Introduction
The renin-angiotensin system (RAS) has been
implicated in the pathophysiology of pancreatitis (1). Evidently, high levels of angiotensin
I converting enzyme 2 (ACE2), angiotensin (Ang) 1–7, and its
corresponding receptor Mas, expression are detected in plasma and
in the pancreas of mice with acute pancreatitis (AP). These factors
serve protective roles during the pathogenesis of pancreatitis
within mice (2). Ang 1–7 has been
reported to serve as an endogenous antagonist of Ang II, possessing
anti-inflammatory and vasodilatory activities, and exerting
protective effects against endothelial injury (3–5). Our
previous study revealed that caerulein (CAE) can stimulate the
ACE2-Ang-1-7-Mas axis and significantly inhibit pancreatitis
development via endothelial nitric oxide synthase activation and
nitric oxide signaling within AR42J cells (6). In addition, further study identified
that in the AR42J cells stimulated by CAE, blocking of Mas receptor
using A779, then enhancement of Ang (1–7),
still can reduce the inflammatory response. It is therefore
hypothesized that Ang (1–7) has other anti-inflammatory pathways
besides the Mas receptor pathway; however, further investigation is
required.
Toll-like receptors (TLRs) are required for the
onset of inflammation, and can initiate inflammatory signaling
(7). TLR4 is a member of the TLR
family and is expressed by pancreatic macrophages, acinar cells and
stellate cells (8). Engagement of
TLR4 with its ligands, such as lipopolysaccharide, can activate the
nuclear factor (NF)-κB signaling pathway and induce the expression
of tumor necrosis factor-α (TNF-α), and other proinflammatory
cytokines that lead to pancreatic inflammation (9–12).
During the pathogenesis of AP, the RAS also regulates the NF-κB
signaling pathway and cytokine production, which contributes to the
pathogenesis of AP (13). Our
previous study reported the expression of Ang 1–7 within AR42J
cells; high levels of circulating Ang 1–7 were detected within mice
with severe AP (SAP) (2). However,
it is unclear whether endogenous and exogenous Ang 1–7 can regulate
TLR4 and NF-κB expression within AR42J cells during the
inflammatory process. In the present study, a cellular model was
employed to investigate the effect of Ang 1–7 on the expression of
TLR4, NF-κB and inflammatory cytokines within AR42J cells.
Materials and methods
Cell culture and grouping
Rat pancreatic acinar AR42J cells [American Type
Culture Collection (ATCC), Manassas, VA, USA] were cultured in
F-12K medium (ATCC) containing 20% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 U/ml
penicillin and 100 µg/ml streptomycin (complete medium) at 37°C in
a humidified atmosphere containing 5% CO2. Cells were
stimulated with 10 nmol/l CAE (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 0.25, 0.5, 2, 6, 12 or 24 h to induce
inflammation (14). All drug
intervention group cells were stimulated at room temperature, and
then incubated at 37°C in a humidified atmosphere containing 5%
CO2. In addition, cells were treated with vehicle [F-12K
medium containing 5% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.)] alone (Control) or 10 nmol/l CAE for 12 h
(Model). Some cells were pretreated with Ang 1–7 (10−7,
10−6 or 10−5 mol/l; Sigma-Aldrich; Merck
KGaA) or Ang 1–7 antagonist A779 (10−7, 10−6
or 10−5 mol/l; Sigma-Aldrich; Merck KGaA) for 12 h and
stimulated with 10 nmol/l CAE for 12 h. The groups of
differentially treated cells were harvested for subsequent
experimentation.
Immunofluorescence assay
Expression levels of TLR4 and NF-κB in the control
and model cell groups were determined by immunofluorescence
analysis. Briefly, cells of the control and model groups were
harvested and washed in phosphate-buffered saline (pH 7.4), fixed
in 4% (v/v) paraformaldehyde at 37°C for 40 min, and were then
treated with 1% bovine serum albumin (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C for 30 min. The cells were incubated with
rabbit anti-TLR4 (ab22048; 1:200; Abcam, Cambridge, UK) and
anti-NF-κB p65 (8242; 1:200; Cell Signaling Technology, Inc.,
Danvers, MA, USA) or control rabbit immunoglobulin G (IgG; ab6730;
1:200; Abcam, Cambridge, UK) at 4°C overnight. Cells were washed
and incubated with fluorescein isothiocyanate-conjugated goat
anti-rabbit IgG (bs-0295M-FITC; 1:100; BIOSS, Beijing, China) for
40 min at 37°C followed by staining with DAPI (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). The cells were examined
under a fluorescent microscope and images were captured.
Western blotting
Harvested cells were lysed using lysis buffer
(BIOSS, Beijing, China) and were centrifuged (25,155 × g for 15 min
at 4°C). Following quantification of protein concentrations using a
Bicinchoninic Acid Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), cell lysate proteins (25
µg/lane) were separated by 10% SDS-PAGE and were transferred onto
polyvinylidene fluoride membranes. The membranes were incubated
with 5% non-fat dry milk in Tris-buffered saline containing 2%
Tween at 37°C for 2 h and were then incubated with monoclonal
rabbit anti-mouse TLR4 (ab22048; 1:800; Abcam), anti-β-actin (4970;
1:500; Cell Signaling Technology, Inc.), or anti-NF-κBp65 (8242;
1:800; Cell Signaling Technology, Inc.) at 4°C overnight. Membranes
were washed and were incubated with peroxidase-conjugated goat
anti-rabbit antibodies (sc-2004; 1:5,000; Santa Cruz Biotechnology,
Inc.) for 1 h at 25°C and were visualized using enhanced
chemiluminescence detection reagents (EMD Millipore, Billerica, MA,
USA). The relative levels of target protein compared with β-actin
were determined via densitometric analysis using Image software
version 3.0 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from different groups of
cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.,) and reverse transcribed into cDNA using the High Capacity
cDNA Reverse Transcription kit (Fermentas; Thermo Fisher
Scientific, Inc., Pittsburgh, PA, USA) according to the
manufacturer's protocol. The resultant cDNA served as templates for
RT-qPCR using the Power SYBR Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) and specific primers in
an Applied Biosystems 7500 fast platform (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The primer sequences were: IL-6
forward, 5′TGCCTTCTTGGGACTGAT3′ and reverse,
5′CTGGCTTTGTCTTTCTTGTTAT3′ (384 bp); IL-10 forward,
5′CCTGGTAGAAGTGATGCC3′ and reverse, 5′CACCTTTGTCTTGGAGCT3 (191 bp);
IL-8 forward, 5′TCGTCCACGCCACAAGTA3′ and reverse,
5′CAGTAGTCCGAAGAATGAAG3′ (117 bp); TNF-α forward,
5′CCACGCTCTTCTGTCTACTG3′ and reverse, 5′GCTACGGGCTTGTCACTC3′ (145
bp); and GAPDH forward, 5′CTCAACTACATGGTCTACATGTTCCA-3′ and
reverse, 5′-CTTCCCATTCTCAGCCTTGACT-3′ (81 bp). The qPCR reactions
were performed in triplicate at 95°C for 10 min, 95°C for 15 sec,
and 60°C for 1 min for 40 cycles. Data were normalized to GAPDH and
analyzed by the 2−ΔΔCq method (15).
Statistical analysis
All cell experiments were repeated three times, and
all data are expressed as the mean ± standard deviation. The
difference among groups was determined by one-way analysis of
variance followed by a Newman-Keuls test using the Statistical
Package for Social Sciences software for Windows, version 16.0
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
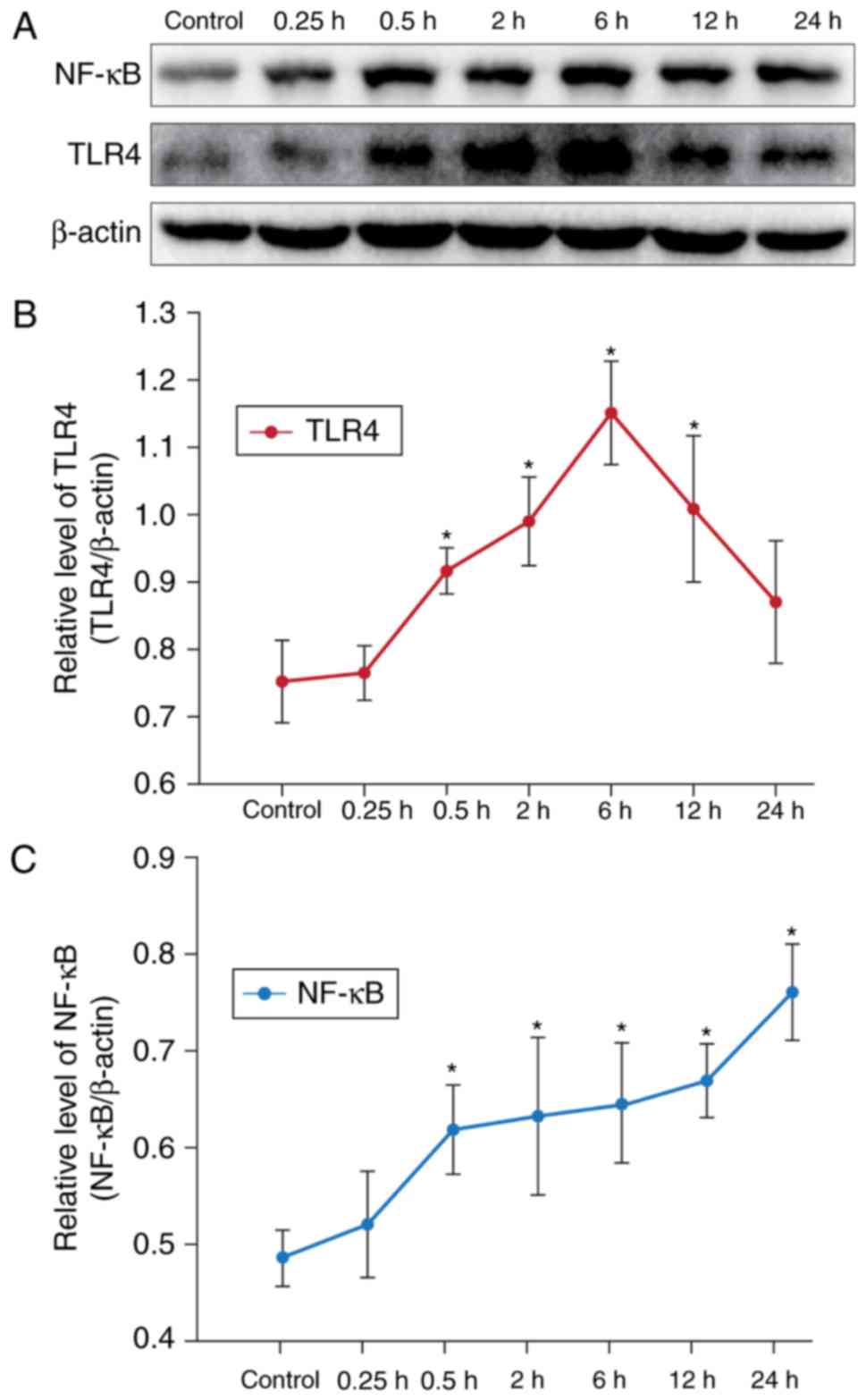
CAE enhances TLR4 and NF-κB expression
in AR42J cells
In order to investigate the effects of CAE on TLR4
and NF-κB, AR42J cells were treated with or without 10 nmol/l CAE
for various durations; TLR4 and NF-κB expression within AR42J cells
was determined by western blotting (Fig. 1A). Compared with the control group,
treatment with CAE for 0.5 h significantly increased the relative
expression levels of TLR4 (Fig.
1B) and NF-κB (Fig. 1C) in
AR42J cells (Fig. 1). The protein
expression levels of TLR4 peaked after 6 h of CAE treatment,
whereas CAE induced NF-κB expression in a time-dependent manner
within AR42J cells. In our previous experiments, it was identified
that the inflammatory response of AR42J cells was evident at 12 h,
and the expression of TLR4 and NF-κB increased markedly at this
time point. Therefore, treatment with CAE for 12 h was selected as
the model of CAE-induced acute inflammation for subsequent
experiments.
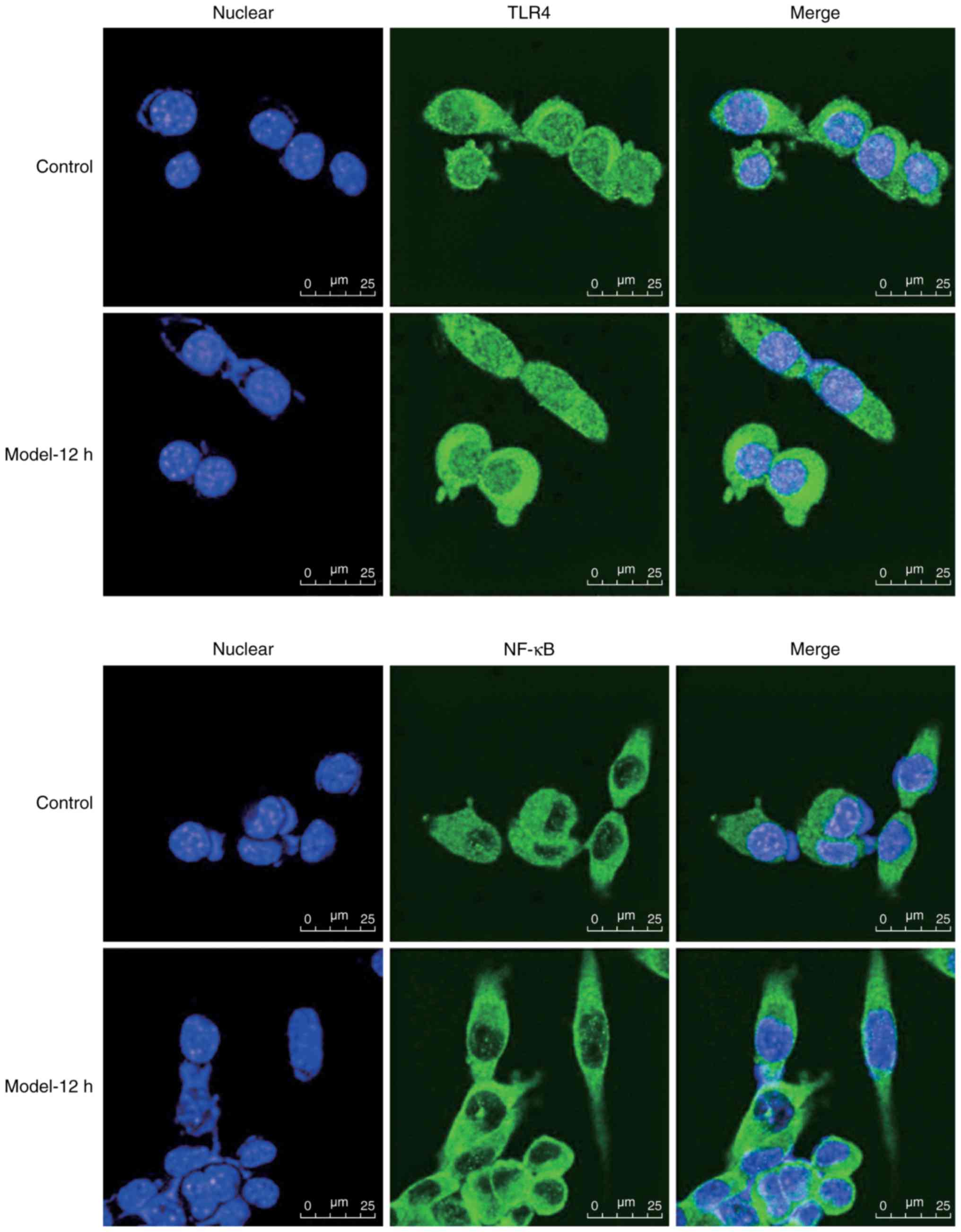
To further determine the effects of CAE on TLR4 and
NF-κB, cells were treated with CAE or vehicle for 12 h, after which
TLR4 and NF-κB expression levels were determined by
immunofluorescence. As presented in Fig. 2, weak anti-TLR4 fluorescence within
the membranes and cytoplasm, and weak anti-NF-κBp65 fluorescence in
the cytoplasm and nuclei was detected within the AR42J control
cells. Conversely, increased anti-TLR4 and anti-NF-κBp65
fluorescence was observed within the CAE-treated cells;
anti-NF-κBp65 fluorescence was greater within the nuclei of
CAE-treated cells. Collectively, these results indicated that CAE
induced TLR4 and NF-κB expression within AR42J cells.
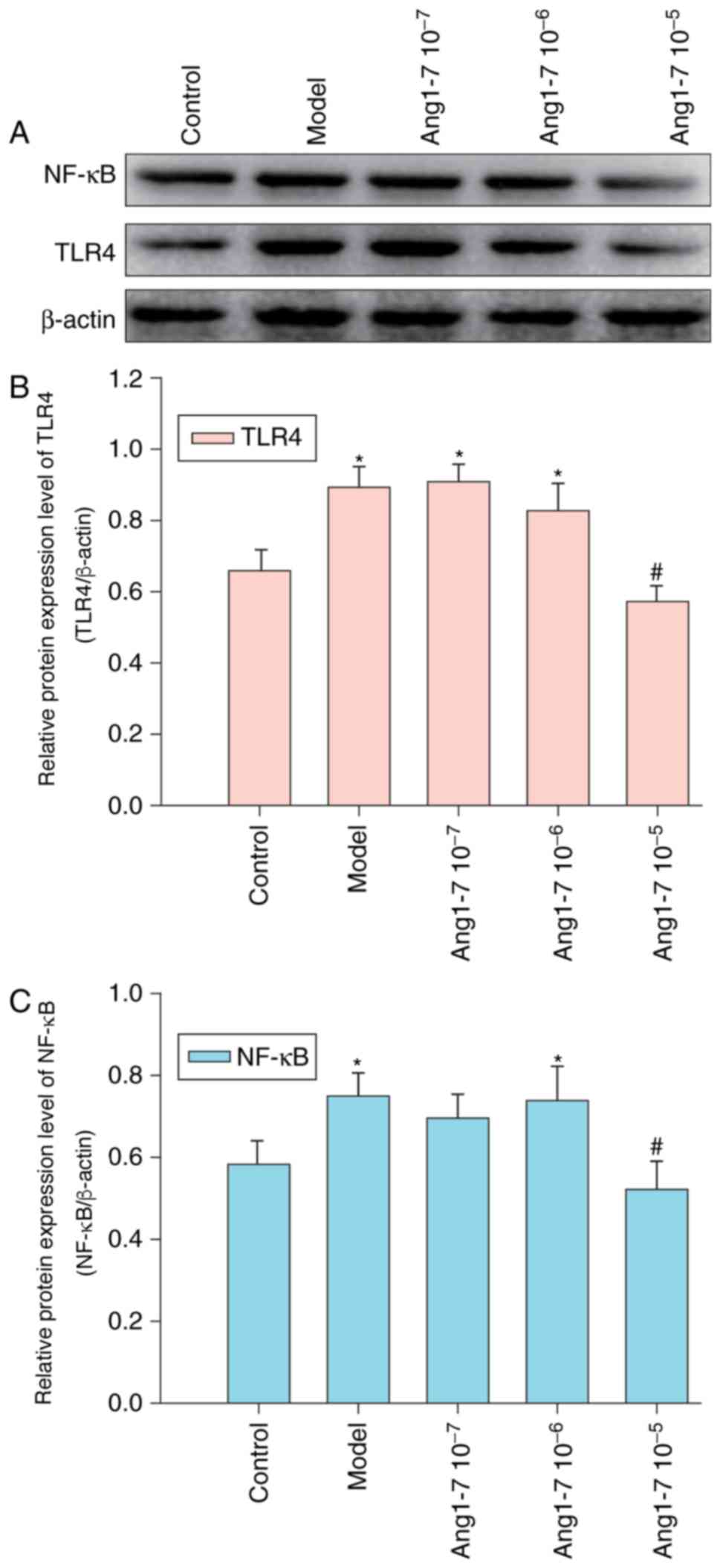
Treatment with Ang 1–7 abrogates
CAE-induced TLR4 and NF-κB expression within AR42J cells
Ang 1–7 has been reported to exhibit
anti-inflammatory activities; the effects of Ang 1–7 on CAE-induced
inflammation were investigated within AR42J cells that were treated
with or without various concentrations of Ang 1–7 and were
stimulated with CAE for 12 h. Control cells were treated with
vehicle alone. Relative protein expression levels of TLR4 and NF-κB
were determined via western blot analysis (Fig. 3). CAE treatment was observed to
significantly enhance TLR4 and NF-κB expression levels; however,
pretreatment with Ang 1–7 (10−6 or 10−7
mol/l) failed to significantly modulate the effect of CAE on TLR4
and NF-κB expression. However, 10−5 mol/l Ang 1–7
significantly abrogated CAE-induced TLR4 and NF-κB expression
within AR42J cells.
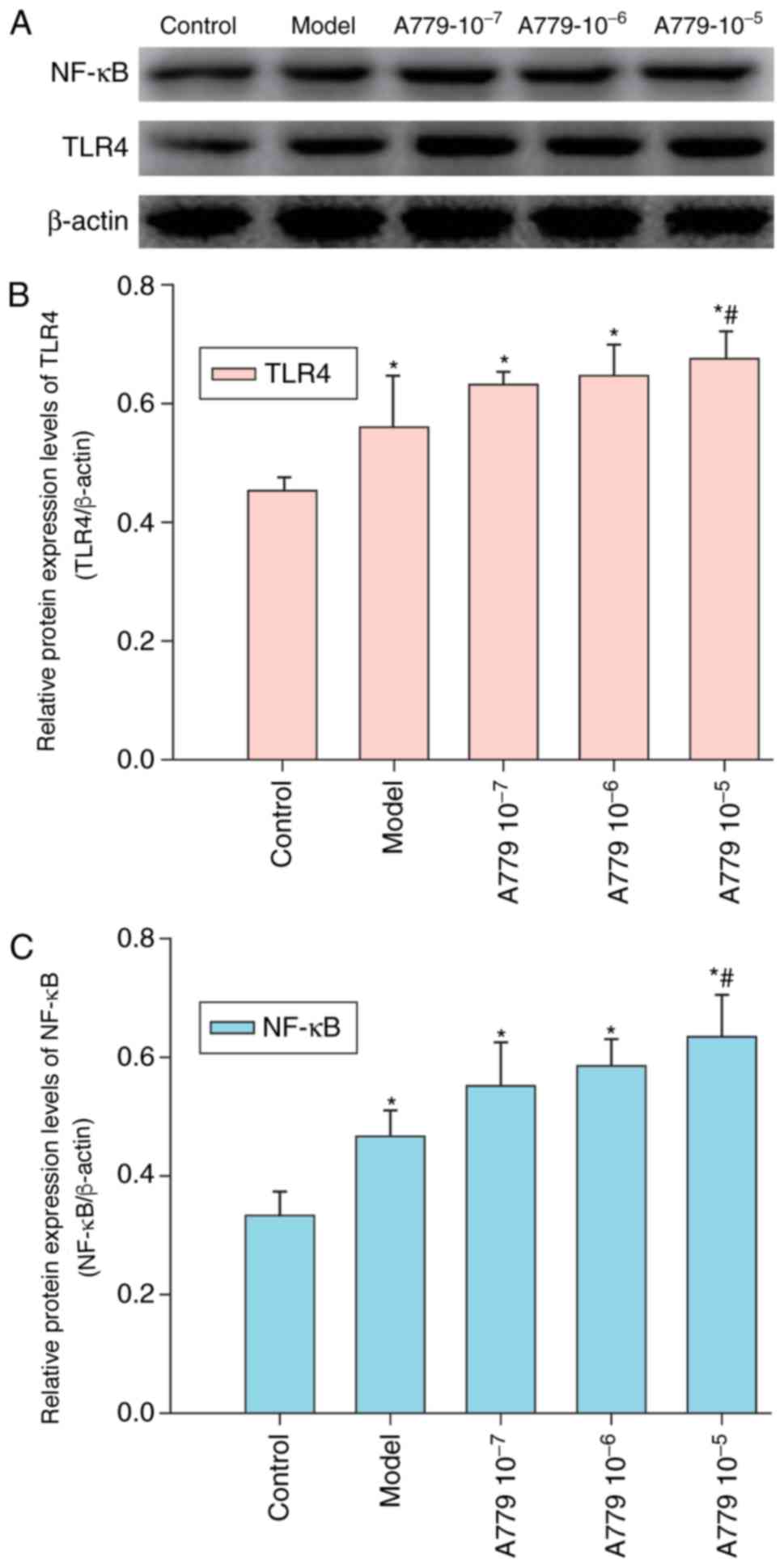
Treatment with Ang 1–7 specific
antagonist A779 enhances CAE-induced TLR4 and NF-κB expression
within AR42J cells
Our previous study demonstrated that Ang 1–7 and its
receptor Mas are expressed within AR42J cells (6). The effect of A779 on CAE-induced TLR4
and NF-κB expression was investigated in AR42J cells pretreated
with various concentrations of A779 followed by CAE treatment for
12 h. Relative expression levels of TLR4 and NF-κB were determined
by western blot analysis. As presented in Fig. 4, treatment with A779 enhanced the
expression levels of TLR4 and NF-κB within AR42J cells in a
dose-dependent manner. Pretreatment with A779 (10−5
mol/l) significantly increased TLR4 and NF-κB expression compared
with in the model group, in which cells were not pretreated with
A779 for 12 h. These results suggested that the reduction of
CAE-induced expression of TLR4 and NF-κB was enhanced by A779 with
AR42J cells.
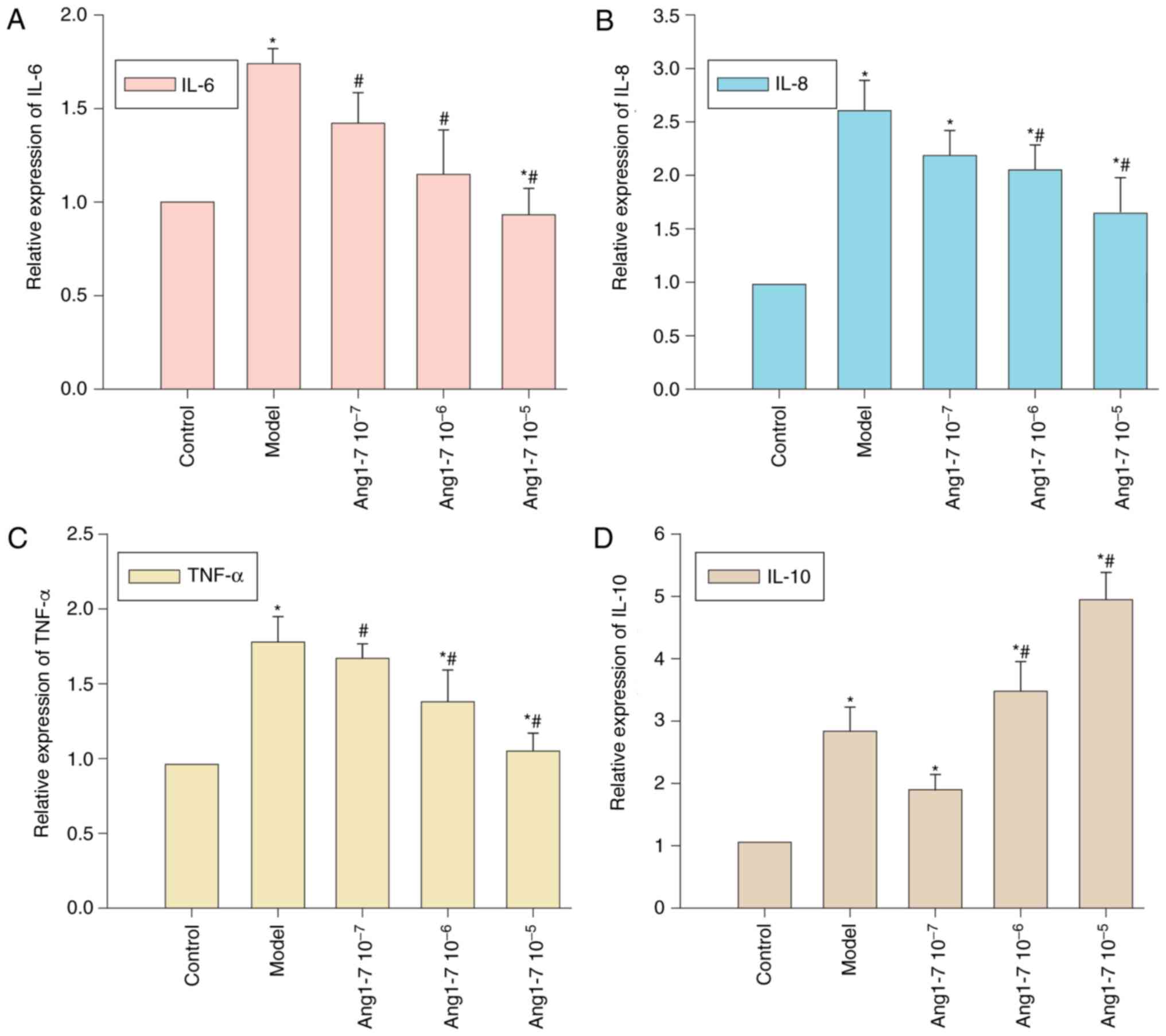
Treatment with Ang 1–7 or A779
modulates CAE-induced cytokine expression within AR42J cells
The effects exerted by Ang 1–7 and A779 on
CAE-induced cytokine expression were investigated. AR42J cells were
pretreated with Ang 1–7 and were stimulated with CAE for 12 h. The
mRNA expression levels of IL-6, IL-8, TNF-α and IL-10 relative to
GAPDH were detected using RT-qPCR. CAE treatment significantly
enhanced IL-6, IL-8, TNF-α and IL-10 expression within AR42J cells
(P<0.05, Fig. 5A-D).
Pretreatment with Ang 1–7 decreased IL-6, IL-8 and TNF-α expression
in a dose-dependent manner. A significant decrease in IL-6 and
TNF-α expression was observed with all concentrations of Ang 1–7,
whereas IL-8 expression was decreased following treatment with
10−6 and 10−5 mol/l Ang 1–7. Conversely,
10−6 and 10−5 mol/l Ang 1–7 pretreatment
increased IL-10 expression. Therefore, the addition of exogenous
Ang 1–7 was associated with a reduction in the expression of
proinflammatory cytokines and an increase in anti-inflammatory
IL-10 expression within AR42J cells.
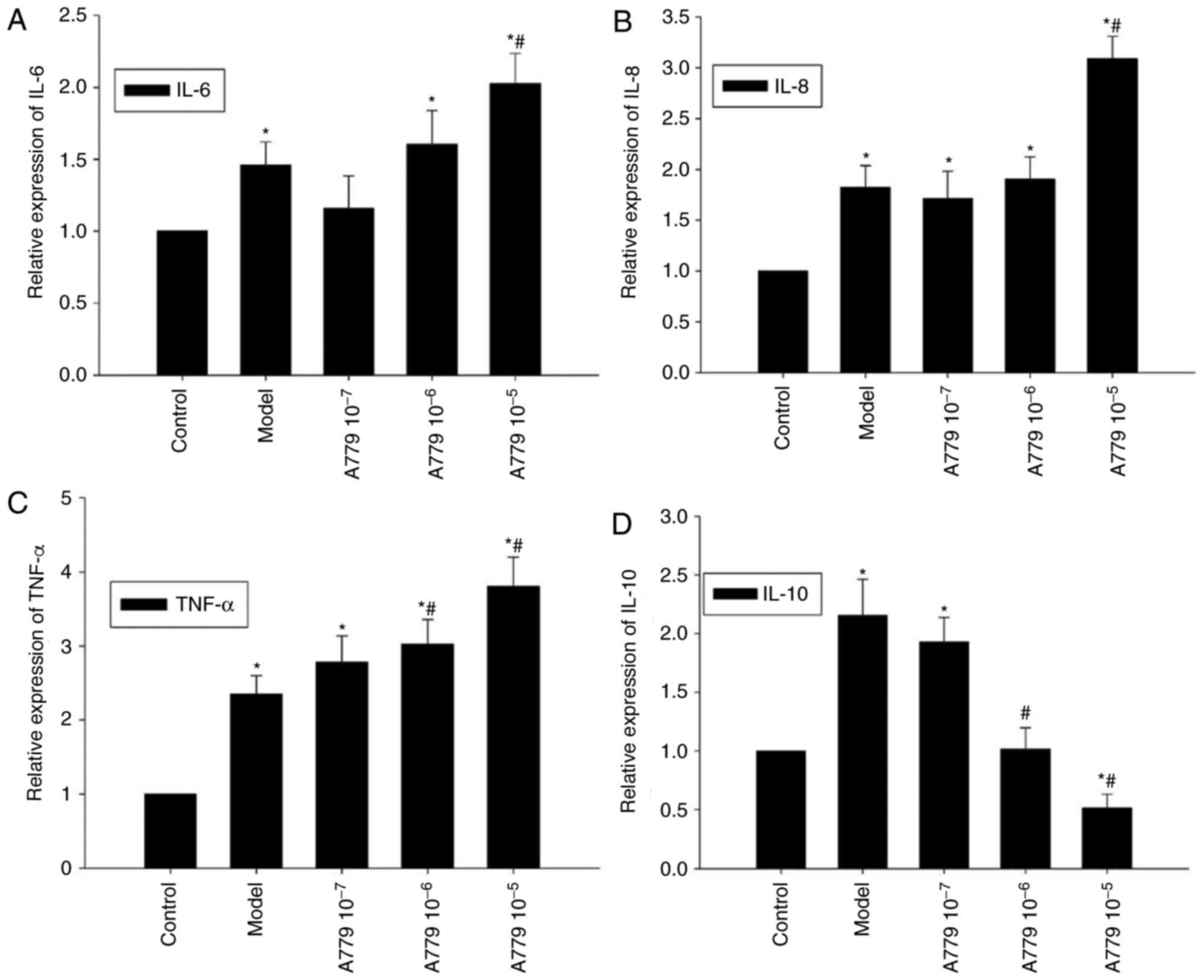
RT-qPCR was employed to investigate whether the
effects of endogenous Ang 1–7 on CAE-induced cytokine expression
may be antagonized by A779 within AR42J cells. Compared with the
model group, pretreatment with 10−5 mol/l A779
significantly increased CAE-induced expression of IL-6 and IL-8;
CAE-induced expression of TNF-α expression within AR42J cells
significantly increased with A779 pretreatment (10−6 and
10−5 mol/l; P<0.05, Fig.
6A-C). Conversely, the mRNA expression levels of IL-10 were
decreased in response to A779 pretreatment, a significant decrease
was observed with the addition of 10−6 and
10−5 mol/l A779, compared with in the control and model
groups (P<0.05; Fig. 6D).
Collectively, antagonism of endogenous Ang 1–7 via A779
significantly enhanced CAE-induced expression of proinflammatory
cytokines and decreased the expression of anti-inflammatory IL-10
within AR42J cells.
Discussion
Ang II and its receptors AT1 and AT2 serve important
roles in the pathogenesis of AP, whereas Ang 1–7 has been reported
to act as an antagonist that inhibits inflammation (16,17).
In addition, recent studies have demonstrated that Ang II can
regulate TLR4 expression in order to modulate inflammation and
other associated functions (18–20).
In the present study, the effects of endogenous and exogenous Ang
1–7 on CAE-induced inflammation within AR42J cells were
investigated. The purpose of the present study was to investigate
the effect of Ang1-7 on TLR4/NF-κB and its possible
anti-inflammatory mechanism by inhibiting TLR4 pathway, so the
effect of ANG1-7 was blocked endogenously following the addition of
the ANG1-7 antagonist A779. Endogenous blocking and exogenous
increase of Ang1-7 was used to explore its role. The results
revealed that CAE upregulated TLR4 and NF-κB expression; however,
high doses of Ang 1–7 abrogated CAE-induced TLR4 and NF-κB
expression in AR42J cells. Notably, Ang 1–7 is expressed within
R42J cells following stimulation by CAE (6). The data of the present study
indicated that endogenous Ang 1–7 may serve as a compensatory
regulator to inhibit inflammation in an autocrine or paracrine
manner during the inflammatory response in AP. This is supported by
a previous report that Ang 1–7 inhibits the TLR4/NF-κB signaling
pathway and ameliorates inflammation of the liver (21); therefore, Ang 1–7 may be considered
an anti-inflammatory factor that serves to downregulate the
TLR4/NF-κB signaling pathway during the pathogenic progression of
AP. These novel findings suggested that Ang 1–7 may serve
importance in the intervention of AP.
Pancreatitis has been recognized to be a result of
the systemic inflammatory response which activate NF-κB and
mitogen-activated protein kinases, which in turn regulate the
expression of inflammatory cytokines, including IL-1β, IL-6,
interleukin-8 and transforming growth factor-β1 in
caerulein-stimulated pancreatic acinar cells (22). Engagement of TLR4 by its ligand can
activate NF-κB and other pathways to stimulate proinflammatory
IL-6, IL-8 and TNF-α production, which also upregulates
anti-inflammatory IL-10 expression during the inflammatory process
of AP (23–25). The findings of the present study
demonstrated that CAE treatment significantly increased the
relative expression levels of IL-6, IL-8, TNF-α and IL-10 within
AR42J cells. A previous study demonstrated that pro-inflammatory
cytokines, such as TNF-α and IL-6, were greatly increased, and the
anti-inflammatory IL-10 was markedly decreased in the circulation
after induction of SAP. The Ace2 KO mice exhibited increased
levels of TNF-α, IL-1β, IL-6, multifocal coagulative necrosis and
inflammatory infiltrate, and lower levels of serum IL-10 and
pancreatic Ang-(1–7) compared with caerulein-treated WT mice at the
same time point (1). Combined with
the current research, these findings suggested that an imbalance
between proinflammatory and anti-inflammatory cytokine responses is
crucial for the pathogenesis of AP. In addition, treatment with
exogenous Ang 1–7 reduced the CAE-induced IL-6, IL-8 and TNF-α
expression, but increased IL-10 production in AR42J cells.
Conversely, antagonism of Ang 1–7 exerted by A779 treatment
increased CAE-induced expression of IL-6, IL-8 and TNF-α, and
decreased IL-10 expression within AR42J cells. Additionally,
endogenous and exogenous Ang 1–7 has been reported to modulate the
imbalance between pro- and anti-inflammatory cytokine responses to
limit inflammation during the pathogenesis of AP (26). A previous study suggested that
inflammatory cytokines may be considered prognostic markers in the
progression of SAP (27). The
findings of the present study indicated that the imbalance between
pro- and anti-inflammatory cytokine responses may serve importance
in the evaluation of AP-associated inflammation reaction. Whether
endogenous and exogenous Ang 1–7 can downregulate TLR4 and NF-κB
expression, and alter the imbalance between pro- and
anti-inflammatory cytokines, in vivo remains to be
elucidated. In addition, the potential mechanisms underlying the
effects of Ang 1–7 on TLR-4 and NF-κB expression during the
progression of AP have yet to be investigated.
In conclusion, the results of the present study
indicated that CAE induced the expression of TLR4 and NF-κB, as
well as pro- and anti-inflammatory cytokines, within AR42J cells,
which was downregulated by endogenous and exogenous Ang 1–7. These
findings may provide novel insights into the pathophysiological
mechanism of pancreatitis and provide a new target for the
treatment of pancreatitis.
Acknowledgements
The authors of the present study would like to thank
their colleagues and the expert panel members for their support and
help. The present study was supported by grants from the National
Natural Science Foundation of China (grant no. 81441060), the
Research Foundation of Beijing Friendship Hospital, Capital Medical
University (grant no. yyqdkt2014-4) and Beijing Municipal
Administration of Hospitals' Youth Programme, (grant no.
QML20150104).
Glossary
Abbreviations
Abbreviations:
|
Ang
|
angiotensin
|
|
CAE
|
caerulein
|
|
RAS
|
renin-angiotensin system
|
|
ACE2
|
angiotensin I converting enzyme 2
|
|
AP
|
acute pancreatitis
|
|
TLRs
|
Toll-like receptors
|
|
LPS
|
lipopolysaccharide
|
|
TNF-α
|
tumor necrosis factor-α
|
References
|
1
|
Liu R, Qi H, Wang J, Wang Y, Cui L, Wen Y
and Yin C: Angiotensin-converting enzyme (ACE and ACE2) imbalance
correlates with the severity of cerulein-induced acute pancreatitis
in mice. Exp Physiol. 99:651–663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Y, Wang J, Liu R, Qi H, Wen Y, Sun F
and Yin C: Severe acute pancreatitis is associated with
upregulation of the ACE2-angiotensin-(1–7)-Mas axis and promotes
increased circulating angiotensin-(1–7). Pancreatology. 12:451–457.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mori J, Patel VB, Ramprasath T, Alrob OA,
DesAulniers J, Scholey JW, Lopaschuk GD and Oudit GY: Angiotensin
1–7 mediates renoprotection against diabetic nephropathy by
reducing oxidative stress, inflammation, and lipotoxicity. Am J
Physiol Renal Physiol. 306:F812–F821. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu CL, Wang Y, Yuan L, Li Y and Li XY: The
angiotensin-converting enzyme 2/angiotensin (1–7)/Mas axis protects
the function of pancreatic β cells by improving the function of
islet microvascular endothelial cells. Int J Mol Med. 34:1293–300.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan L, Lu CL, Wang Y, Li Y and Li XY: Ang
(1–7) protects islet endothelial cells from palmitate-induced
apoptosis by AKT, eNOS, p38 MAPK, and JNK pathways. J Diabetes Res.
2014:3914762014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang J, Liu R, Qi H, Wang Y, Cui L, Wen Y,
Li H and Yin C: The ACE2-angiotensin-(1–7)-Mas axis protects
against pancreatic cell damage in cell culture. Pancreas.
44:266–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gordon S: Pattern recognition receptors:
Doubling up for the innate immune response. Cell. 111:927–930.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan LF, Yu L, Wang LM, He JT, Sun JL, Wang
XB, Bai ZH, Wang H, Yan TL and Pei HH: The Toll-like receptor 4
antagonist TAK-242 protects against chronic pancreatitis in rats.
Mol Med Rep. 16:3863–3868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li G, Wu X, Yang L, He Y, Liu Y, Jin X and
Yuan H: [Corrigendum] TLR4-mediated NF-κB signaling pathway
mediates HMGB1-induced pancreatic injury in mice with severe acute
pancreatitis. Int J Mol Med. 38:13132016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Awla D, Abdulla A, Regnér S and Thorlacius
H: TLR4 but not TLR2 regulates inflammation and tissue damage in
acute pancreatitis induced by retrograde infusion of taurocholate.
Inflamm Res. 60:1093–1098. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xue J and Habtezion A: Carbon
monoxide-based therapy ameliorates acute pancreatitis via TLR4
inhibition. J Clin Invest. 124:437–447. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li S, Lu H, Hu X, Chen W, Xu Y and Wang J:
Expression of TLR4-MyD88 and NF-κB in the iris during
endotoxin-induced uveitis. Mediators Inflamm. 2010:7482182010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chan YC and Leung PS: Angiotensin II type
1 receptor-dependent nuclear factor-kappaB activation-mediated
proinflammatory actions in a rat model of obstructive acute
pancreatitis. J Pharmacol Exp Ther. 323:10–18. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu JH, Lim JW and Kim H: Altered gene
expression in cerulein-stimulated pancreatic acinar cells:
Pathologic mechanism of acute pancreatitis. Korean J Physiol
Pharmacol. 13:409–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Furukawa H, Shinmura A, Tajima H, Tsukada
T, Nakanuma S, Okamoto K, Sakai S, Makino I, Nakamura K, Hayashi H,
et al: Concentration of tissue angiotensin II increases with
severity of experimental pancreatitis. Mol Med Rep. 8:335–338.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shimizu K: Mechanisms of pancreatic
fibrosis and applications to the treatment of chronic pancreatitis.
J Gastroenterol. 43:823–832. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dange RB, Agarwal D, Masson GS, Vila J,
Wilson B, Nair A and Francis J: Central blockade of TLR4 improves
cardiac function and attenuates myocardial inflammation in
angiotensin II-induced hypertension. Cardiovasc Res. 103:17–27.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lv J, Chen Q, Shao Y, Chen Y and Shi J:
Cross-talk between angiotensin-II and toll-like receptor 4 triggers
a synergetic inflammatory response in rat mesangial cells under
high glucose conditions. Biochem Biophys Res Commun. 459:264–269.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wolf G, Bohlender J, Bondeva T, Roger T,
Thaiss F and Wenzel UO: Angiotensin II upregulates toll-like
receptor 4 on mesangial cells. J Am Soc Nephrol. 17:1585–1593.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Santos SH, Andrade JM, Fernandes LR,
Sinisterra RD, Sousa FB, Feltenberger JD, Alvarez-Leite JI and
Santos RA: Oral Angiotensin-(1–7) prevented obesity and hepatic
inflammation by inhibition of resistin/TLR4/MAPK/NF-κB in rats fed
with high-fat diet. Peptides. 46:47–52. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ju KD, Lim JW, Kim KH and Kim H: Potential
role of NADPH oxidase-mediated activation of Jak2/Stat3 and
mitogen-activated protein kinases and expression of TGF-β1 in the
pathophysiology of acute pancreatitis. Inflamm Res. 60:791–800.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Johnson GB, Brunn GJ and Platt JL: Cutting
edge: An endogenous pathway to systemic inflammatory response
syndrome (SIRS)-like reactions through Toll-like receptor 4. J
Immunol. 172:20–24. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lai JL, Liu YH, Liu C, Qi MP, Liu RN, Zhu
XF, Zhou QG, Chen YY, Guo AZ and Hu CM: Indirubin inhibits
LPS-induced inflammation via TLR4 abrogation mediated by the NF-κB
and MAPK signaling pathways. Inflammation. 40:1–12. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu M, Wang KN, Wu K and Wang XP:
Pyrrolidine dithiocarbamate inhibits nuclear factor κB and
Toll-like receptor 4 expression in rats with acute necrotizing
pancreatitis. Gut Liver. 9:411–416. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Simões e Silva AC, Silveira KD, Ferreira
AJ and Teixeira MM: ACE2, angiotensin-(1–7) and Mas receptor axis
in inflammation and fibrosis. Br J Pharmacol. 169:477–492. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aoun E, Chen J, Reighard D, Gleeson FC,
Whitcomb DC and Papachristou GI: Diagnostic accuracy of
interleukin-6 and interleukin-8 in predicting severe acute
pancreatitis: A meta-analysis. Pancreatology. 9:777–785. 2009.
View Article : Google Scholar : PubMed/NCBI
|