-1β | F:
5′-TGCTGATGTACCAGTTGGGG-3′ |
|
| R:
5′-CTCCATGAGCTTTGTACAAG-3′ |
| IL-6 | F:
5′-GCCCTTCAGGAACAGCTATG-3′ |
|
| R:
5′-CAGAATTGCCATTGCACAAC-3 |
| GAPDH | F:
5′-CCCCCAATGTATCCGTTGTG-3′ |
|
| R:
5′-TAGCCCAGGATGCCCTTTAGT-3′ |
Western blotting
Rats were sacrificed and T10-T12 spinal cord
sections were rapidly harvested and frozen on dry ice. Samples were
quickly micro-dissected and homogenized using a hand-held pestle
with ice-cold SDS sample lysis buffer (Roche Diagnostics, Basel,
Switzerland; 10 ml/mg tissue), containing a cocktail of proteinase
inhibitors. The homogenates were centrifuged at 10,000 × g for 10
min at 4°C. Subsequent to the protein concentration being measured
using a bicinchoninic acid protein assay kit (cat. no. 23225;
Pierce; Thermo Fisher Scientific, Inc.), the homegenates were
heated at 100°C for 5 min. Proteins (50 µg) were separated using
SDS-PAGE on a 10% gel with standard Laemmli solutions (Bio-Rad
Laboratories, Hercules, CA, USA) and subsequently transferred onto
a polyvinylidene difluoride membrane (Immobilon-P, EMD Millipore).
The membranes were washed with Tris-buffered saline with 0.02%
Tween (TBST) and blocked with 5% non-fat dry milk for 1 h at room
temperature, and incubated overnight with gentle agitation with
rabbit anti-JNK (cat. no. sc-7345; 1:1,000; Santa-Cruz
Biotechnology, Inc.) and anti-β-actin (cat. no. A0483; 1:1,000;
Sigma-Aldrich; Merck KGaA) primary antibodies. Subsequently,
membranes were incubated overnight at 4°C with a goat anti-rabbit
horseradish peroxidase-conjugated secondary antibody (cat. no.
RPN4301; 1:10,000; GE Healthcare, Chicago, IL, USA). Between each
of these steps, the membranes were rinsed with TBST. All reactions
were detected through the enhanced chemiluminescence (ECL)
detection method (GE Healthcare). The densities of protein blots
were analyzed using Labworks Software (version 4.5; UVP, Inc.,
Upland CA, USA) and normalized against the values of β-actin.
Statistical analysis
After the images were captured, the optical density
of the same areas of the superficial dorsal horn (lamina I and II)
of the five spinal sections were calculated. All data are presented
as mean ± standard error and were analyzed by one-way analysis of
variance. Multiple comparisons between the groups were performed
using the Student-Newman-Keuls method. All statistical analyses
were performed using SPSS version 18.0 software (SPSS Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Upregulated HDAC2 expression is
associated with suppressed MOR activity in the thoracic spinal
dorsal horn after CP induction by TNBS
The rats were sacrificed at different time points to
investigate the expression changes of HDAC2 and MOR. Double
immunofluorescent staining was performed to reveal the association
between HDAC2 and MOR in the thoracic spinal cord. Colocalization
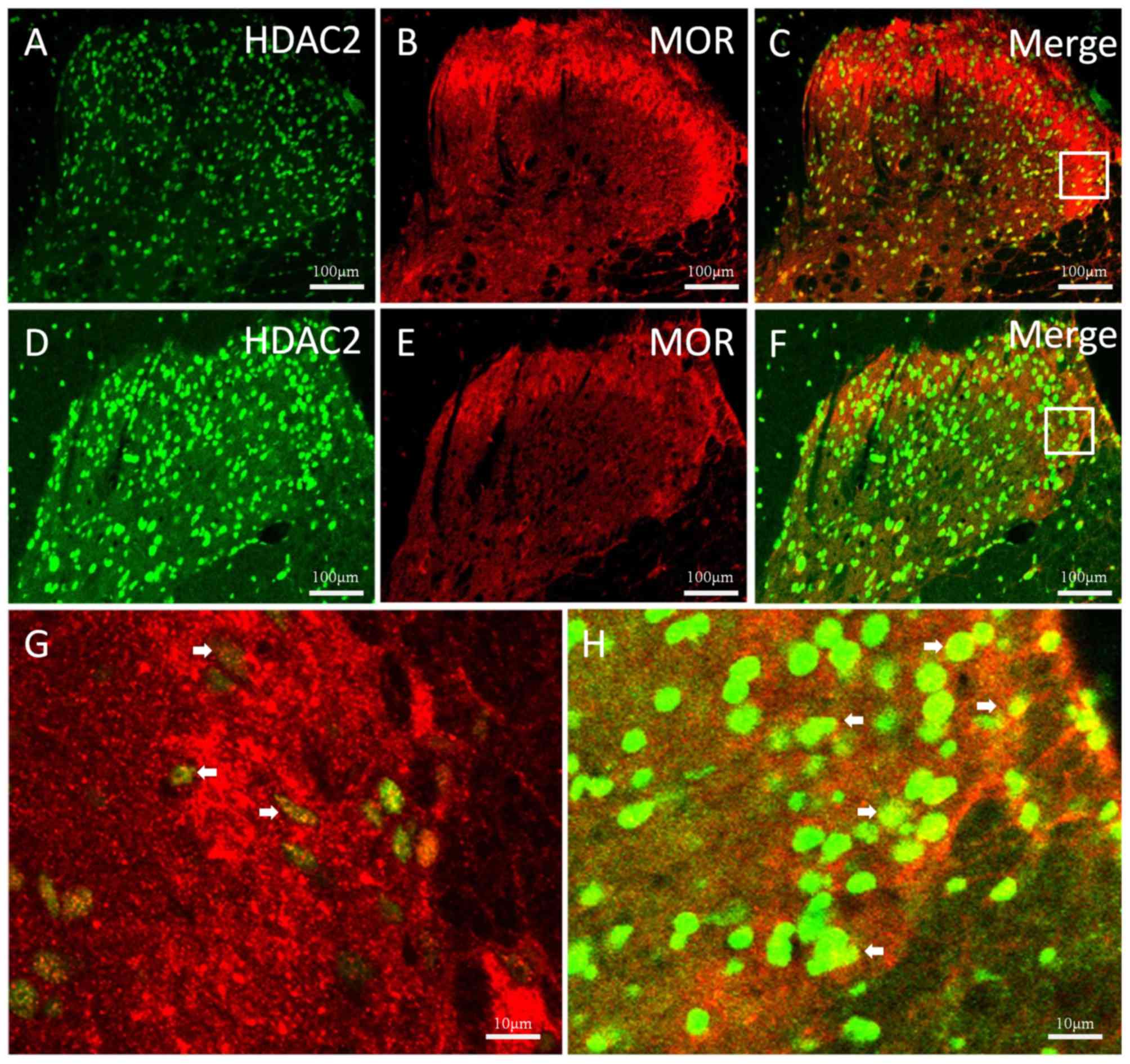
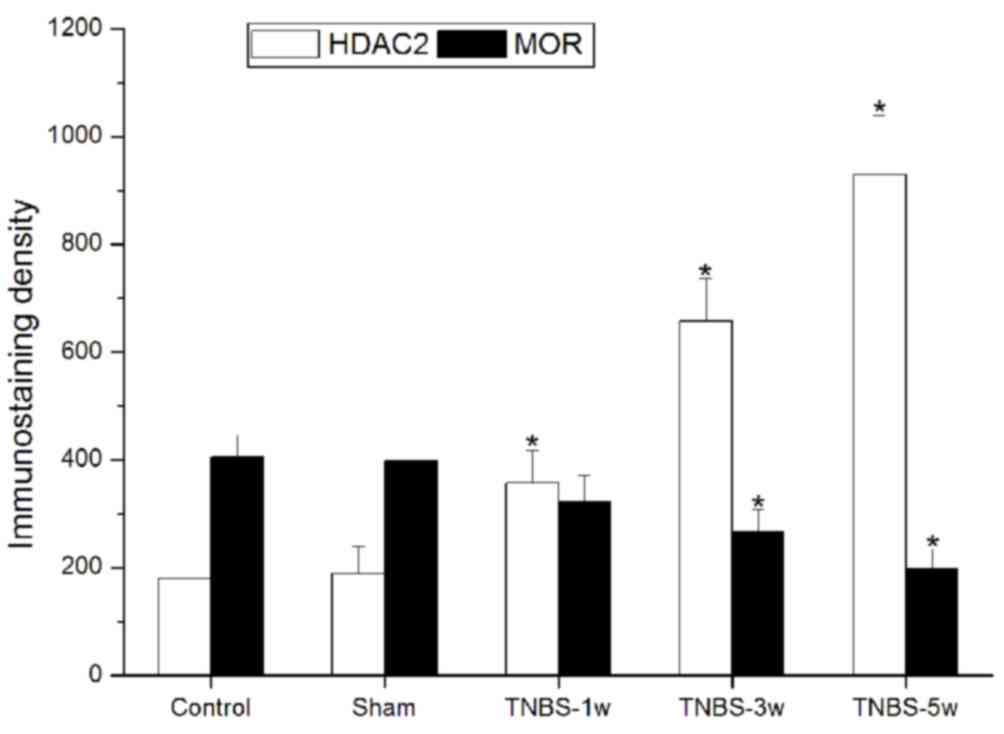
of HDAC2 and MOR were observed both in the control (Fig. 1A-C) and TNBS-treated (Fig. 1D-H) groups. In the TNBS-treated
group, HDAC2 expression levels were significantly elevated and
persistent from 1 to 5 weeks. Suppressed MOR activity was
simultaneously observed and maintained at a low level.
Immunostaining density statistical analysis was used to confirm
these changes (P<0.05 vs. control group; Fig. 2).
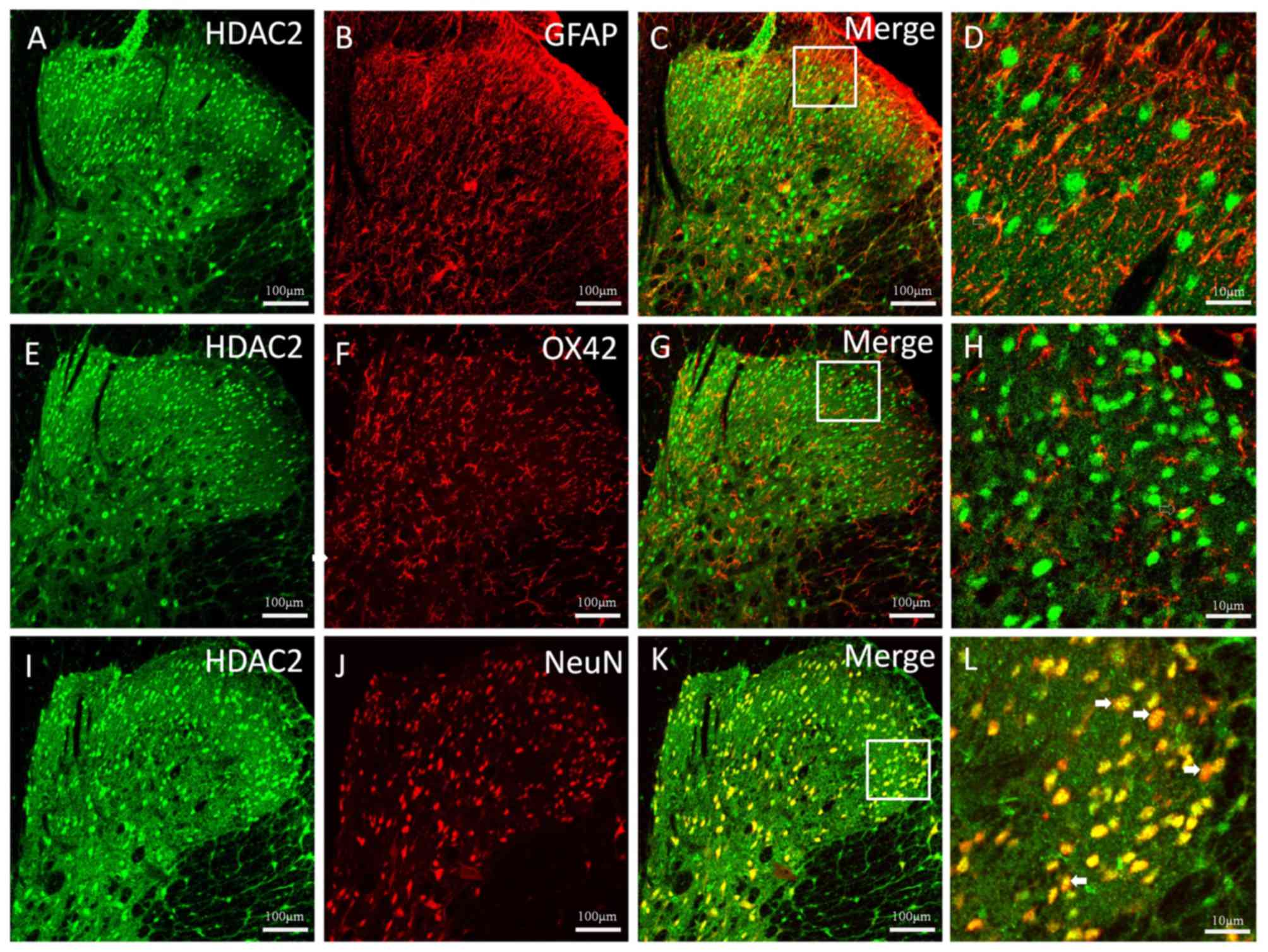
The cellular localization of HDAC2 in the thoracic
spinal dorsal horn 5 weeks after CP induction was observed
(Fig. 3). Double immunofluorescent
staining of HDAC2 with the astrocytic specific marker GFAP
(Fig. 3A-C), the microglial marker
OX42 (Fig. 3E-G) and the neuronal
marker NeuN (Fig. 3I-K) was
conducted. The results revealed a clear colocalization between
HDAC2 and NeuN (Fig. 3 K and L);
however, HDAC2 rarely colocalized with GFAP or OX42 (Fig. 3D and H).
Intrathecal infusion of HDACi
significantly attenuates CP-induced mechanical allodynia
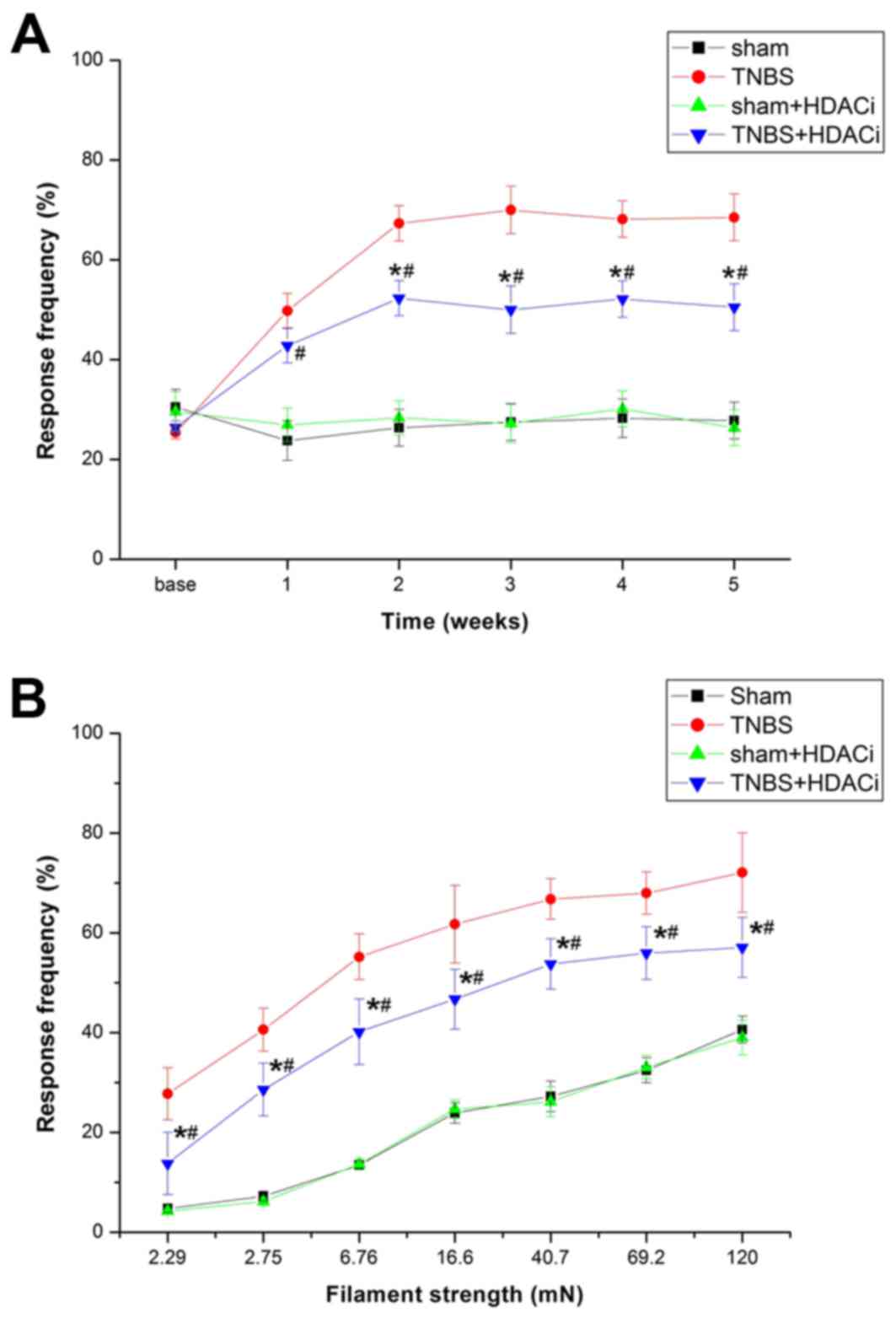
To assess whether upregulated HDAC2 contributes to
CP-induced pain, AR-42 was used to selectively suppress HDAC2, and
thus investigate the nociceptive behavioral consequences and
cellular and molecular alterations. The results demonstrated that
AR-42 significantly attenuated mechanical allodynia (P<0.05 vs.
TNBS; Fig. 4A). However, AR-42
exerted no influence on the response frequency (RF) of sham
controlled rats (P<0.05 vs. sham; Fig. 4A). Different stimulations on RFs
were tested, and it was observed that RFs of rats were
significantly reduced after AR-42 treatment compared with
non-treated CP model rats (Fig.
4B). Furthermore, rats treated with only AR-42 did not exhibit
differences in RF compared with sham rats. Therefore, AR-42 may
attenuate CP-induced chronic pain without interfering with the
basic pain behavior.
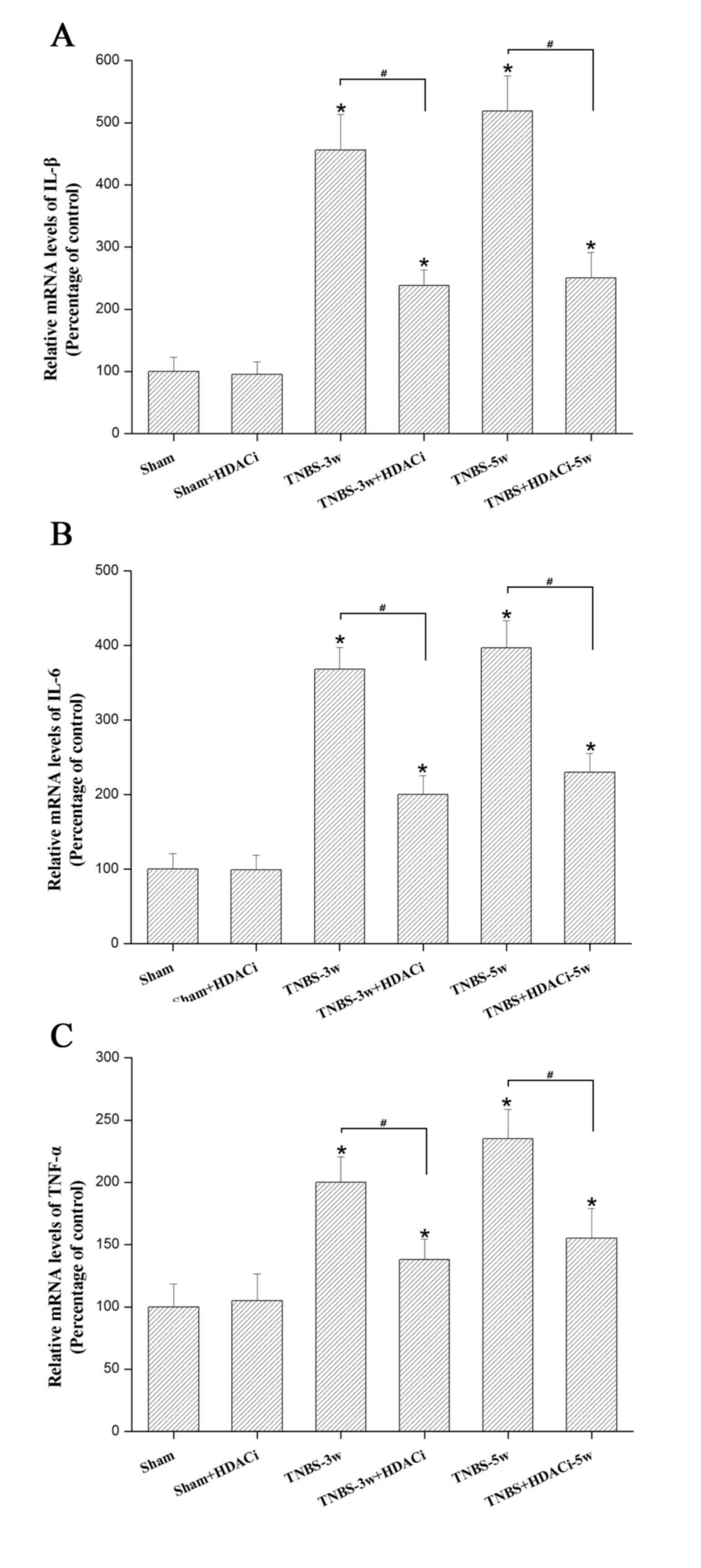
HDAC2 facilitates the release of the
pro-inflammatory cytokines IL1-β, IL-6 and TNF-α
Our previous study demonstrated that
pro-inflammatory cytokines such as IL-1β IL-6, and TNF-α in the
spinal dorsal horn increase the status of CP (18). Using RT-qPCR, it was observed that
the mRNA expression levels of these cytokines were significantly
reduced by AR-42 at 3 and 5 weeks in CP rats (P<0.05 vs. TNBS
control; Fig. 5), though this
effect was not completely reversed (P<0.05 vs. sham; Fig. 5). In addition, AR-42 administration
did not significantly affect the sham group (P<0.05 vs. sham
control; Fig. 5). These data
indicated that HDAC2 may promote the release of pro-inflammatory
mediators under chronic CP allodynia.
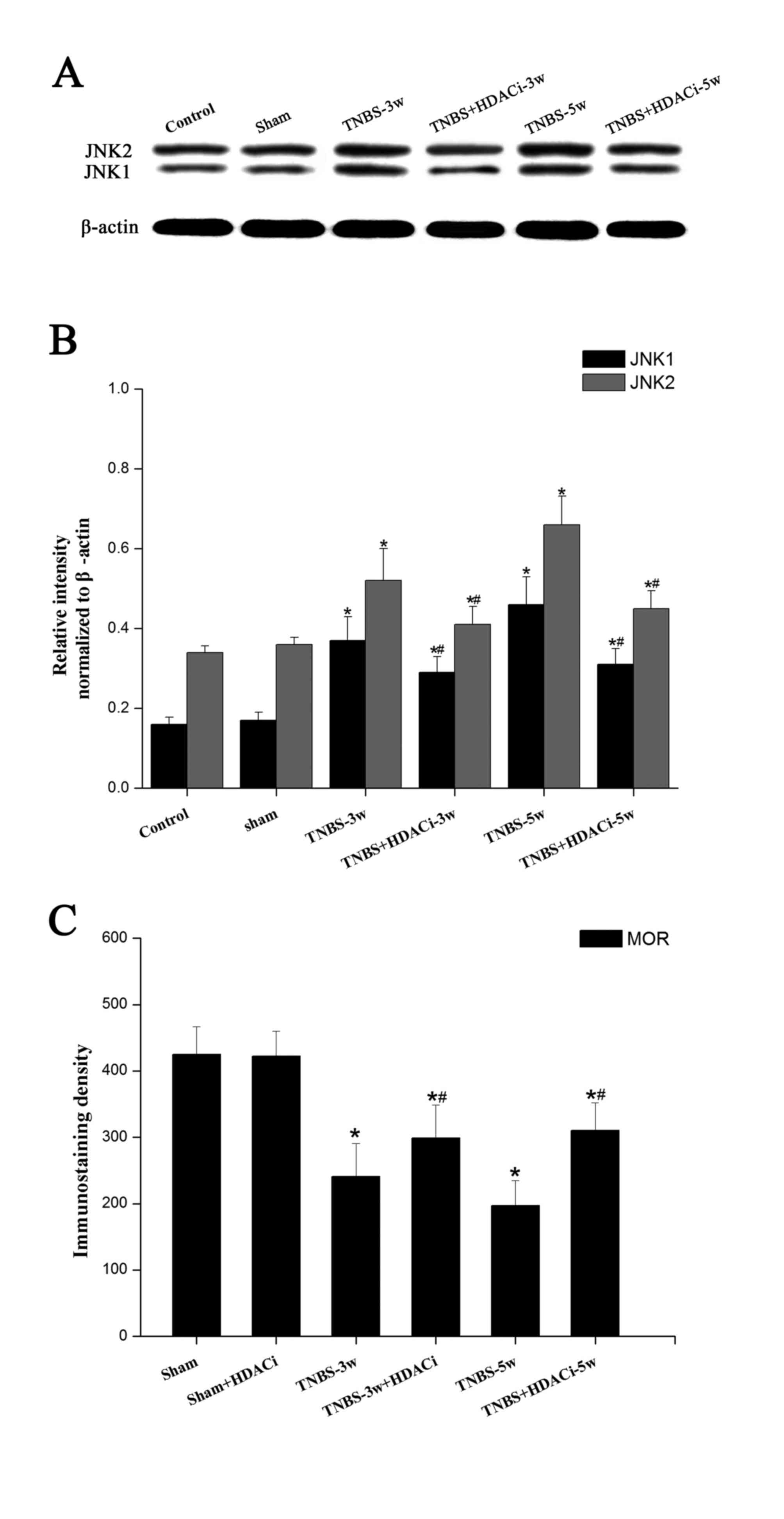
Effects of AR-42 on JNK signaling
pathways in CP allodynia
As MOR expression is mainly negatively regulated by
the JNK signaling pathway (17),
the present study selected to further detected the expression
levels of this intracellular kinase. Western blotting data
indicated that JNK has two subtypes, JNK1 and JNK2, and that the
protein expression levels of both were significantly increased
after TNBS infusion (P<0.05 vs. control; Fig. 6A and B). However, HDACi AR-42
administration reversed the overexpression of JNK1/2 in CP rats
(P<0.05, vs. TNBS; Fig. 6A-B),
although the expression level remained higher than the control and
sham groups. As presented in Fig.
6C, following a TNBS-induced reduction in MOR immunostaining
density, HDACi AR-42 significantly rescued MOR activity in CP rats.
No significant differences were observed between the control and
sham group. In addition, no significant differences were observed
on basic MOR expression levels following AR-42 treatment (Fig. 6C). These results suggested that
HDAC2 may suppress MOR activity via regulating JNK signaling
pathways.
Discussion
The present study demonstrated that HDAC2 was
upregulated and MOR activity was reduced in the spinal cord after
CP induction, and HDAC2 was primarily upregulated in neurons rather
than in glial cells in the spinal dorsal horn. As MOR is mainly
expressed in neurons (16), it was
hypothesized that the contrary expression between HDAC2 and MOR
during CP might be involved in CP-induced allodynia. In the present
study, it was demonstrated that HDAC2 increased in the spinal
dorsal horn after CP induction. This was also confirmed by
immunostaining analysis. Furthermore, the molecular mechanisms
underlying the inflammatory process in the CNS following CP
induction were investigated. Intrathecal injection of AR-42
significantly attenuated CP-induced mechanical allodynia and
pro-inflammatory cytokine expression levels, following suppressed
JNK expression and rescued MOR activity. Though these effects were
not completely reversed, these results somewhat elucidated the
underlying mechanisms supporting that HDAC2 serves important roles
in suppressing MOR activity, thus inducing endogenous analgesia
that is disrupted after CP induction.
In chronic pain, studies on HDACs and their
inhibitors are only just beginning to emerge (21). HDACi have attracted increasing
interest in recent years due to their potential usage in enhancing
neuroplasticity and synaptic activity (22), and reducing neuronal damage
(23,24) and neuroinflammation (25). Previous studies have demonstrated
that HDAC inhibitors can alleviate inflammatory pain and attenuate
the development of hypersensitivity in models of neuropathic pain.
Thus, HDACi could be a valid alternative to traditional therapeutic
agents by eliminating the side effect of pain (9). However, a clear demonstration that
selective interference with HDAC activity can affect CP-induced
chronic pain remains lacking. After the HDAC2 selective inhibitor
AR-42 was intrathecally administrated, mechanical allodynia was
significantly in TNBS treated rats, though not completely. In
addition, it was confirmed that AR-42 could attenuate CP-induced
chronic pain without interfering with basic pain behavior. This
suggested the potential role of HDAC2 in CP chronic pain
induction.
It has been reported that the overexpression of
pro-inflammatory cytokines during CP induction, such as TNF-, IL-1β
and IL-6 may promote the allodynia of CP rats (18,26),
and these mediators may induce the activation of JNK and
extracellular signal-regulated kinase (ERK) signaling pathways,
which serve important roles in the maintenance of chronic pain.
IL-1β is the most important inflammatory mediator that causes
pancreatic islet dysfunction and destruction, which may promote
activation of JNK (27–29); previous studies have also
demonstrated positive effect of TNF-α and IL-6 on JNK activation
(30–32). MOR expression is mainly regulated
by signaling pathways such as JNK (17). However, ERK exhibits no
significantly effect on MOR expression (33,34).
Further investigation into the JNK signaling pathway may help
reveal the underlying mechanisms involved in CP pain induction. The
results of the present study demonstrated that the mRNA expression
levels of these cytokines were significantly reduced by AR-42 at 3
and 5 weeks after CP induction, indicating that HDAC2 may promote
the release of pro-inflammatory mediators, maintaining CP
allodynia. Though this effect was not completely reversed by AR-42,
no significant differences were induced by AR-42 in the sham group.
These data are consistent with previous studies that HDACi
possesses promising anti-inflammatory activities (35,36).
Western blot analysis demonstrated that JNK
expression levels were upregulated after CP induction, and that
inhibition of HDAC2 with AR-42 could significantly reverse JNK1/2
overexpression. Nevertheless, MOR activity was significantly
rescued by AR-42 treatment. Furthermore, AR-42 did not effect on
basic MOR activity. Additionally, the cellular localization of
HDAC2 was observed in the thoracic spinal dorsal horn, which
primarily localized to the cell bodies of neurons and rarely on
glial cells, indicating that neuroglia may not directly express
HDAC2. These results indicated that HDAC2 may be important in
regulating neuroactivity, and is involved in MOR deactivation,
causing central sensitization and persistent pain. The underlying
mechanisms may facilitate the release of inflammatory cytokines,
thus activating JNK signaling pathways, and finally suppressing MOR
activity.
In conclusion, pain induced by CP can be as limiting
as the disease itself (37), and
requires medical treatment; however, treatment options are limited.
Previous studies on humans and animal experimental models have
implicated several mechanisms involved in pain, including
neuroimmune alterations (38,39).
To the best of our knowledge, the present study investigated
epigenetic regulation in CP-induced allodynia for the first time,
and observed increased HDAC2 activity in the spinal cord and
behavioral allodynia after TNBS treatment. Therefore, HDAC2 may be
facilitating the release of inflammatory cytokines, activating the
JNK signaling pathway, and suppressing MOR activity, causing
sensitization and pain induction. These results may facilitate the
development of novel therapeutics for patients suffering from CP
pain. Further studies are required to establish drugs with maximum
efficacy and fewer side effects.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81400906, 81370928,
81620108008, 31100861 and 2012YQ0302609) and the China Postdoctoral
Science Foundation (grant no. 2012M521864).
References
|
1
|
Keefe MD, Wang H, De La OJP, Khan A, Firpo
MA and Murtaugh LC: β-catenin is selectively required for the
expansion and regeneration of mature pancreatic acinar cells in
mice. Dis Model Mech. 5:503–514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Braganza JM, Lee SH, McCloy RF and McMahon
MJ: Chronic pancreatitis. Lancet. 377:1184–1197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Olesen SS, Juel J, Graversen C, Kolesnikov
Y, Wilder-Smith OH and Drewes AM: Pharmacological pain management
in chronic pancreatitis. World J Gastroenterol. 19:7292–7301. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Drewes AM, Krarup AL, Detlefsen S,
Malmstrøm ML, Dimcevski G and Funch-Jensen P: Pain in chronic
pancreatitis: The role of neuropathic pain mechanisms. Gut.
57:1616–1627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Feng W, Teng R, Zhao Y, Gao J and Chu H:
Epigenetic modulation of Wnt signaling contributes to neuropathic
pain in rats. Mol Med Rep. 12:4727–4733. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gáranton SM: Targeting epigenetic
mechanisms for pain relief. Curr Opin Pharmacol. 12:35–41. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lessans S and Dorsey SG: The role for
epigenetic modifications in pain and analgesia response. Nurs Res
Pract 2013. 9614932013.
|
|
8
|
Gilbert RE, Huang Q, Thai K, Advani SL,
Lee K, Yuen DA, Connelly KA and Advani A: Histone deacetylase
inhibition attenuates diabetes-associated kidney growth: Potential
role for epigenetic modification of the epidermal growth factor
receptor. Kidney Int. 79:1312–1321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Capasso KE, Manners MT, Quershi RA, et al:
Effect of histone deacetylase inhibitor JNJ-26481585 in pain. J Mol
Neurosci. 55:570–578. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bai G, Wei D, Zou S, Ren K and Dubner R:
Inhibition of class II histone deacetylases in the spinal cord
attenuates inflammatory hyperalgesia. Mol Pain. 6:512010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chiechio S, Copani A, Zammataro M,
Battaglia G, Gereau RW IV and Nicoletti F: Transcriptional
regulation of type-2 metabotropic glutamate receptors: An
epigenetic path to novel treatments for chronic pain. Trends
Pharmacol Sci. 31:153–160. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chiechio S, Zammataro M, Morales ME,
Busceti CL, Drago F, Gereau RW IV, Copani A and Nicoletti F:
Epigenetic modulation of mGlu2 receptors by histone deacetylase
inhibitors in the treatment of inflammatory pain. Mol Pharmacol.
75:1014–1020. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Z, Cai YQ, Zou F, Bie B and Pan ZZ:
Epigenetic suppression of GAD65 expression mediates persistent
pain. Nat Med. 17:1448–1455. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harrison C: Analgesia: Unravelling
epigenetic mechanisms of chronic pain. Nat Rev Drug Discov.
10:9002011.PubMed/NCBI
|
|
15
|
Jamil MF, Subki MF, Lan TM, Majid MI and
Adenan MI: The effect of mitragynine on cAMP formation and mRNA
expression of mu-opioid receptors mediated by chronic morphine
treatment in SK-N-SH neuroblastoma cell. J Ethnopharmacol.
148:135–143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hwang CK, Kim CS, Kim DK, Law PY, Wei LN
and Loh HH: Up-regulation of the mu-opioid receptor gene is
mediated through chromatin remodeling and transcriptional factors
in differentiated neuronal cells. Mol Pharmacol. 78:58–68. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wagley Y, Hwang CK, Lin HY, Kam AF, Law
PY, Loh HH and Wei LN: Inhibition of c-Jun NH2-terminal kinase
stimulates mu opioid receptor expression via p38 MAPK-mediated
nuclear NF-κB activation in neuronal and non-neuronal cells.
Biochim Biophys Acta. 1833:1476–1488. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qian NS, Liao YH, Feng QX, Tang Y, Dou KF
and Tao KS: Spinal toll like receptor 3 is involved in chronic
pancreatitis-induced mechanical allodynia of rat. Mol Pain.
7:152011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ravillah D, Mohammed A, Qian L, Brewer M,
Zhang Y, Biddick L, Steele VE and Rao CV: Chemopreventive effects
of an HDAC2-selective inhibitor on rat colon carcinogenesis and
APCmin/+ mouse intestinal tumorigenesis. J Pharmacol Exp Ther.
348:59–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Denk F and McMahon SB: Chronic pain:
Emerging evidence for the involvement of epigenetics. Neuron.
73:435–444. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haggarty SJ and Tsai LH: Probing the role
of HDACs and mechanisms of chromatin-mediated neuroplasticity.
Neurobiol Learn Mem. 96:41–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dietz KC and Casaccia P: HDAC inhibitors
and neurodegeneration: At the edge between protection and damage.
Pharmacol Res. 62:11–17. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hashioka S, Klegeris A and McGeer PL: The
histone deacetylase inhibitor suberoylanilide hydroxamic acid
attenuates human astrocyte neurotoxicity induced by interferon-γ. J
Neuroinflammation. 9:1132012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Suh HS, Choi S, Khattar P, Choi N and Lee
SC: Histone deacetylase inhibitors suppress the expression of
inflammatory and innate immune response genes in human microglia
and astrocytes. J Neuroimmune Pharmacol. 5:521–532. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu PY, Lu CL, Wang CC, Lee IH, Hsieh JC,
Chen CC, Lee HF, Lin HC, Chang FY and Lee SD: Spinal microglia
initiate and maintain hyperalgesia in a rat model of chronic
pancreatitis. Gastroenterology. 142:165–173.e2. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng H, Xie Z, Jones WP, Wei XT, Liu Z,
Wang D, Kulp SK, Wang J, Coss CC, Chen CS, et al: Preclinical
pharmacokinetics study of R- and S-enantiomers of the histone
deacetylase inhibitor, AR-42 (NSC 731438), in rodents. AAPS J.
18:737–745. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shakespear MR, Halili MA, Irvine KM,
Fairlie DP and Sweet MJ: Histone deacetylases as regulators of
inflammation and immunity. Trends Immunol. 32:335–343. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu Y, Wang D, Zhuang Z, Jin K, Zheng L,
Yang Q and Guo K: Hypericin-mediated photodynamic therapy induces
apoptosis in K562 human leukemia cells through JNK pathway
modulation. Mol Med Rep. 12:6475–6482. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Herrmann JL, Weil BR, Abarbanell AM, Wang
Y, Poynter JA, Manukyan MC and Meldrum DR: IL-6 and TGF-α
costimulate mesenchymal stem cell vascular endothelial growth
factor production by ERK-, JNK-, and PI3K-mediated mechanisms.
Shock. 35:512–516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu ZY, Chen WC, Li YH, Li L, Zhang H, Pang
Y, Xiao ZF, Xiao HW and Xiao Y: TNF-α enhances vascular cell
adhesion molecule-1 expression in human bone marrow mesenchymal
stem cells via the NF-κB, ERK and JNK signaling pathways. Mol Med
Rep. 14:643–648. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liang R, Zhang W and Song YM: Levels of
leptin and IL-6 in lungs and blood are associated with the severity
of chronic obstructive pulmonary disease in patients and rat
models. Mol Med Rep. 7:1470–1476. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim DK, Hwang CK, Wagley Y, Law PY, Wei LN
and Loh HH: p38 mitogen-activated protein kinase and PI3-kinase are
involved in up-regulation of mu opioid receptor transcription
induced by cycloheximide. J Neurochem. 116:1077–1087. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bian JM, Wu N, Su RB and Li J:
Phosphatidylethanolamine-binding protein is not involved in
µ-opioid receptor-mediated regulation of extracellular
signal-regulated kinase. Mol Med Rep. 11:3368–3374. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang ZY and Schluesener HJ: HDAC
inhibitor MS-275 attenuates the inflammatory reaction in rat
experimental autoimmune prostatitis. Prostate. 72:90–99. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang B, West EJ, Van KC, Gurkoff GG, Zhou
J, Zhang XM, Kozikowski AP and Lyeth BG: HDAC inhibitor increases
histone H3 acetylation and reduces microglia inflammatory response
following traumatic brain injury in rats. Brain Res. 1226:181–191.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang S, Suvannasankha A, Crean CD, White
VL, Chen CS and Farag SS: The novel histone deacetylase inhibitor,
AR-42, inhibits gp130/Stat3 pathway and induces apoptosis and cell
cycle arrest in multiple myeloma cells. Int J Cancer. 129:204–213.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lucas DM, Alinari L, West DA, Davis ME,
Edwards RB, Johnson AJ, Blum KA, Hofmeister CC, Freitas MA, Parthun
MR, et al: The novel deacetylase inhibitor AR-42 demonstrates
pre-clinical activity in B-cell malignancies in vitro and in vivo.
PLoS One. 5:e109412010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ji RR: Neuroimmune interactions in itch:
Do chronic itch, chronic pain, and chronic cough share similar
mechanisms? Pulm Pharmacol Ther. 35:81–86. 2015. View Article : Google Scholar : PubMed/NCBI
|