Introduction
Clinical and preclinical studies have shown that
chronic stress has an impact on tumor growth and progression
(1–4). As important stress hormones,
glucocorticoids (GCs) influence tumor biology not only through
their systematical immunosuppressive and anti-inflammatory effects
(5,6), but also by changing the tumor
microenvironment and playing a direct role in regulating the
proliferation, metabolism, differentiation and apoptosis of tumor
cells (7). Moreover, synthetic
GCs, such as dexamethasone (Dex), have been widely used as
concomitant medications to reduce acute toxicity and alleviate side
effects, such as hyperemesis induced by chemotherapy or
radiotherapy in non-hematologic cancer therapy (7,8). GCs
are also given to patient before, during and after chemotherapy of
solid malignant tumors to protect normal tissue, e.g., bone marrow
progenitor cells, against the long-term effects of genotoxic drugs
(9). Recently, emerging evidence
has shown that GCs exert inhibitory effects on tumor apoptosis
induced by chemotherapeutics not only in established cancer cell
lines and tumor xenografts, but also in freshly isolated cells from
surgical specimens, such as breast, ovary, prostate, liver and skin
(10–13). Therefore, it is important to
consider the clinical relevance of the survival-promoting effects
of GCs when they interfere with chemotherapeutics.
The effects of GCs are mediated by the
glucocorticoid receptor (GR), which is ubiquitously expressed in
all cell, and exerts its biological effects by regulating the
expression of genes and cross-talking with multiple trans-membrane
signaling pathways (14). An
increasing number of studies have reported that GCs/GR promote
cancer cell survival in an unfavorable microenvironment and enhance
the resistance of solid tumors to chemotherapy by regulating the
expression of genes and activating trans-membrane signaling
pathways, which is very pivotal for the cancer progression
(15). The pro-survival and
anti-apoptotic effects of GCs are meditated by GR through
regulation of the expression of genes, such as inhibitors of
apoptosis (cIAP-2, X-IAP, Bcl-XL, and Bcl-2), mitogen-activated
protein kinase phosphatase-1 (MKP-1), as well as serum- and
glucocorticoid-inducible kinase 1 (SGK-1) (16,17).
Cell adhesion to the extracellular matrix (ECM) is
pivotal for survival and growth of most of solid cancer cells and
is mediated by several cell surface adhesion molecules such as
integrin β1 and CD44 and their ligands, which are ECM components,
such as collagens, fibronectin (FN) and laminin (18,19).
Binding of the key ECM protein FN to cell surface adhesion
molecules not only supports cell adhesion, but also brings
cytoplasmic molecules together to form protein-rich focal complexes
that activate focal adhesion kinase (FAK) and several intracellular
signaling molecules and pathways, such as Rho GTPases, Ras GTPase,
Src and the PI3K-Akt pathway, that regulate cell proliferation,
survival, spreading and migration (18–22).
So far, studies on the regulation of FN by GCs and its role in
GC-induced pathophysiological process are limited. Ahadome et
al reported that Dex increases FN expression in ocular
trabecular meshwork cells, and this increase in FN expression is
involved in the steroid-induced glaucoma (23), while another study reported Dex
negatively regulates FN expression in cytotrophoblasts isolated
from human placenta (24).
However, due to limited data it is unclear whether the regulation
of FN expression by GCs is cell type-dependent.
Melanoma is characterized by frequent recurrence and
high mortality in skin cancers. Human melanoma cells express
high-affinity GR (25). Previous
studies have shown that GCs have no significant direct effects on
the proliferation of most human melanoma cells in vitro, but
giving liposomal prednisolone phosphate (PLP) for prolonged periods
of time reduces the melanoma growth by inhibiting tumor
angiogenesis in mice (25,26). Recently a clinicopathological study
demonstrated that the subcellular distribution of GR in cutaneous
melanoma specimens is associated with tumor thickness and Clark
level, the level of anatomical invasion of melanoma in the skin
(27). However, it is still
unclear whether GCs affect the adhesion and survival of melanoma
cells.
In this study, we found that Dex, a synthetic GC,
significantly upregulated FN expression and increased its secretion
in melanoma cells. We further investigated the mechanism and
biological significance of FN upregulation by Dex in melanoma
cells. This study facilitates understanding the mechanism by which
GCs affect melanoma biology, especially the adhesion and survival
of melanoma cells.
Materials and methods
Cell culture
Human A375 melanoma cells and murine B16F10 melanoma
cells were obtained from the Cell Bank of the Chinese Academy of
Sciences (Shanghai, China). The two cell lines were routinely
cultured in RPMI-1640 (Gibco, USA) containing 10% fetal bovine
serum (FBS, Bioind, Israel) and maintained in an incubator with a
humidified atmosphere of 5% CO2 at 37°C. Wortmannin,
cisplatin, cycloheximide (CHX) and RU486 were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and dissolved in
dimethyl sulfoxide (DMSO) or ethanol. Recombinant human FN was
purchased from R&D Systems, Inc. (Minneapolis, MN, USA). For
Dex treatment, cells were cultured in medium containing 10%
Dextran-coated charcoal (DCC)-treated FBS to avoid possible
interference from serum steroids and incubated with 100 nM Dex
(Sigma-Aldrich; Merck KGaA) for different periods of time. Control
cells were incubated with ethanol vehicle (<1‰ v/v).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and
2 µg total RNA was reverse transcribed using Reverse Transcription
Reagents (Takara Bio, Inc., Otsu, Japan) following the
manufacturer's protocol. RT-qPCR was performed in triplicate using
SYBR Green PCR Master Mix (Toyobo Life Science, Osaka, Japan) on a
Mastercycler ep realplex (Eppendorf, Hamburg, Germany). The primer
sequences used were as follows. FN (human):
5′-CGGTGGCTGTCAGTCAAAG-3′ (forward) and 5′-AAACCTCGGCTTCCTCCATAA-3′
(reverse). FN (mouse): 5′-GTCAGTGTCTCCAGTGTCTAC-3′ (forward) and
5′-TGGCTTGCTGGCCAATCAGT-3′ (reverse). GAPDH (human):
5′-CATGAGAAGTATGACAACAGCCT-3′ (forward) and
5′-AGTCCTTCCACGATACCAAAGT-3′ (reverse). β-actin (mouse):
5′-CTGTATGCCTCTGGTCGTAC-3′ (forward) and 5′-TGATGTCACGCACGATTTCC-3′
(reverse). Thermal cycling conditions consisted of an initial
denaturing step (95°C, 2 min) followed by 40 cycles of denaturing
(95°C, 15 sec), annealing (60°C, 15 sec) and extending (72°C, 45
sec). The level of FN mRNA was normalized to GAPDH or β-actin
(internal control), and relative quantification was done using the
2ΔΔCq formula. Changes in gene expression were expressed
as the relative fold-increase in mRNA compared with a control.
Western blotting
Total cell lysates were prepared with 1× SDS lysis
buffer with 100 mM Dithiothreitol and 2 µg/ml protease inhibitor
solution containing 0.1 mM each leupeptin, aprotinin, and
pepstatin. After electrophoresis, proteins were transferred to
nitrocellulose membrane, blocked with 5% (v/v) nonfat milk in
tris-buffered saline Tween-20 (TBST), and probed overnight at 4°C
with primary antibodies against FN (sc-6953, 1:500; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), β-actin (1:10,000,
Sigma-Aldrich; Merck KGaA), or p-Akt1/2/3 (Ser 473) (sc-7985,
1:1,000; Santa Cruz Biotechnology, Inc.), Akt1/2/3 (sc-8312,
1:1,000; Santa Cruz Biotechnology, Inc.). Then the membranes were
washed three times and incubated with HRP-conjugated secondary
antibodies (1:5,000; Rockland Immunochemicals, Inc., Gilbertsville,
PA, USA) for 2 h. Finally HRP was detected using enhanced
chemiluminescence (Pierce; Thermo Fisher Scientific, Inc.). Protein
bands were quantified with ImageJ software (National Institutes of
Health, Bethesda, MD, USA) using β-actin as an internal
control.
ELISA
A375 melanoma cells were treated with or without 100
nM Dex for the indicated times, and then the conditioned media were
collected and analyzed using human FN ELISA kits (R&D Systems,
Inc.) according to the manufacturer's instructions. Absorbance of
samples was read at 450 nm in a UV-visible spectrophotometer. The
protein concentration was calibrated from a dose response curve
based on reference standards.
RNA interference
SiRNAs were manufactured by GenePharma Co., Ltd.
(Shanghai, China). The sequences were as follows. FN siRNA:
5′-GCAGUGGCUGAAGACACAAGGAAAU-3′; Control siRNA:
5′-CGCTTACCGATTCAGAATGG-3′. A375 cells were transfected with a
final concentration of 10 nmol/l FN siRNA or control siRNA using
INTERFERin™ (Polyplus Transfection, Strasbourg, France) following
the manufacturer's instructions.
Cell adhesion assay
Cell adhesive ability was determined by cell
adhesion assay. After cells were treated with Dex or transiently
transfected with siRNA for the indicated times, cells were digested
into single cell suspension and 8×104 cells were seeded
onto non-coated 96-well plates in triplicate and incubated at 37°C
for 60 min. The plates were gently washed thrice with 1X PBS to
remove the non-adherent cells. The number of remaining cells in the
96-well plates was determined by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Briefly, the remaining cells were incubated in medium
supplemented with 50 µg/ml methylthiazole tetrazolium for 3 h.
Cells were then solubilized by adding 200 µl DMSO. The absorbance
was measured at 570 nm in a UV-visible spectrophotometer.
Cell adhesion ability was also determined in 96-well
plates coated with or without human FN purchased from CORNING
(USA). Before the cell adhesion assay, cells were pre-treated with
a CD44 primary antibody (103014; BioLegend, Inc., San Diego, CA,
USA) and Con IgG antibody (400622; BioLegend, Inc.) for 1 h.
Analysis of cell viability
Cells were seeded in 96-well culture plates at a
density of 3×103 cells per well in triplicate, allowed
to attach overnight, and then treated with the indicated chemicals
or reagents. Cisplatin, a chemotherapeutic drug applied in melanoma
therapy, was used to induce cell death. At the indicated time, cell
viability was evaluated using the Cell Counting Kit-8 (CCK-8;
Dojindo Molecular Technologies, Inc., Kumamoto, Japan) following
the standard procedures provided by the manufacturer. The optical
density (OD) was measured at a wavelength of 450 nm using a
Labsystem multiskan microplate reader (Merck Eurolab, Dietikon,
Switzerland).
Statistical analysis
Statistical significances between multiple
experimental groups were analyzed by one-way analysis of variance
and Tukey's post hoc tests. The Student's t-test was used to
compare the difference between two different groups. P<0.05 was
considered to indicate a statistically significant difference.
Quantitative data were expressed as the mean ± standard deviation
of at least three determinations.
Results
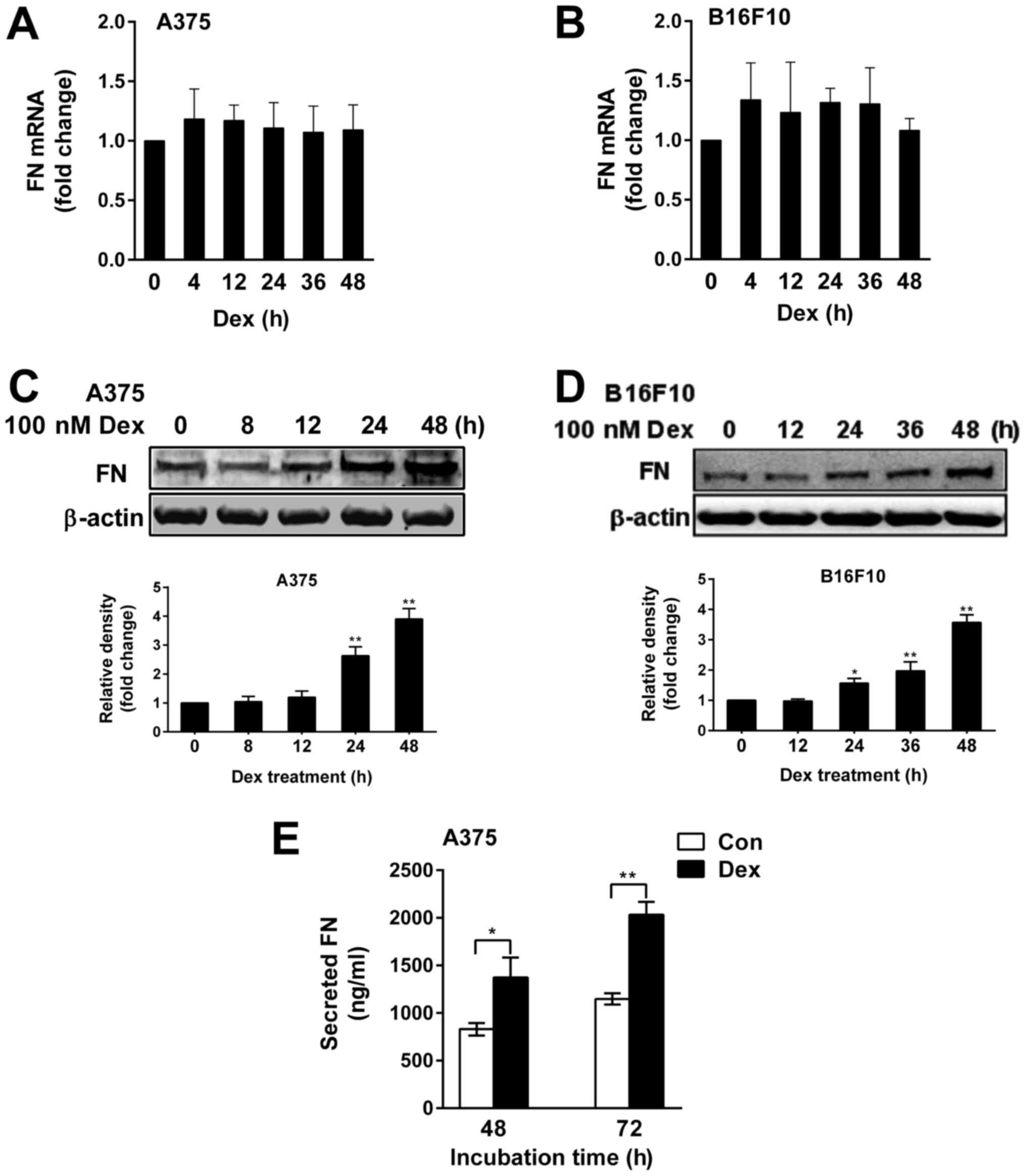
Dex increased the expression and
secretion of FN in melanoma cells
We first examined the expression of FN in A375 and
B16F10 melanoma cells treated with 100 nM Dex for different periods
of times by RT-qPCR and western blotting. We did not observe a
significant change in the level of FN mRNA (Fig. 1A and B), but we found that Dex
significantly upregulated FN protein in A375 and B16F10 cells in a
time-dependent fashion, with the maximal induction of FN protein
(3.9-fold of control and 3.5-fold of control, respectively,
P<0.01) observed at 48 h after Dex treatment (Fig. 1C and D). A significant increase in
the level of secreted FN was also observed at 48 h and 72 h after
Dex treatment in A375 cells (Fig.
1E).
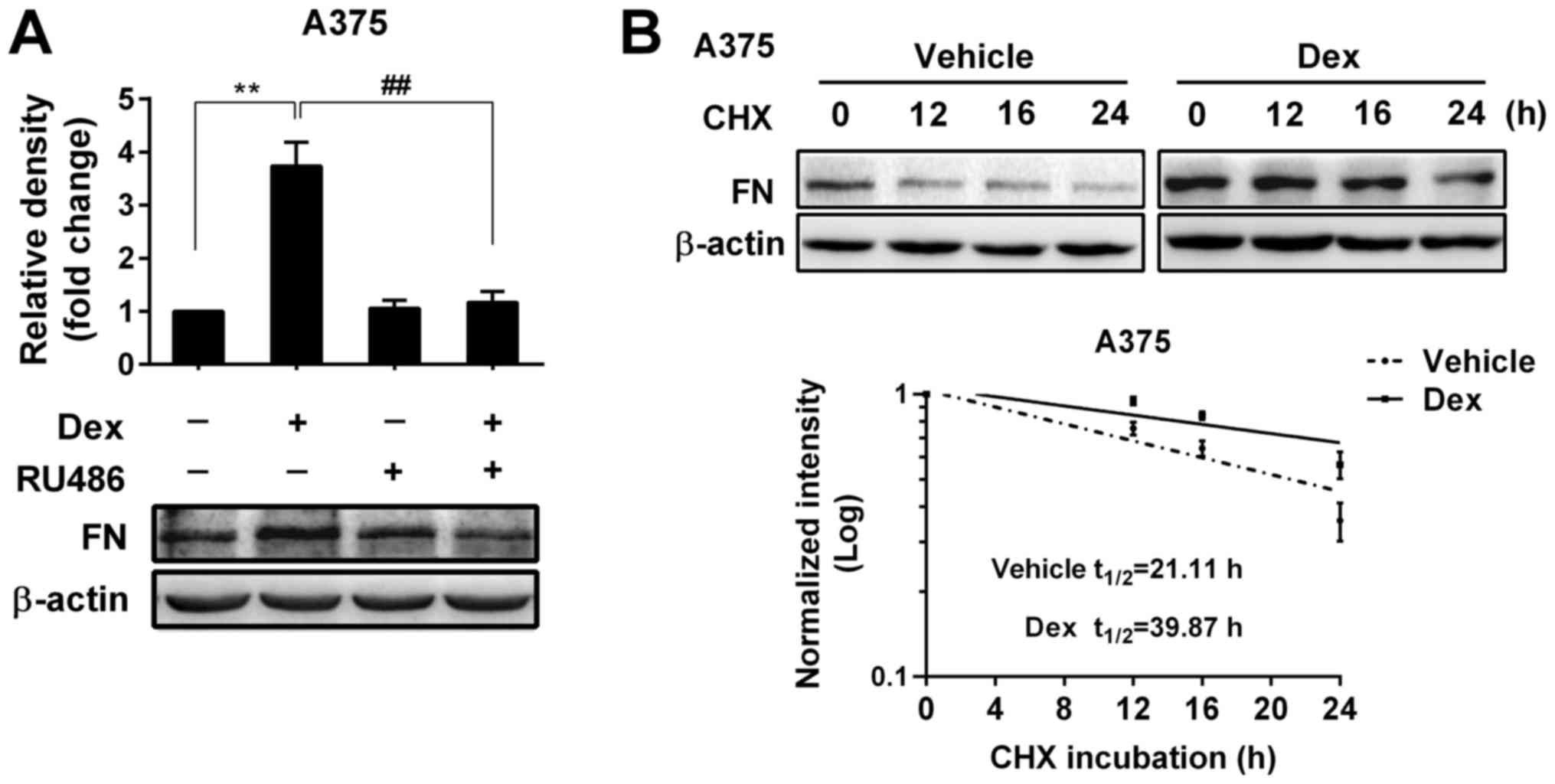
Upregulation of FN expression by Dex
was due to a GR-mediated increase in protein stability
Since upregulation of FN by Dex occurred at the
post-transcriptional level, we investigated whether Dex-induced
upregulation of FN was mediated through GR and caused by an
increase in the protein stability in A375 melanoma cells. As shown
in Fig. 2A, RU486, an antagonist
of GR, dramatically reversed the upregulation of FN protein,
indicating that the effect of Dex was mediated through GR.
We next examined the protein stability of FN in the
presence and absence of Dex by western blotting. A375 cells were
pre-treated with Dex or vehicle for 48 h and further treated with
100 µg/ml cycloheximide (CHX, a protein synthesis inhibitor) for
different amounts of times. As shown in Fig. 2B, Dex significantly extended the
half-life of FN protein (from 21.11 to 39.87 h, a 1.89-fold
increase, P<0.01). These results indicate that Dex-induced
upregulation of FN was achieved by preventing protein
degradation.
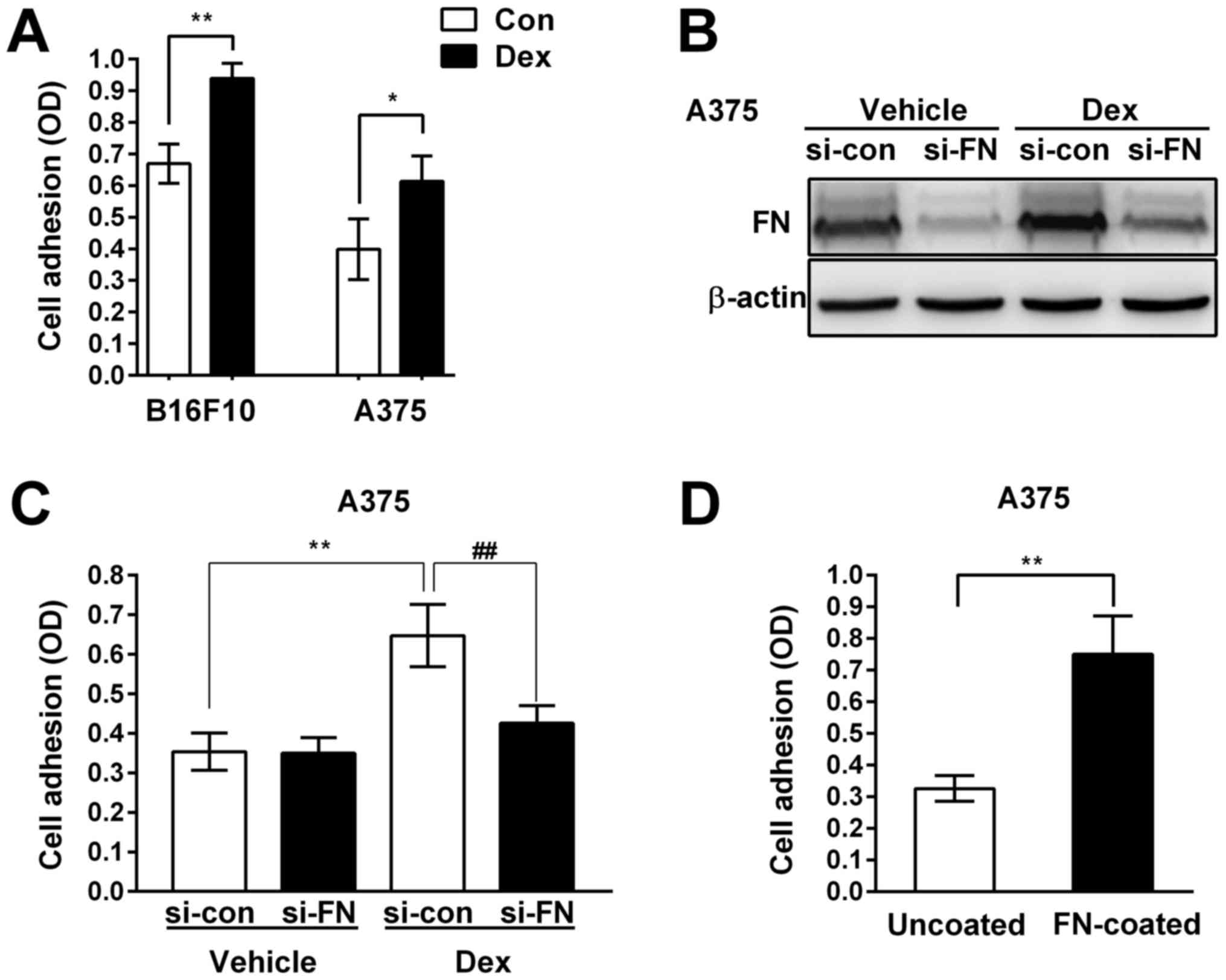
Upregulation of FN contributed to the
pro-adhesive effect of Dex in melanoma cells
As a multifunctional ECM glycoprotein and a core
component of many extracellular matrices, FN plays an important
role in regulating intracellular signal transduction and tumor
behaviors, including cell adhesion and cell survival (28–30).
Therefore, we investigated whether the effect of Dex on melanoma
biology was due to upregulation of FN. We silenced the expression
of FN using specific small RNA interference (si-FN) and determined
the effects of knock-down of FN on melanoma cell adhesion. We found
that 100 nM Dex significantly enhanced the adhesive ability of
B16F10 and A375 cells (Fig. 3A).
Western blotting confirmed that knock-down of FN expression with
si-FN almost abolished the Dex-induced expression of FN in A375
cells (Fig. 3B). As shown in
Fig. 3C, Dex significantly
enhanced the adhesive ability of A375 cells transfected with
control small RNA interference (si-con). However, the pro-adhesive
effect of Dex in FN knock-down cells was almost completely
inhibited, indicating that upregulation of FN contributed to the
pro-adhesive effect of Dex in melanoma cells.
In order to further evaluate the role of FN in the
adhesion of A375 cells, we examined cell adhesive capacity on
uncoated and FN-coated wells (to imitate the over-expression of
extracellular FN). The number of adhering cells in the FN-coated
group was significantly higher than the uncoated control group
(Fig. 3D), indicating that
augmentation of extracellular FN enhanced A375 cell adhesive
capacity.
Upregulation of FN contributed to the
pro-survival effect of Dex by enhancing melanoma cell adhesion
Studies have shown that cell adhesion to the ECM is
pivotal for survival and growth of most of solid malignant cells,
so we further investigated the role of FN upregulation in the
effect of Dex on melanoma cell survival in the presence of the
chemotherapeutic agent cisplatin. We found that treatment with
cisplatin for 24 h decreased A375 cell viability in a
dose-dependent manner (Fig. 4A).
The survival rate of A375 cells treated with 40 µM cisplatin was
decreased to 39% (P<0.01) (Fig.
4B). However, we found that 100 nM Dex significantly weakened
the cytotoxic effect of cisplatin and increased the cell survival
rate to 68% (P<0.01) (Fig. 4B).
Similar results were observed for B16F10 cells (Fig. 4C), indicating that Dex exerted a
pro-survival effect on melanoma cells under unfavorable conditions
(Fig. 4B and C). Next, we observed
the effect of Dex and cisplatin treatment on survival in cells
where FN expression was silenced by si-FN. We found that Dex also
enhanced cell resistance to cisplatin in A375 cells transfected
with si-con (Fig. 4D), but
inhibiting FN expression with si-FN significantly reduced the
pro-survival effect of Dex in the presence of cisplatin (Fig. 4D). These results indicate that
upregulation of FN contributes to Dex-induced melanoma cell
survival and chemo-resistance.
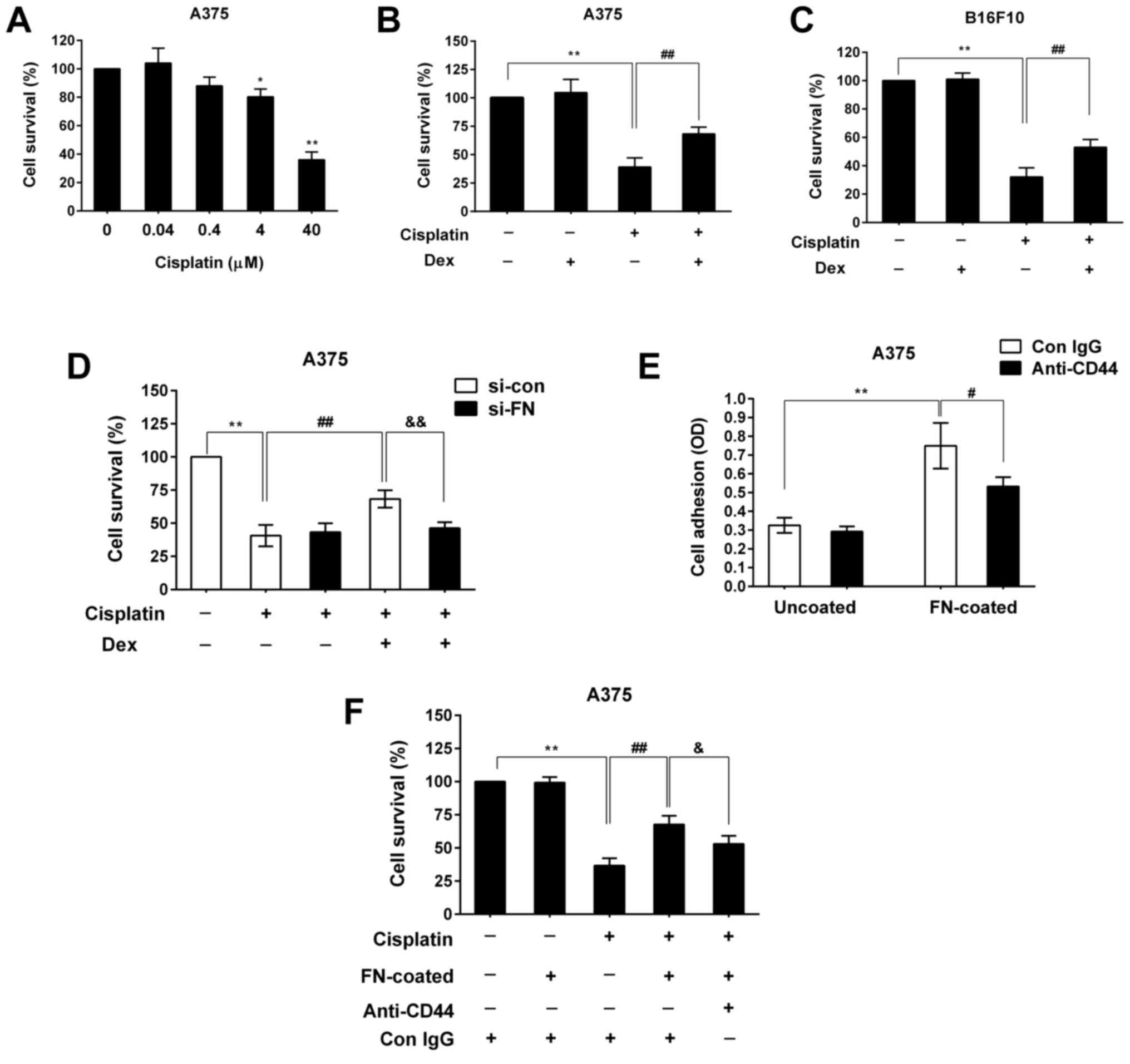 | Figure 4.Upregulation of FN mediated the
pro-survival effect of Dex by enhancing the adhesion of melanoma
cells. (A) A375 cells were treated with different dose of cisplatin
for 24 h, and then cell viability was analyzed using the CCK-8 kit.
(B) A375 cells, (C) B16F10 cells and (D) transfected A375 cells
were pre-incubated with or without Dex (100 nM) for 24 h and then
cultured continuously in the presence or absence of cisplatin (40
µM) for another 24 h. Cell viability was analyzed as described in
the Materials and Methods. (E) Following pre-incubation with a
CD44-blocking antibody (anti-CD44, 40 µg/ml) or Con IgG antibody
for 1 h, A375 cells were seeded onto 96-well plates coated with or
without human FN (10 µg/ml). Cell adhesion was assayed as described
in the Materials and methods. (F) Cells were further treated with
cisplatin (40 µM) for 24 h, and cell viability was measured. The
values represent means ± standard deviation of three separate
experiments. *P<0.05, **P<0.01 vs. the vehicle-treated
control, ##P<0.01 vs. cisplatin-treated cells,
#P<0.05 vs. the FN-coated control (E),
&P<0.05, &&P<0.01 vs.
cisplatin-treated cells with Dex or cisplatin-treated cells with
FN. |
The interaction of extracellular FN with one of its
receptors, such as CD44, mediates cell adhesion by triggering
several signaling pathways, and adhesion consequently regulates
cell behaviors, including cell survival (31,32).
Therefore, we further investigated the correlation between
FN-enhanced cell adhesion and FN-enhanced melanoma cell survival
and resistance to chemotherapeutics. We used FN-coated wells to
mimic extracellular FN. As shown in Fig. 4E, blocking CD44 signaling with an
anti-CD44 antibody significantly abrogated the effect of
FN-enhanced cell adhesion in A375 cells, suggesting the partial
involvement of extracellular FN/CD44 signaling in enhanced cell
adhesion. Furthermore, extracellular FN protected A375 cells
against chemotherapeutics, further validating the effect of FN on
enhanced cell survival under unfavorable conditions (Fig. 4F). Inhibiting extracellular FN
signaling with an anti-CD44 antibody not only reduced cell
adhesion, but also significantly reduced the FN-mediated
pro-survival effect in the presence of cisplatin (Fig. 4F). These data indicate that cell
adhesion is positively linked to melanoma cell survival and that
Dex-induced survival and chemo-resistance is mediated through FN
upregulation and enhancement of cell adhesion.
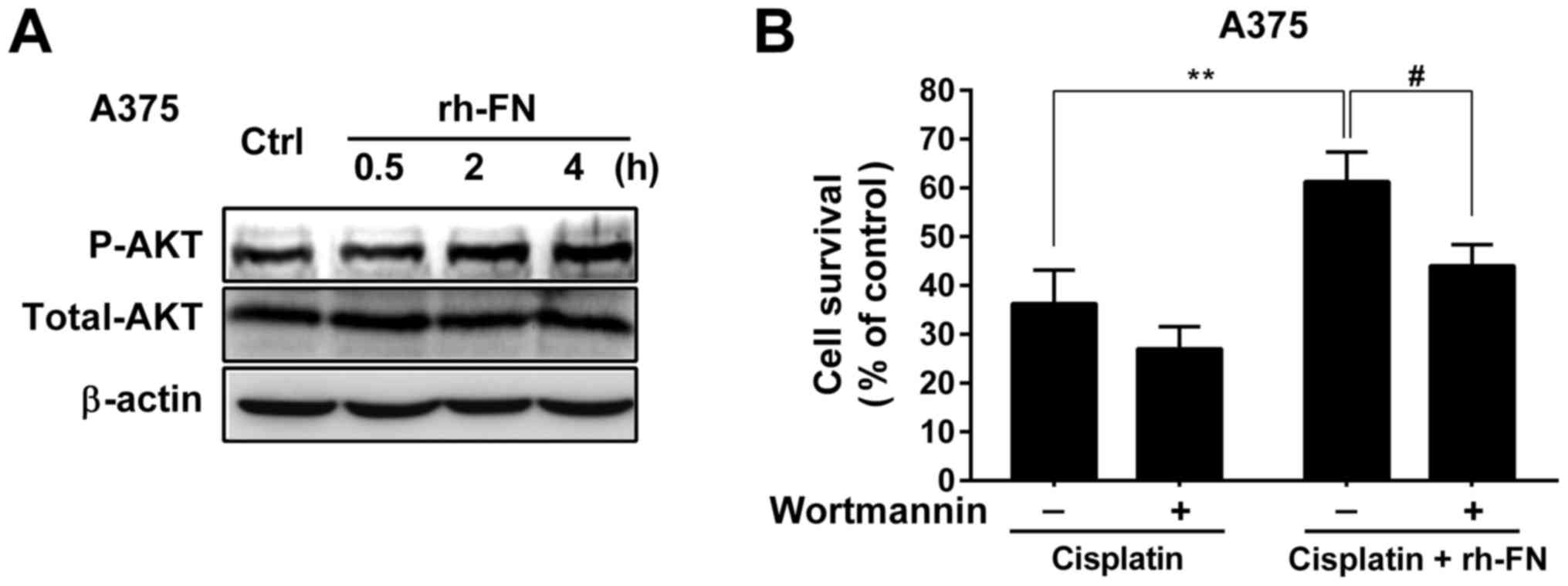
PI3K/AKT activation contributed to
FN-mediated melanoma cell survival
The PI3K/AKT pathway plays a critical role in
modulating tumor cell proliferation, adhesion and survival
(33–35). Therefore, we examined whether the
PI3K/AKT pathway was involved in the pro-survival effect of FN on
melanoma cells. We found that treatment with recombinant human FN
(rh-FN; 10 µg/ml) significantly increased the level of
phosphorylated AKT at S473 in A375 cells in a time-dependent manner
(Fig. 5A). Treatment with rh-FN
also significantly increased cell viability from 36 to 61%
(P<0.01) (Fig. 5B). However,
inhibiting the PI3K/AKT pathway with wortmannin almost abolished
the pro-survival effect of rh-FN in the presence of cisplatin in
A375 cells (Fig. 5B). These
results indicate that activation of PI3K/AKT by extracellular FN is
involved in the survival and enhancement of cisplatin resistance in
melanoma cells.
Discussion
GCs are important stress hormones and are used as a
concomitant medication during malignant tumor chemotherapy. Current
studies have suggested a positive relationship between GCs and
melanoma progression (27,36–38).
The mechanism by which GC influences melanoma biology is still
unclear. In this study, we investigated the role of Dex in
regulating FN expression, and the biological significance of this
regulation in melanoma cells. We found that Dex, a synthetic GC,
significantly increased the level of FN protein in melanoma cells,
including its secreted form. Upregulation of FN protein by Dex was
mediated through GR, which acted post-transcriptionally by
increasing FN protein stability. GC was previously reported to
increase FN synthesis and induce FN matrix assembly in normal human
fibroblasts, HT-1080 fibrosarcoma cells and chick hepatocytes, but
GC was found to negatively regulate FN expression in placenta
(24,39–41).
These contrasting results suggest that the regulation of FN
expression by GC is dependent on cell type.
FN is a multifunctional ECM glycoprotein and a core
component of many extracellular matrices, and plays an essential
role in regulating epithelial cell adhesion to the ECM (42). In the present study, we found that
Dex significantly increased the levels of both intracellular and
secreted FN protein in melanoma cells and promoted adhesion.
Knock-down of FN expression in melanoma cells significantly reduced
the pro-adhesive effect of Dex. Furthermore, we demonstrated that
the addition of extracellular FN enhanced the adhesive capacity of
melanoma cells. These results indicate that upregulation of FN
mediates the pro-adhesive effect of Dex on melanoma cells.
Cell adhesion to the ECM is pivotal for survival and
growth of most solid malignant cells (18,19).
Consistent with this, we demonstrated that in addition to
regulating the adhesion of melanoma cells, Dex increased the
survival and chemo-resistance of melanoma cells during
cisplatin-induced cell death. Knock-down of FN expression not only
reduced cell adhesive capacity, but also abrogated the pro-survival
effect of Dex; therefore, we hypothesize that upregulation of FN by
Dex enhances cell adhesion, thereby enhancing cell survival under
unfavorable conditions. CD44, a broadly distributed transmembrane
glycoprotein, mediates cell-matrix interactions through binding to
some ECM components, such as FN, collagens and laminin. To test our
hypothesis, an anti-CD44 antibody was used to block the
extracellular FN signaling and inhibit FN-enhanced cell adhesion.
We found that anti-CD44 antibody treatment only partially blocked
the FN-mediated increase in melanoma cell survival in the presence
of cisplatin, indicating that FN binding to other receptors in
addition to CD44 may contribute to the pro-survival effect of
Dex.
It is known that increased adhesion mediated by
FN-receptor interaction plays an essential role in regulating cell
proliferation, survival and migration by triggering several
signaling pathways, especially the PI3K/AKT pathway, which is the
most important pathway promoting cell survival (43). Interaction of CD44 with its
ligands, including FN, can also enhance proliferation, survival and
invasion by activating PI3K/AKT pathway (31,32,44).
It is unclear whether the PI3K/AKT pathway is involved in the
FN-enhanced melanoma cell survival. Here, we demonstrated that
extracellular FN activated PI3K/AKT signaling in a time-dependent
manner and inhibiting PI3K/AKT signaling almost abrogated the
pro-survival effect of FN, indicating that activation of this
pathway contributed to the FN-mediated melanoma cell survival.
In summary, we found that Dex upregulated the
expression of FN protein in melanoma cells through a GR-mediated
increase in protein stability. In melanoma cells, upregulation of
FN contributed to the adhesion-promoting effect of Dex, thereby
promoting cell survival and enhancing cell resistance to
chemotherapy through activation of PI3K/AKT pathway. These new
findings increase our understanding of the mechanism responsible
for GC promotion of melanoma cell adhesion and survival.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81472690) and the Scientific
Research and Technology Development Program of Guilin (no.
2016012702-3).
Glossary
Abbreviations
Abbreviations:
|
GC
|
glucocorticoid
|
|
GR
|
glucocorticoid receptor
|
|
Dex
|
dexamethasone
|
|
FN
|
fibronectin
|
|
CHX
|
cycloheximide
|
|
ECM
|
extracellular matrix
|
References
|
1
|
Andersen BL, Yang HC, Farrar WB,
Golden-Kreutz DM, Emery CF, Thornton LM, Young DC and Carson WE
III: Psychologic intervention improves survival for breast cancer
patients: A randomized clinical trial. Cancer. 113:3450–3458. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chida Y, Hamer M, Wardle J and Steptoe A:
Do stress-related psychosocial factors contribute to cancer
incidence and survival? Nat Clin Pract Oncol. 5:466–475. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim-Fuchs C, Le CP, Pimentel MA,
Shackleford D, Ferrari D, Angst E, Hollande F and Sloan E: Chronic
stress accelerates pancreatic cancer growth and invasion: A
critical role for beta-adrenergic signaling in the pancreatic
microenvironment. Brain Behav Immun. 40:40–47. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sloan EK, Priceman SJ, Cox BF, Yu S,
Pimentel MA, Tangkanangnukul V, Arevalo JM, Morizono K, Karanikolas
BD, Wu L, et al: The sympathetic nervous system induces a
metastatic switch in primary breast cancer. Cancer Res.
70:7042–7052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Powell ND, Tarr AJ and Sheridan JF:
Psychosocial stress and inflammation in cancer. Brain Behav Immun.
30 Suppl:S41–S47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beck IM, Vanden Berghe W, Vermeulen L,
Yamamoto KR, Haegeman G and De Bosscher K: Crosstalk in
inflammation: The interplay of glucocorticoid receptor-based
mechanisms and kinases and phosphatases. Endocr Rev. 30:830–882.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin KT and Wang LH: New dimension of
glucocorticoids in cancer treatment. Steroids. 111:84–88. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rutz HP: Effects of corticosteroid use on
treatment of solid tumours. Lancet. 360:1969–1970. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kriegler AB, Bernardo D and Verschoor SM:
Protection of murine bone marrow by dexamethasone during cytotoxic
chemotherapy. Blood. 83:65–71. 1994.PubMed/NCBI
|
|
10
|
Herr I, Ucur E, Herzer K, Okouoyo S,
Ridder R, Krammer PH, von Knebel Doeberitz M and Debatin KM:
Glucocorticoid cotreatment induces apoptosis resistance toward
cancer therapy in carcinomas. Cancer Res. 63:3112–3120.
2003.PubMed/NCBI
|
|
11
|
Sui M, Chen F, Chen Z and Fan W:
Glucocorticoids interfere with therapeutic efficacy of paclitaxel
against human breast and ovarian xenograft tumors. Int J Cancer.
119:712–717. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang C, Beckermann B, Kallifatidis G, Liu
Z, Rittgen W, Edler L, Büchler P, Debatin KM, Büchler MW, Friess H
and Herr I: Corticosteroids induce chemotherapy resistance in the
majority of tumour cells from bone, brain, breast, cervix, melanoma
and neuroblastoma. Int J Oncol. 29:1295–1301. 2006.PubMed/NCBI
|
|
13
|
Zhang C, Marmé A, Wenger T, Gutwein P,
Edler L, Rittgen W, Debatin KM, Altevogt P, Mattern J and Herr I:
Glucocorticoid-mediated inhibition of chemotherapy in ovarian
carcinomas. Int J Oncol. 28:551–558. 2006.PubMed/NCBI
|
|
14
|
Schoneveld OJ, Gaemers IC and Lamers WH:
Mechanisms of glucocorticoid signalling. Biochim Biophys Acta.
1680:114–128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herr I, Gassler N, Friess H and Büchler
MW: Regulation of differential pro- and anti-apoptotic signaling by
glucocorticoids. Apoptosis. 12:271–291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Warny M, Keates AC, Keates S, Castagliuolo
I, Zacks JK, Aboudola S, Qamar A, Pothoulakis C, LaMont JT and
Kelly CP: p38 MAP kinase activation by Clostridium difficile toxin
A mediates monocyte necrosis, IL-8 production and enteritis. J Clin
Invest. 105:1147–1156. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paul A, Wilson S, Belham CM, Robinson CJ,
Scott PH, Gould GW and Plevin R: Stress-activated protein kinases:
Activation, regulation and function. Cell Signal. 9:403–410. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zaidel-Bar R and Geiger B: The switchable
integrin adhesome. J Cell Sci. 123:1385–1388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ponta H, Sherman L and Herrlich PA: CD44:
From adhesion molecules to signalling regulators. Nat Rev Mol Cell
Biol. 4:33–45. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hynes RO: Integrins: Bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Winograd-Katz SE, Fässler R, Geiger B and
Legate KR: The integrin adhesome: From genes and proteins to human
disease. Nat Rev Mol Cell Biol. 15:273–288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zaidel-Bar R, Itzkovitz S, Ma'ayan A,
Iyengar R and Geiger B: Functional atlas of the integrin adhesome.
Nat Cell Biol. 9:858–867. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ahadome SD, Zhang C, Tannous E, Shen J and
Zheng JJ: Small-molecule inhibition of Wnt signaling abrogates
dexamethasone-induced phenotype of primary human trabecular
meshwork cells. Exp Cell Res. 357:116–123. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guller S, Wozniak R, Leibman MI and
Lockwood CJ: Negative regulation of placental fibronectin
expression by glucocorticoids and cyclic adenosine
3′,5′-monophosphate. Ann N Y Acad Sci. 734:132–142. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dobos J, Kenessey I, Tímár J and Ladányi
A: Glucocorticoid receptor expression and antiproliferative effect
of dexamethasone on human melanoma cells. Pathol Oncol Res.
17:729–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Banciu M, Metselaar JM, Schiffelers RM and
Storm G: Liposomal glucocorticoids as tumor-targeted
anti-angiogenic nanomedicine in B16 melanoma-bearing mice. J
Steroid Biochem Mol Biol. 111:101–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lai S, Piras F, Spiga S, Perra MT, Minerba
L, Piga M, Mura E, Murtas D, Demurtas P, Corrias M, et al: Nestin
and vimentin colocalization affects the subcellular location of
glucocorticoid receptor in cutaneous melanoma. Histopathology.
62:487–498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakagawa Y, Nakayama H, Nagata M, Yoshida
R, Kawahara K, Hirosue A, Tanaka T, Yuno A, Matsuoka Y, Kojima T,
et al: Overexpression of fibronectin confers cell adhesion-mediated
drug resistance (CAM-DR) against 5-FU in oral squamous cell
carcinoma cells. Int J Oncol. 44:1376–1384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schmidt S and Friedl P: Interstitial cell
migration: Integrin-dependent and alternative adhesion mechanisms.
Cell Tissue Res. 339:83–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leiss M, Beckmann K, Girós A, Costell M
and Fässler R: The role of integrin binding sites in fibronectin
matrix assembly in vivo. Curr Opin Cell Biol. 20:502–507. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li XP, Zhang XW, Zheng LZ and Guo WJ:
Expression of CD44 in pancreatic cancer and its significance. Int J
Clin Exp Pathol. 8:6724–6731. 2015.PubMed/NCBI
|
|
32
|
McFarlane S, McFarlane C, Montgomery N,
Hill A and Waugh DJ: CD44-mediated activation of α5β1-integrin,
cortactin and paxillin signaling underpins adhesion of basal-like
breast cancer cells to endothelium and fibronectin-enriched
matrices. Oncotarget. 6:36762–36773. 2015.PubMed/NCBI
|
|
33
|
Clark AS, West K, Streicher S and Dennis
PA: Constitutive and inducible Akt activity promotes resistance to
chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol
Cancer Ther. 1:707–717. 2002.PubMed/NCBI
|
|
34
|
Larue L and Bellacosa A:
Epithelial-mesenchymal transition in development and cancer: Role
of phosphatidylinositol 3′kinase/AKT pathways. Oncogene.
24:7443–7454. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Polivka J Jr and Janku F: Molecular
targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol
Ther. 142:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Valles SL, Benlloch M, Rodriguez ML, Mena
S, Pellicer JA, Asensi M, Obrador E and Estrela JM: Stress hormones
promote growth of B16-F10 melanoma metastases: An interleukin 6-
and glutathione-dependent mechanism. J Transl Med. 11:722013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Collinson FJ, Lam TK, Bruijn WM, de Wilt
JH, Lamont M, Thompson JF and Kefford RF: Long-term survival and
occasional regression of distant melanoma metastases after adrenal
metastasectomy. Ann Surg Oncol. 15:1741–1749. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Flaherty DC, Deutsch GB, Kirchoff DD, Lee
J, Huynh KT, Lee DY, Foshag LJ, Bilchik AJ and Faries MB:
Adrenalectomy for metastatic melanoma: Current role in the age of
nonsurgical treatments. Am Surg. 81:1005–1009. 2015.PubMed/NCBI
|
|
39
|
McKeown-Longo PJ and Etzler CA: Induction
of fibronectin matrix assembly in human fibrosarcoma cells by
dexamethasone. J Cell Biol. 104:601–610. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nimmer D, Bergtrom G, Hirano H and Amrani
DL: Regulation of plasma fibronectin biosynthesis by
glucocorticoids in chick hepatocyte cultures. J Biol Chem.
262:10369–10375. 1987.PubMed/NCBI
|
|
41
|
Oliver N, Newby RF, Furcht LT and
Bourgeois S: Regulation of fibronectin biosynthesis by
glucocorticoids in human fibrosarcoma cells and normal fibroblasts.
Cell. 33:287–296. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zollinger AJ and Smith ML: Fibronectin,
the extracellular glue. Matrix Biol. 60-61:1–37. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Benbrook DM and Masamha CP: The
pro-survival function of Akt kinase can be overridden or altered to
contribute to induction of apoptosis. Curr Cancer Drug Targets.
11:586–599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Onodera Y, Teramura T, Takehara T and
Fukuda K: Hyaluronic acid regulates a key redox control factor Nrf2
via phosphorylation of Akt in bovine articular chondrocytes. FEBS
Open Bio. 5:476–484. 2015. View Article : Google Scholar : PubMed/NCBI
|