Introduction
The retinal pigment epithelium (RPE) is composed of
a simple layer of pigmented cells and is an important element of
the blood-retina barrier. Dysfunction of the RPE may result in
retinal degeneration, visual function loss and blindness (1). The epithelial-mesenchymal transition
(EMT) of the RPE is implicated in various intraocular fibrotic
disorders, including proliferative vitreoretinopathy (2–5),
proliferative diabetic retinopathy (6) and wet age-related macular
degeneration (AMD) (7). Fibrotic
lesions induce retinal detachment and result in severe visual
impairment. However, the complex underlying mechanisms of the EMT
process in the RPE (RPE-EMT) are yet to be fully determined.
Numerous studies have reported that autophagy is
involved in the EMT process in numerous tissue and cell types,
including human malignant glioma cells, atrial myofibroblasts and
annulus fibrosus cells (8–10). Furthermore, autophagy has been
demonstrated to exert beneficial and non-beneficial effects on EMT,
which may be dependent on the associated cell type (11–14).
However, to the best of our knowledge, it has not previously been
determined whether autophagy activity is involved in the process of
RPE-EMT.
Autophagy is an intracellular homeostatic pathway
that assists in the degradation and recycling of proteins and
cellular organelles (15).
Autophagy is activated in response to metabolic stress and other
variations within the microenvironment (8). Autophagy is crucial for the
maintenance of RPE homeostasis. Autophagy aids the degradation of
cytotoxic protein aggregates in RPE cells that result from
stimulation by photo-oxidative stress and inflammation (16). In RPE cells, autophagy is involved
in the prevention of cytotoxic protein aggregates; however, this
capacity may be decreased in senescent or stressed RPE cells, and
lipofuscin accumulation into lysosomes may be induced by decreased
autophagy. The formation of lipofuscin impairs the autophagic
clearance of protein aggregates and induces further RPE damage,
which may contribute to AMD progression or alternative pathological
development (17–19).
RPE-EMT is an important pathological process in
intraocular fibrotic disorders that results in severe
non-reversible retinal pathological development. Therefore, it is
important to determine the role of autophagy in the process of
RPE-EMT. The present study aimed to investigate whether autophagy
deficiency may induce or modulate the EMT process in RPE cells. It
is well established that transforming growth factor (TGF)-β2 is a
potent inducer of EMT; TGF-β2-induced EMT has been reported by
numerous studies using in vitro EMT models (20–22).
In the present study, the role of autophagy in TGF-β2-induced EMT
in human RPE (ARPE-19 cell line) cells was investigated. The
results demonstrated that the administration of TGF-β2 induced
autophagy in ARPE-19 cells. Furthermore, the activation of
autophagy enhanced the RPE-EMT process, while inhibition of
autophagy attenuated the RPE-EMT process, in ARPE-19 cells.
Materials and methods
Cell culture and cell treatment
The ARPE-19 human RPE cell line was obtained from
State Key Laboratory of Ophthalmology (Zhongshan Ophthalmic Center,
Sun Yat-sen University, Guangzhou, China). The cells were cultured
in complete medium composed of Dulbecco's modified Eagle's medium
(DMEM; GIBCO; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 1% penicillin-streptomycin solution (GIBCO;
Thermo Fisher Scientific, Inc.) and 10% fetal bovine serum (GIBCO;
Thermo Fisher Scientific, Inc.) at 37°C in 5% CO2
humidified atmosphere. The cells were routinely passaged at a
confluence between 80–90% for ~3–4 days and dissociated with 0.25%
trypsin-EDTA solution (GIBCO; Thermo Fisher Scientific, Inc.). The
ARPE-19 cells were subsequently transferred to a 6-well plate.
After reaching 70–80% confluence, the medium was replaced with
fresh medium prior to drug treatment. Following this, recombinant
human TGF-β2 (2, 5 or 10 ng/ml; R&D Systems, Inc., Minneapolis,
MN, USA) was added to the cells with or without chloroquine (50 µM;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), 3-methyladenine
(3-MA; 5 mM; Sigma-Aldrich; Merck KGaA) and rapamycin (200 nM;
Sigma-Aldrich; Merck KGaA) for 0–36 h. The control cells were
treated with medium only without drugs. All the cells were cultured
at 37°C in 5% CO2 humidified atmosphere.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cultures using the
RNeasy Mini kit (Qiagen, Inc., Valencia, CA, USA), in accordance
with the manufacturer's protocol. RNA concentration was quantified
using the NanoDrop 2000 spectrophotometer (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Following this, total RNA (2
µg) was reverse transcribed using the Maxima First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Inc.). The reaction
mixture was incubated for 10 min at 25°C followed by 30 min at
50°C, and the reaction was terminated by heating at 85°C for 5 min.
qPCR was subsequently performed using the StepOnePlus Real-Time PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.) using
the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The reaction is as
follows: 50°C for 2 min for UDG activation and 95°C for 2 min for
the activation of dual-lock DNA polymerase. The PCR amplification
was then performed for 40 cycles by denaturing the cDNA template at
95°C for 3 sec and annealing/extension at 60°C for 30 sec. The
dissociation curve includes 95°C for 15 sec, 60°C for 1 min and
95°C for 15 sec. The primers (Thermo Fisher Scientific, Inc.) used
were as follows: Human neural (N)-cadherin,
5′-AGCCAACCTTAACTGAGGAGT-3′ (forward) and
5′-GGCAAGTTGATTGGAGGGATG-3′ (reverse); human vimentin,
5′-AGTCCACTGAGTACCGGAGAC-3′ (forward) and
5′-CATTTCACGCATCTGGCGTTC-3′ (reverse); human fibronectin,
5′-CGGTGGCTGTCAGTCAAAG-3′ (forward) and 5′-AAACCTCGGCTTCCTCCATAA-3′
(reverse); human GAPDH, 5′-CTGGGCTACACTGAGCACC-3′ (forward) and
5′-AAGTGGTCGTTGAGGGCAATG-3′ (reverse). All samples were tested and
normalized to the reference gene GAPDH. All experiments were
performed in triplicate. Results were normalized to GAPDH according
to the 2−∆∆Cq relative quantification method (23).
Western blot analysis
Total protein was extracted from cells using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China) with 1 mM phenylmethylsulfonyl
fluoride (Beyotime Institute of Biotechnology) on ice for 20 min.
Protein was subsequently quantified using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology). Following protein
quantification, 20 µg of each sample was run on a 12 or 8% SDS-PAGE
gel and proteins were transferred to polyvinylidene difluoride
membranes (Merck KGaA, Darmstadt). Following blocking with 5%
bovine serum albumin (MP Biomedicals, LLC., Santa Ana, CA, USA) for
1 h at room temperature, the membranes were incubated overnight at
4°C with the following primary antibodies: Rabbit
anti-microtubule-associated protein 1 light chain (LC3) 3α/β
(1:1,000; cat. no. 12741S; Cell Signaling Technology, Inc.,
Danvers, MA, USA; differences in molecular weight was used to
distinguish between the cytosolic and membrane bound forms), rabbit
anti-beclin 1 (1:1,000; cat. no. 3495S, Cell Signaling Technology,
Inc.), rabbit anti-p62 (also termed sequestosome 1; 1:1,000; cat.
no. 8025S; Cell Signaling Technology, Inc.), rabbit anti-vimentin
(1:1,000; cat. no. 5741; Cell Signaling Technology, Inc.), mouse
anti-β-actin (1:1,000; cat. no. 3700; Cell Signaling Technology,
Inc.) and rabbit anti-fibronectin (1:1,000; cat. no. ab6328; Abcam,
Cambridge, MA, USA); and rabbit anti-N-cadherin (1:1,000; cat. no.
04–1126; Merck KGaA, Darmstadt, Germany). Following washing with
Tris-buffered saline solution containing 0.05% Tween-20 buffer, the
membranes were incubated with anti-mouse or anti-rabbit horseradish
peroxidase-conjugated secondary antibodies (1:2,000) for 1 h at
room temperature (anti-mouse secondary antibody cat. no. sc-2005;
anti-rabbit secondary antibody cat. no. sc-2004; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). Luminol reagent was used as
the visualization reagent (Merck KGaA) and blots were analyzed
using the FluorChem Q system (ProteinSimple, San Jose, CA, USA).
ImageJ v10.2 (National Institutes of Health, Bethesda, MD, USA) was
used for quantification of the bands relative to β-actin expression
and normalization to total protein loaded in each lane.
Transwell migration and Matrigel
invasion assays
Transwell Chambers with 8.0-µm pore inserts (Corning
Incorporated, Corning, NY, USA) were used to investigate RPE cell
migration and invasion. To perform Transwell migration assays,
1×105 ARPE-19 cells were placed in the upper chamber
with a volume of 200 µl serum-free DMEM. Subsequently, 5 ng/ml
TGF-β2, 50 µM chloroquine, 5 ng/ml TGF-β2 + 50 µM chloroquine, 200
nM rapamycin or 5 ng/ml TGF-β2 + 200 nM rapamycin was added to the
upper chamber in each group. A total of 600 µl DMEM with 10% fetal
bovine serum was added to the lower chamber of each well. Following
24 h incubation at 37°C, the non-migrating cells were scraped off
with a cotton swab, and the migrated cells on the lower surface
were fixed with 4% paraformaldehyde for 30 min at room temperature
and stained with 0.1% crystal violet for 1 h at room temperature.
To assess the average number of migrating cells, images
(magnification, ×100) were captured with an inverted light
microscope (Zeiss Observer A1, Oberkochen, Germany) and cells were
counted in five randomly selected fields with Adobe Photoshop CC
2015 (Adobe Systems, Inc., San Jose, CA, USA). All experiments were
repeated three times. In order to perform the Transwell invasion
assays, Matrigel was melted at 4°C and diluted using serum-free
DMEM (1:3). Subsequently, 40 µl diluted Matrigel was added to the
Transwell chamber inserts, which were then placed in the incubator
at 37°C for 4 h to coagulate. The remainder of the assay was
performed in accordance with the aforementioned migration assay
protocol.
Statistical analysis
Data presented in the figures are representative of
three repetitions. Data were analyzed by one-way analysis of
variance with Tukey's post-hoc test using GraphPad Prism version
7.0 (GraphPad Software, Inc., La Jolla, CA, USA). Data are
presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
TGF-β2 induces autophagy in cultured
ARPE-19 cells
To determine whether autophagy is modulated during
EMT, the effects of TGF-β2 on autophagy were investigated by
measuring the protein expression of LC3, beclin-1 and p62 using
western blot analysis (Fig. 1).
LC3-phosphatidylethanolamine conjugate (LC3-II) is the lipidated
form of the cystolic form of LC3 (LC3-I), and the conversion from
LC3-I to LC3-II represents the formation of autophagosomes
(24). p62 combines
polyubiquitinated proteins and forms the completed autophagosomes,
which are subsequently degraded into autolysosomes; therefore, the
quantity of p62 may be considered an index of autophagic
degradation (24). As demonstrated
in Fig. 1A and B, the
administration of TGF-β2 increased the expression of LC3-II at 12,
24 and 36 h post-treatment (Fig.
1B), compared with the control group. However, compared with
the control group, the protein expression of p62 was significantly
decreased following TGF-β2 administration in a time-dependent
manner between 0 and 36 h (Fig. 1A and
C). The protein expression of LC3-II also increased following
treatment of cells with different concentrations of TGF-β2,
compared with the control group (2, 5 and 10 ng/ml; Fig. 1D and E), while p62 expression was
significantly decreased following administration of TGF-β2 in a
dose-dependent manner between 2 and 10 ng/ml, compared with the
control group (Fig. 1D and F). No
marked alterations in beclin-1 expression were observed across
treatment groups (Fig. 1A and D).
These results indicate that administration of TGF-β2 induced
autophagy in ARPE-19 cells.
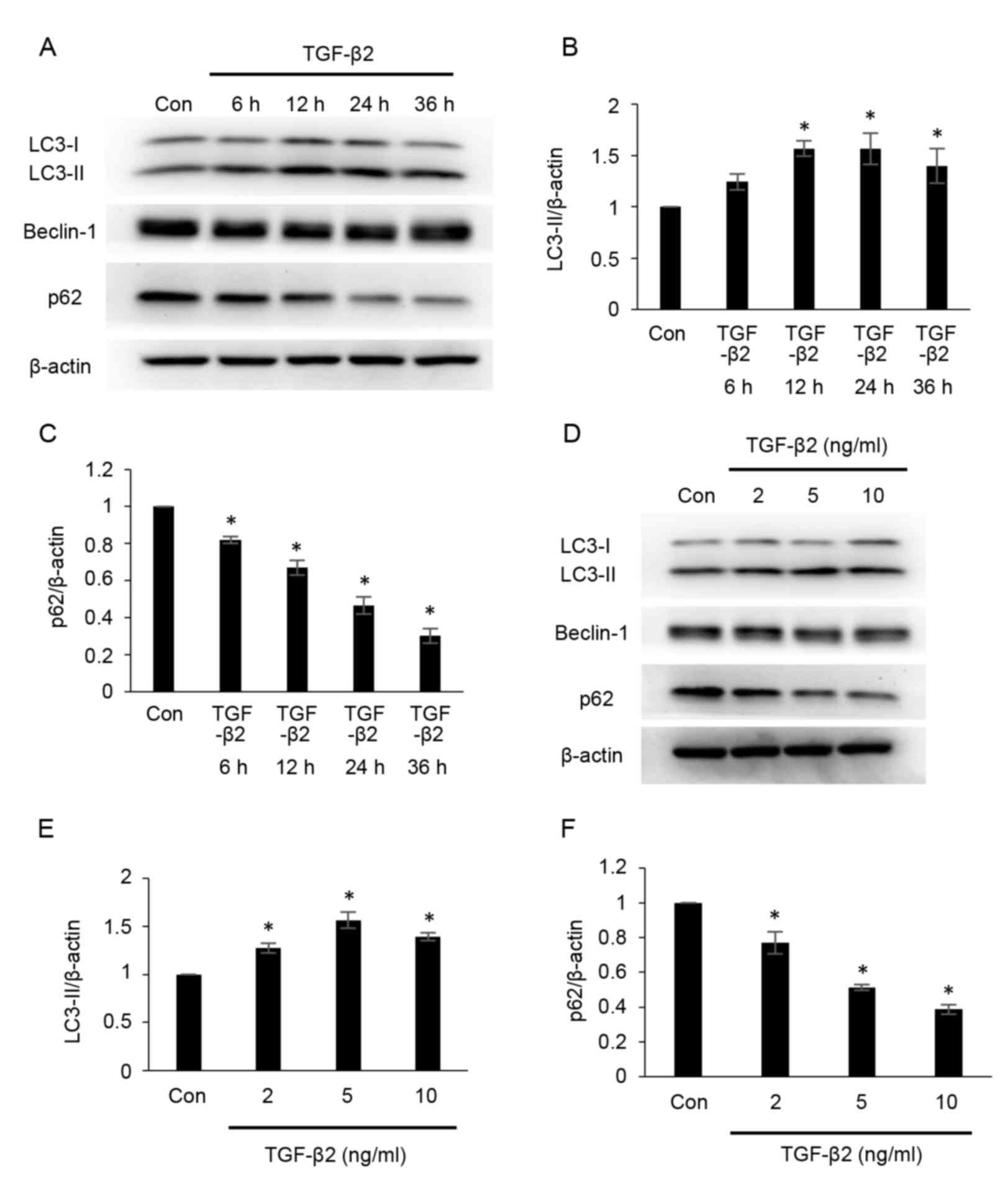 | Figure 1.TGF-β2 induces autophagy in cultured
ARPE-19 cells. (A) ARPE-19 cells were treated with TGF-β2 (5 ng/ml)
for 0–36 h. Protein expression levels of LC3-II, beclin-1 and p62
were analyzed by western blotting. Protein loading was confirmed
using β-actin. Densitometric analysis of (B) LC3-II and (C) p62
protein expression levels in ARPE-19 cells treated with TGF-β2 (5
ng/ml) for 0–36 h. (D) ARPE-19 cells were cultured with various
concentrations of TGF-β2 (2, 5 and 10 ng/ml) for 12 h. LC3-II,
beclin-1 and p62 protein expression levels were analyzed by western
blotting. Densitometric analysis of (E) LC3-II and (F) p62 protein
expression levels in ARPE-19 cells treated with TGF-β2 (2, 5 and 10
ng/ml) for 12 h. For each experiment, LC3-II and p62 expression
levels were normalized to corresponding β-actin expression levels.
Bars represent the mean ± standard deviation of three independent
experiments. *P<0.05 vs. Con. TGF-β2, transforming growth
factor-β2; LC3, microtubule-associated protein 1 light chain;
LC3-II, LC3-phosphatidylethanolamine conjugate; p62, sequestosome
1; LC3-I, cytosolic form of LC3; Con, control. |
Effects of autophagy inhibitors and
inducers on autophagy-associated protein expression in cultured
ARPE-19 cells
To validate the effects of autophagy inhibitors and
inducers on autophagy-associated protein expression in ARPE-19
cells, ARPE-19 cells were treated with TGF-β2 (5 ng/ml) with or
without chloroquine (50 µM), 3-MA (5 mM) or rapamycin (200 nM) for
24 h, and the protein expression levels of LC3 and p62 were
analyzed by western blotting. As demonstrated in Fig. 2, administration of chloroquine
resulted in the accumulation of LC3-II and the inhibition of p62
degradation, thus demonstrating an inhibitory effect with regards
to autophagy. Although administration of chloroquine led to LC3-II
accumulation rather than reduced LC3-II expression, accumulation of
LC3-II occurs as the autophagosome-lysosome fusion at the
post-sequestration step following LC3-II formation is inhibited by
chloroquine, which represents inhibition of autophagy (24). 3-MA is a selective
phosphatidylinositol 3-kinase inhibitor. The effect of 3-MA on
autophagy is conditional and depends on the time and the
environment of application (24).
In the present study, administration of 3-MA decreased the
expression levels of LC3-II and p62, therefore demonstrated the
increase of autophagic flux and demonstrated the activation of
autophagy (Fig. 2). Rapamycin is a
mechanistic target of rapamycin inhibitor and predominantly
functions as an autophagy inducer (24). Administration of rapamycin
increased LC3-II expression and p62 degradation, therefore
indicating that rapamycin activated autophagy (Fig. 2).
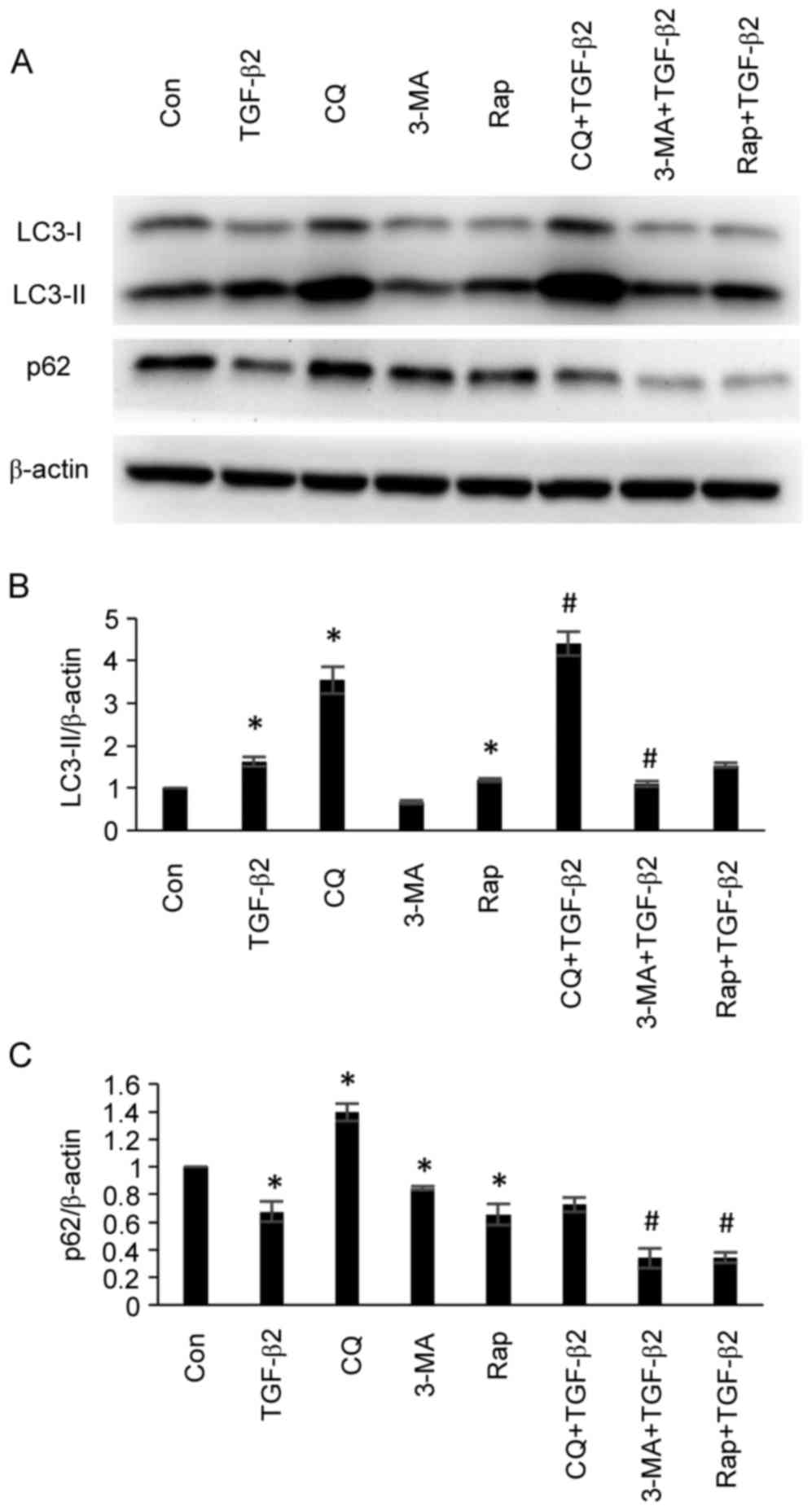 | Figure 2.Effects of the administration of
chloroquine, 3-MA and rapamycin on the expression levels of
autophagy-associated proteins in ARPE-19 cells. (A) ARPE-19 cells
were treated with TGF-β2 (5 ng/ml) with or without chloroquine (50
µM), 3-MA (5 mM) and rapamycin (200 nM) for 24 h. The protein
expression levels of LC3-II and p62 were analyzed by western
blotting. Administration of chloroquine did not decrease LC3-II
expression, however, accumulation of LC3-II was observed as
chloroquine interrupted the autophagosome-lysosome fusion at the
post-sequestration step in autophagy following LC3-II formation.
Chloroquine inhibited the degradation of p62 and demonstrated an
inhibitory effect with regards to autophagy. Administration of 3-MA
decreased the expression levels of LC3-II and p62, which indicated
that autophagy was induced. Administration of rapamycin increased
LC3-II expression and p62 degradation, thus indicating that
rapamycin administration induced the activation of autophagy.
Densitometric analysis of (B) LC3-II and (C) p62 protein expression
levels in ARPE-19 cells. Bars represent the mean ± standard
deviation of three independent experiments. *P<0.05 vs. Con;
#P<0.05 vs. TGF-β2 group. 3-MA, 3-methyladenine;
TGF-β2, transforming growth factor-β2; LC3, microtubule-associated
protein 1 light chain; LC3-II, LC3-phosphatidylethanolamine
conjugate; p62, sequestosome 1; LC3-I, cytosolic form of LC3; Con,
control; CQ, chloroquine, Rap, rapamycin. |
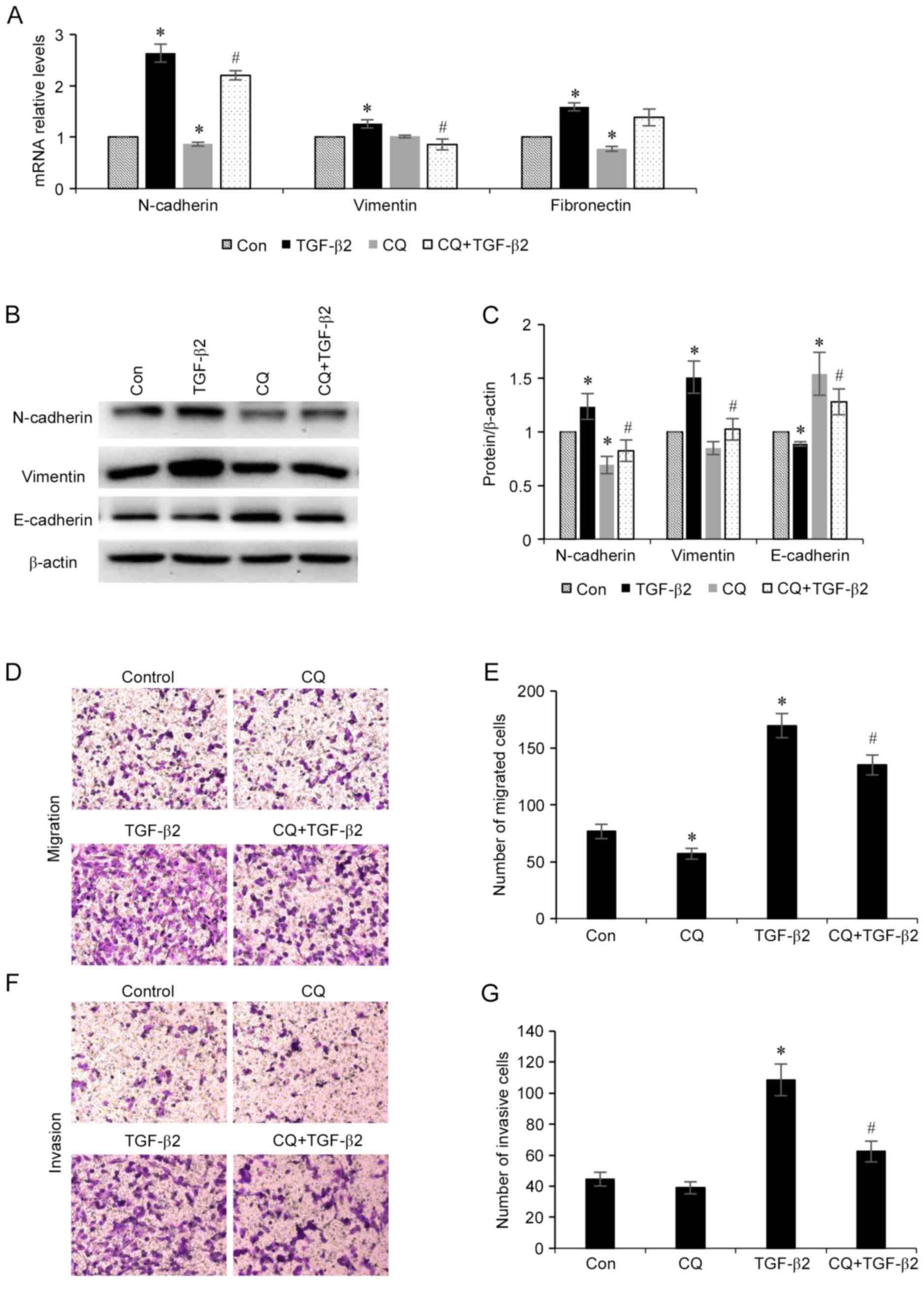
Inhibition of autophagy suppresses
TGF-β2-induced EMT
As autophagy is activated during TGF-β2-induced EMT
in ARPE-19 cells, the association between the EMT process and
autophagy level was investigated. Furthermore, whether the
modulation of autophagy may influence the EMT process was also
investigated. ARPE-19 cells were treated with TGF-β2 (5 ng/ml) with
or without chloroquine (50 µM) for 24 h, total RNA was collected
and the mRNA expression levels of N-cadherin, vimentin and
fibronectin mesenchymal markers were determined. As revealed in
Fig. 3A, chloroquine
administration decreased the mRNA expression levels of all three of
these mesenchymal markers. Furthermore, the protein expression
levels of mesenchymal markers (N-cadherin and vimentin) and the
epithelial marker epithelial (E)-cadherin in ARPE-19 cells were
investigated (Fig. 3B and C). It
was demonstrated that chloroquine administration decreased the
expression levels of N-cadherin and vimentin, and increased the
expression level of E-cadherin. Additionally, the effect of
chloroquine administration on RPE cell migration and invasion was
also investigated (Fig. 3D-G). It
was demonstrated that administration of TGF-β2 enhanced the
migratory and invasive potential of the RPE cells compared with
control cells, and treatment with chloroquine significantly reduced
TGF-β2-induced RPE cell migration and invasion. These results
indicated that inhibition of autophagy may suppress the EMT process
that is induced by TGF-β2 in RPE cells.
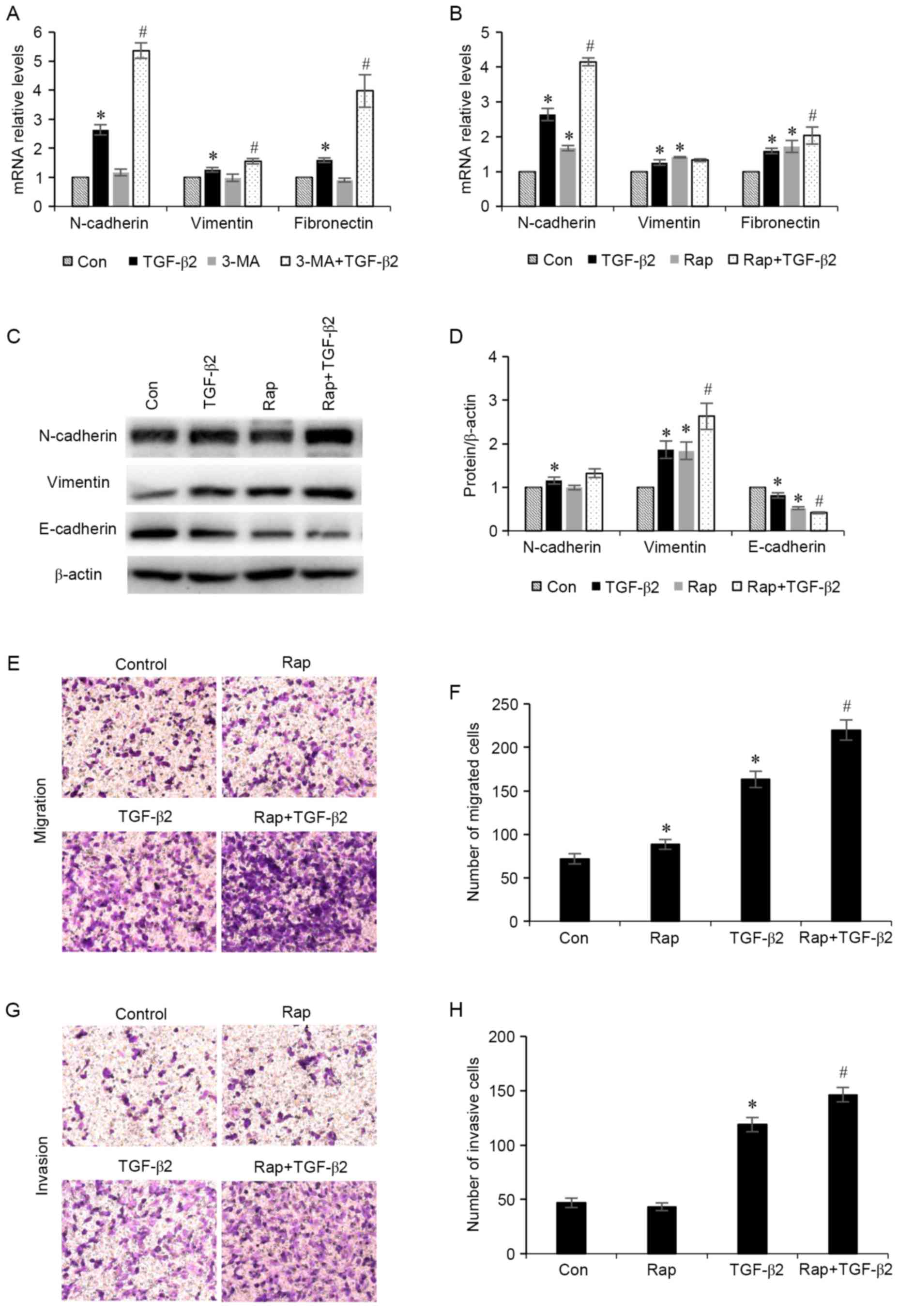
Increased levels of autophagy
exacerbates TGF-β2-induced EMT
To determine whether stimulation of autophagy
affects EMT, the effects of TGF-β2 stimulation in ARPE-19 cells in
the presence of autophagy inducers were investigated. ARPE-19 cells
were treated with TGF-β2 (5 ng/ml) with or without 3-MA (5 mM), an
autophagy stimulator, or rapamycin (200 nM), for 24 h. The results
demonstrated that administration of 3-MA and rapamycin increased
the mRNA expression levels of N-cadherin, vimentin and fibronectin
mesenchymal markers (Fig. 4A and
B). Furthermore, cells treated with rapamycin demonstrated
increased protein expression levels of N-cadherin and vimentin
mesenchymal markers, and decreased expression levels of the
epithelial marker E-cadherin (Fig. 4C
and D). In addition, the effect of rapamycin administration on
RPE cell migration and invasion was investigated (Fig. 4E-H). It was revealed that treatment
with rapamycin significantly enhanced TGF-β2-induced increases in
RPE cell migration and invasion. These results indicated that
increased autophagy may induce the EMT process in ARPE-19
cells.
Discussion
Autophagy has an important role in numerous
biological processes, including cell differentiation and
metabolism, and response to drug administration, radiation and
mechanical stress. It is well established that EMT is an important
mechanism in the pathogenesis associated with fibrotic disease.
Furthermore, a number of previous studies have reported the
occurrence of autophagy in cells undergoing EMT (8–12).
Administration of TGF-β1 simultaneously induced fibrosis and
autophagy in primary human atrial myofibroblasts, and autophagy
markers were previously demonstrated to be elevated in scar tissue
isolated from post-myocardial infarction rats (9). It was also reported that autophagy
may be induced by administration of TGF-β1 during the EMT process
in malignant glioma cells, as demonstrated by the upregulated
expression of LC3-II, beclin 1 and lysosomal-associated membrane
protein 1 (8). In the present
study, LC3-phosphatidylethanolamine conjugate (LC3-II) is the
lipidated form of the cystolic form of LC3 (LC3-I), and the
conversion from LC3-I to LC3-II represents the formation of
autophagosomes (24). p62 combines
polyubiquitinated proteins and forms the completed autophagosomes,
which are subsequently degraded into autolysosomes; therefore, the
quantity of p62 may be considered an index of autophagic
degradation (24). Although
beclin-1 is a regulatory protein of autophagy, its expression was
not markedly altered when autophagy was induced by TGF-β2
administration. Following induction of autophagy by various
stimuli, beclin-1 is released from B-cell lymphoma 2 and combines
with phosphatidylinositol 3-kinase catalytic subunit type 3
(PIK3C3) to generate multiple complexes (24). Thus, it is possible that the
expression of beclin-1 expression did not demonstrate a marked
alteration in the present study due to binding to PIK3C3. Similar
results have been reported by Lee et al (25). It is well established that TGF-β2
has been well established is a potent inducer of EMT in RPE cells.
In the present study, the TGF-β2-induced EMT model was established
in ARPE-19 cells in vitro, and an increased autophagic flux
during the process of EMT in RPE cells was demonstrated, thus
indicating that autophagy may participate in the EMT process of RPE
cells.
The effect of autophagy on the EMT process in RPE
cells was investigated further in the present study. Chloroquine,
3-MA and rapamycin pharmacological modulators, with the ability to
regulate autophagy activity in ARPE-19 cells, were employed.
Chloroquine, which possesses the ability to raise lysosomal pH, is
generally used as an autophagy inhibitor, and rapamycin is commonly
used as an autophagy inducer. 3-MA is a selective phosphoinositide
3-kinase inhibitor and its effect on autophagy was reported to be
conditional. Rapamycin is an autophagy inducer by inhibiting
mammalian target of rapamycin (mTOR) (24). Consistent with previous studies
(26–28), the present study demonstrated that
administration of chloroquine inhibited autophagy activity, and
administration of rapamycin increased autophagy activity in ARPE-19
cells. Furthermore, it was also demonstrated that administration of
3-MA promoted the induction of autophagy. Therefore, future studies
aiming to investigate the role of autophagy activity associated
with RPE-EMT may use the aforementioned pharmacological modulators.
The expression of E-cadherin, N-cadherin, fibronectin and vimentin
was examined. E-cadherin and N-cadherin are classical members of
the cadherin superfamily, which are a type of cell adhesion
molecule that is important in the formation of adherens junctions
to bind cells with each other (29). Fibronectin is a high molecular
weight glycoprotein of the extracellular matrix that binds to
integrins (30). Vimentin is a
type III intermediate filament protein that is expressed in
mesenchymal cells (31).
Epithelial cells express high levels of E-cadherin, whereas
mesenchymal cells express those of N-cadherin, fibronectin and
vimentin (32). The results in the
present study indicated that the inhibition of autophagy following
chloroquine administration may significantly attenuate
TGF-β2-induced EMT in ARPE-19 cells. Furthermore, induction of
autophagy following administration of either 3-MA or rapamycin in
ARPE-19 cells resulted in enhanced expression of mesenchymal
markers.
However, the effect of autophagy on the EMT process
is complex. The diverse effects of autophagy on EMT have been
reported at different experimental conditions. Researchers
demonstrated that induced autophagy increased EMT in some cells or
tissues and inhibited EMT in others (8–14).
Kim et al (11)
investigated the role of autophagy in primary mouse mesangial cells
and revealed that reduced levels of autophagy, via gene knockdown
using specific small interfering RNA, led to enhanced expression of
type I collagen, indicating that autophagy may exacerbate EMT.
Furthermore, Kim et al (11) also demonstrated that, following
treatment with trifluoperozine, which is an inducer of autophagy,
the expression level of type I collagen was decreased (11). However, numerous previous studies
(8,9,14)
have also demonstrated that autophagy was able to activate EMT,
including a study by Li et al (14), which demonstrated that
starvation-induced autophagy promoted the activation of EMT in
hepatocellular carcinoma cells (14). Therefore, the exact involvement of
autophagy activity in RPE-EMT remains to be determined and requires
further investigation, such as through use of alternative EMT
models and performing alternative experiments in order to further
investigate autophagy activity. The complex effects of autophagy on
fibrosis also highlights the importance for further investigation
to determine the role autophagy in intraocular fibrotic
disorders.
Numerous studies have reported the roles of
autophagy in RPE, demonstrating that autophagy may aid the
elimination of cytotoxic protein aggregates in RPE cells. Mitter
et al (18) investigated
RPE cells, retina samples from patients suffering from AMD and
rodent models with an AMD phenotype, and demonstrated that
autophagy was dysregulated. RPE cell damage resulting from
autophagy dysregulation may induce more severe pathophysiological
alterations, such as EMT and choroidal neovascularization (18). EMT in RPE cells is reported to
exhibit an important role in retinal fibrotic diseases, which are
predominantly irreversible and result in severe damage to vision
(4,5). To the best of our knowledge, the
present study demonstrated for the first time that autophagy may
function as a regulator of EMT in RPE cells, thus indicating that
autophagy may have an important function in the process of EMT in
RPE cells. A potential mechanism underlying this process is that
autophagy may provide adenosine triphosphate for the biosynthesis
of profibrotic proteins (9).
Future studies should investigate the potential underlying
mechanisms associated with the effects of autophagy on EMT
formation, which may provide new perspectives for the determination
of the function of autophagy associated with the EMT process and
the development of novel therapeutic agents for the treatment of
retinal fibrotic pathogenesis.
In the present study, it was revealed that autophagy
activity was enhanced in ARPE-19 cells treated with TGF-β2.
Furthermore, it was demonstrated that autophagy activation
exacerbated RPE-EMT, and inhibition of autophagy attenuated EMT. In
addition, the results of the present study revealed that autophagy
may be a regulator of EMT in RPE cells. The present study may
contribute to an enhanced understanding of the role of autophagy in
the pathophysiology of intraocular fibrotic disorders, which
frequently result from EMT. Furthermore, these results indicate
that autophagy may be a potential novel therapeutic target for the
attenuation of EMT in fibro-proliferative disease.
Acknowledgements
The authors thank Professor Fu Shang (State Key
Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun
Yat-sen University, Guangzhou, China) for kindly providing the
ARPE-19 human RPE cell line. The current study was supported by the
National Natural Science Foundation of China (grant nos. 81300749
and 81320108008).
References
|
1
|
Strauss O: The retinal pigment epithelium
in visual function. Physiol Rev. 85:845–881. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Priglinger CS, Obermann J, Szober CM,
Merl-Pham J, Ohmayer U, Behler J, Gruhn F, Kreutzer TC, Wertheimer
C, Geerlof A, et al: Epithelial-to-mesenchymal transition of rpe
cells in vitro confers increased beta1,6-N-glycosylation and
increased susceptibility to galectin-3 binding. PLoS One.
11:e01468872016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kampik A, Green WR, Quigley HA and Pierce
LH: Scanning and transmission electron microscopic studies of two
cases of pigment dispersion syndrome. Am J Ophthalmol. 91:573–587.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Machemer R: Proliferative
vitreoretinopathy (PVR): A personal account of its pathogenesis and
treatment. Proctor lecture Invest Ophthalmol Vis Sci. 29:1771–1783.
1988.PubMed/NCBI
|
|
5
|
Hiscott P, Sheridan C, Magee RM and
Grierson I: Matrix and the retinal pigment epithelium in
proliferative retinal disease. Prog Retin Eye Res. 18:167–190.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Ye S, Xiao W, Luo L and Liu Y:
Differentially expressed microRNAs in TGFβ2-induced
epithelial-mesenchymal transition in retinal pigment epithelium
cells. Int J Mol Med. 33:1195–1200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirasawa M, Noda K, Noda S, Suzuki M,
Ozawa Y, Shinoda K, Inoue M, Ogawa Y, Tsubota K and Ishida S:
Transcriptional factors associated with epithelial-mesenchymal
transition in choroidal neovascularization. Mol Vis. 17:1222–1230.
2011.PubMed/NCBI
|
|
8
|
Zou M, Zhu W, Wang L, Shi L, Gao R, Ou Y,
Chen X, Wang Z, Jiang A, Liu K, et al: AEG-1/MTDH-activated
autophagy enhances human malignant glioma susceptibility to
TGF-β1-triggered epithelial-mesenchymal transition. Oncotarget.
7:13122–13138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ghavami S, Cunnington RH, Gupta S, Yeganeh
B, Filomeno KL, Freed DH, Chen S, Klonisch T, Halayko AJ, Ambrose
E, et al: Autophagy is a regulator of TGF-β1-induced fibrogenesis
in primary human atrial myofibroblasts. Cell Death Dis.
6:e16962015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ni BB, Li B, Yang YH, Chen JW, Chen K,
Jiang SD and Jiang LS: The effect of transforming growth factor β1
on the crosstalk between autophagy and apoptosis in the annulus
fibrosus cells under serum deprivation. Cytokine. 70:87–96. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ and
Choi ME: Autophagy promotes intracellular degradation of type I
collagen induced by transforming growth factor (TGF)-β1. J Biol
Chem. 287:11677–11688. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grassi G, Di Caprio G, Santangelo L, Fimia
GM, Cozzolino AM, Komatsu M, Ippolito G, Tripodi M and Alonzi T:
Autophagy regulates hepatocyte identity and
epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions
promoting Snail degradation. Cell Death Dis. 6:e18802015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bertrand M, Petit V, Jain A, Amsellem R,
Johansen T, Larue L, Codogno P and Beau I: SQSTM1/p62 regulates the
expression of junctional proteins through epithelial-mesenchymal
transition factors. Cell Cycle. 14:364–374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo
Y, Song Z, Zheng Q and Xiong J: Autophagy promotes hepatocellular
carcinoma cell invasion through activation of
epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sinha D, Valapala M, Shang P, Hose S,
Grebe R, Lutty GA, Zigler JS Jr, Kaarniranta K and Handa JT:
Lysosomes: Regulators of autophagy in the retinal pigmented
epithelium. Exp Eye Res. 144:46–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hyttinen JM, Petrovski G, Salminen A and
Kaarniranta K: 5′-Adenosine monophosphate-activated protein
kinase-mammalian target of rapamycin axis as therapeutic target for
age-related macular degeneration. Rejuvenation Res. 14:651–660.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mitter SK, Song C, Qi X, Mao H, Rao H,
Akin D, Lewin A, Grant M, Dunn W Jr, Ding J, et al: Dysregulated
autophagy in the RPE is associated with increased susceptibility to
oxidative stress and AMD. Autophagy. 10:1989–2005. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johansson I, Monsen VT, Pettersen K,
Mildenberger J, Misund K, Kaarniranta K, Schønberg S and Bjørkøy G:
The marine n-3 PUFA DHA evokes cytoprotection against oxidative
stress and protein misfolding by inducing autophagy and NFE2L2 in
human retinal pigment epithelial cells. Autophagy. 11:1636–1651.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen X, Xiao W, Wang W, Luo L, Ye S and
Liu Y: The complex interplay between ERK1/2, TGFβ/Smad and
Jagged/Notch signaling pathways in the regulation of
epithelial-mesenchymal transition in retinal pigment epithelium
cells. PLoS One. 9:e963652014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Asaria RH, Kon CH, Bunce C, Sethi CS, Limb
GA, Khaw PT, Aylward GW and Charteris DG: Silicone oil concentrates
fibrogenic growth factors in the retro-oil fluid. Br J Ophthalmol.
88:1439–1442. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baudouin C, Fredj-Reygrobellet D, Brignole
F, Negre F, Lapalus P and Gastaud P: Growth factors in vitreous and
subretinal fluid cells from patients with proliferative
vitreoretinopathy. Ophthalmic Res. 25:52–59. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee SY, Oh JS, Rho JH, Jeong NY, Kwon YH,
Jeong WJ, Ryu WY, Ahn HB, Park WC, Rho SH, et al: Retinal pigment
epithelial cells undergoing mitotic catastrophe are vulnerable to
autophagy inhibition. Cell Death Dis. 5:e13032014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu YD, Wang ZB, Han G and Zhao P:
Hyperbaric oxygen treatment attenuates neuropathic pain by
elevating autophagy flux via inhibiting mTOR pathway. Am J Transl
Res. 9:2629–2638. 2017.PubMed/NCBI
|
|
27
|
Zhou Q, Zhang H, Wu Q, Shi J and Zhou S:
Pharmacological manipulations of autophagy modulate
paraquat-induced cytotoxicity in PC12 cells. Int J Biochem Mol
Biol. 8:13–22. 2017.PubMed/NCBI
|
|
28
|
Cui D, Sun D, Wang X, Yi L, Kulikowicz E,
Reyes M, Zhu J, Yang ZJ, Jiang W and Koehler RC: Impaired
autophagosome clearance contributes to neuronal death in a piglet
model of neonatal hypoxic-ischemic encephalopathy. Cell Death Dis.
8:e29192017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alimpeti S and Andreadis ST: CDH2 and
CDH11 act as regulators of stem cell fate decisions. Stem Cell Res.
14:270–282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pankov R and Yamada KM: Fibronectin at a
glance. J Cell Sci. 115:3861–3863. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eriksson JE, Dechat T, Grin B, Helfand B,
Mendez M, Pallari HM and Goldman RD: Introducing intermediate
filaments: From discovery to disease. J Clin Invest. 119:1763–1771.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|