Introduction
Hepatitis B virus (HBV) is a small DNA virus with a
genome size of 3.2 kb. HBV has only four overlapping open reading
frames. The 3.5 kb pre-genomic mRNAs (pgRNAs) and pre-core mRNAs
(pcRNAs) are produced from juxtaposed promoter sequences (1). The pgRNA serves as the template for
viral DNA synthesis and the mRNA for HBV core and polymerase
translation. The pcRNA is utilized for the translation of the HBe
antigen (Ag) and polymerase. The 2.4 kb pre-S1/S mRNAs encode the
large surface envelope protein of HBV (LHBs). The pre-S1 region of
the large HBsAg is responsible for viral binding and entry
processes. The 2.1 kb pre-S2/S mRNAs encode middle and small
envelope proteins (MHBs/SHBs). The concentration of the pre-S1/S
mRNA is lower compared with pre-S2/S mRNA in infected hepatocytes
(2). The 0.7 kb X mRNAs encode the
HBx protein, which is the most cryptic protein of HBV with diverse
functions (3,4). HBsAg loss and anti-HBs seroconversion
are regarded to be a functional cure for chronic HBV infection.
HBsAg interacts with plasmacytoid dendritic cells (pDCs) and
contributes to pDC impairment (5).
High HBV surface antigen concentrations additionally promote
HBV-specific T-cell dysfunction by affecting the phenotype and
function of peripheral and intrahepatic T cells (6). HBsAg clearance occurs in only a
minority of cases, not exceeding 3% following treatment with
nucleoside and nucleotide analogues, and not more than 7.8%
following treatment with pegylated interferon-α (7,8).
There is an urgent medical requirement to reduce the serum HBsAg
level and thus reverse T-cell dysfunction.
A number of viruses have been widely reported to be
regulated by class I phosphatidylinositol 3-kinase (PI3K) (9,10).
Hepatitis C virus utilizes the PI3K-RAC-α serine/threonine-protein
kinase (AKT) signaling pathway to facilitate viral entry (11). Vaccinia virus and cowpox virus, two
members of the poxvirus family, activate the PI3K-AKT pathway to
prevent apoptosis, ensuring host survival and viral replication
(12). Contradictory to the
beneficial role of the PI3K-AKT pathway for the majority of
viruses, the PI3K-AKT-serine/threonine-protein kinase mTOR (mTOR)
pathway suppresses the replication of HBV and may be partially
responsible for the elimination of HBV replication from tumor cells
(13). The LHBsAg has been
demonstrated to activate the PI3K-AKT pathway and to act as a
tumorigenesis factor (14). HBx
was reported to activate the PI3K-AKT pathway and act as a
balancing factor between HBV replication and cell survival
(15–17). LY294002 and wortmannin are first
generation compounds with highly potent PI3K-inhibiting properties.
The AKT1/2 inhibitor (AKTi) specifically inhibits pleckstrin
homology domain-containing kinases. Rapamycin is a prototypical
inhibitor of mTOR complex 1 (mTORC1) (18). However, prolonged treatment with
high concentrations of rapamycin inhibits mTOR2 and AKT activation
(19). In the present study, the
role of PI3K-AKT-mTOR signaling inhibitors in the infection and
replication of HBV was investigated.
Materials and methods
Cell culture
HepG2 (American Type Culture Collection, Manassas,
VA, USA; HB-8065) and Huh7 (JCRB Cell Bank, National Institutes of
Biomedical Innovation, Health and Nutrition, Osaka, Japan; 0403)
cells were cultured in Dulbecco's modified Eagle's medium/F12 (Life
Technologies; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Life Technologies; Thermo
Fisher Scientific, Inc.), 2 mM L-glutamine, 100 U/ml penicillin,
and 100 µg/ml streptomycin, at 37°C in humidified air containing 5%
CO2. For HepG2.2.15, 300 µg/ml geneticin (Invitrogen;
Thermo Fisher Scientific, Inc.) was added. HepAD38 (American Type
Culture Collection; CRL-12077) is tetracycline-inducible
HBV-producing cell line, in which HBV expression is suppressed by
tetracycline. Tetracylcine (cat no. 250163) and entecavir (cat no.
SML1103) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). The cell culture of HepaRG cells (cat no. HPR101;
Biopredic International, Saint Grégoire, France) and primary human
hepatocytes (Bioreclamation IVT, Hicksville, NY, USA) was performed
as previously described (20,21).
The cells seeded into 12-well and 96-well plates were at a density
of 2×105 and 3×104, respectively, if not
explicitly stated otherwise. The cells were infected with 50 genome
equivalents of purified HBV virus from HepAD38 cells, constituted
in 4% polyethylene glycol (PEG; cat no. P1458, Sigma-Aldrich, Merck
KGaA) and 1% dimethyl sulfoxide (DMSO). After 16 h incubation at
37°C, cells were washed three times with PBS and replenished with
William's E medium (cat no. 32551-020; Life Technologies, Thermo
Fisher Scientific, Inc.) supplemented with primary hepatocyte
maintenance supplements (cat no. CM4000; Life Technologies; Thermo
Fisher Scientific, Inc.) for primary human hepatocyte cell culture,
or additives (cat no. ADD710; Biopredic International, Saint
Grégoire, France) for HepaRG cells. DMSO, (cat no. D2650) and
PI3K-AKT-mTOR inhibitors Ly294002 (cat no. L9908), AKTi (cat no.
A6730) and rapamycin (cat no. V900930) were purchased from
Sigma-Aldrich (Merck KGaA). The inhibitors were dissolved in DMSO
and diluted in the William's E or DMEM/F12 medium. The final
concentration of DMSO was 1%. Control cells were treated with DMSO
alone. In HBV natural infection, Ly294002 was either added 30 h
prior to infection or 30 h subsequently. In HBV replicating cells,
the inhibitors were treated for 4–5 days to monitor the effect of
PI3K-AKT-mTOR inhibitors on HBV replication.
Plasmid construction and transient
transfection
Plasmid pBR322-HBV1.3ayw, which was kindly provided
by Dr. Qiang Deng in Pasteur Institute of Shanghai, contained
1.3-mer length of HBV genome. Small HBV surface antigen (SHBs;
spanning nucleotide 157 to nucleotide 837 of HBV ayw genome) were
cloned into pcDNA3.1 (+) vector (cat no. V79020; Invitrogen, Thermo
Fisher Scientific, Inc.) with KpnI and XhoI. HBx
(spanning nucleotide 1376 to nucleotide 1840 of HBV ayw genome)
were cloned into pLVX-IRES-tdTomato vector (cat no. 631238;
Clontech Laboratories, Inc., Mountainview, CA, USA) with
XhoI and BamHI. The restriction enzymes were
purchased from New England BioLabs, Inc. (Ipswich, MA, USA). To
transduce HBx, the HBx-pLVX-IRES-tdTomato plasmid was used to
produce lentivirus by NovoBio (Novobio Scientific, Inc., Shanghai,
China). One day before transfection, 1×105 cells were
seeded into each well of 12-well plates. Plasmids (1 µg) and 3 µl
X-tremeGENE™ HP DNA transfection reagent (cat no. 06366236001;
Roche Diagnostics GmbH, Mannheim, Germany) were diluted in 200 µl
Opti-MEM medium (cat no. 31985062; Life Technologies, Thermo Fisher
Scientific, Inc.). Incubated for 15 min at room temperature and
added the transfection complex to the cells in a dropwise manner.
After 6 h incubation at 37°C, cells were replenished with 1 ml cell
culture medium. The cells were incubated for 5 days post
transfection with or without the presence of PI3K-AKT-mTOR
inhibitors.
DNA/RNA extraction and
quantification
The DNA and RNA were extracted using a DNA Blood
Mini kit (Qiagen GmbH, Hilden, Germany) and TRIzol reagent (Thermo
Fisher Scientific, Inc.), respectively. The RNAs were reverse
transcribed using a cDNA synthesis kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Plasmid pBR322-HBV1.3ayw, which contained
1.3mer HBV genome, was used as HBV DNA standard for PCR.
Quantitative polymerase chain reaction (qPCR) analysis was
performed using TaqMan qPCR Master Mix reagent (Roche Diagnostics,
Basel, Switzerland). The primers used for HBV DNA quantification
were: HBX forward, 5′-cccgtctgtgccttctcatc-3′; HBX reverse,
5′-tcggtcgttgacattgctga-3′; HBX probe,
5′-FAM+tcgcttcacctctgcacgtcgc+Black Hole Quencher®-3′.
The PCR cycling program was performed as follows: Denaturation at a
temperature of 95°C for 10 min, followed by 45 cycles of
amplification at a denaturation temperature of 95°C for 10 sec, an
annealing temperature of 60°C for 10 sec, and an extension
temperature of 72°C for 15 sec. The cycle of quantification (Cq)
was obtained to determine the absolute quantification of HBV DNAs
using a standard curve. The standard curve was generated by
plotting Cq values vs. the concentrations of HBV DNA standard with
the analysis method ‘Abs Quant/2nd Derivative Max for All Samples’
of the LightCycler480 software (Roche Diagnostics GmbH, Mannheim,
Germany). HBV DNA copy numbers were calculated from Cq values fit
on the HBV plasmid DNA standard curve by the LightCycler480
software.
Agarose gel electrophoresis of viral
particles
Cells seeded in 12-well plates were lysed in 0.5 ml
lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% CA-630,
supplemented with a protease & phosphatase inhibitor cocktail,
cat no. 78440, Thermo Fisher Scientific, Inc.) and treated with
same procedure as previously described (22). The HBV capsids were blotted to
enhanced chemiluminescence membranes (GE Healthcare, Chicago, IL,
USA) by capillary transfer. The membranes were blocked with 10%
non-fat milk in TBS-0.1% Tween 20 for 30 min. HBV capsids were
detected with primary anti-core antibody (cat no. B0586; Dako,
Agilent Technologies, Inc., Santa Clara, CA, USA). The membrane was
incubated with 1:1,000 diluted primary antibody at 4°C with gentle
shaking overnight. Following washing three times with TBST, the
membrane was incubated with 1:2,000 diluted anti-rabbit antibody
(cat no. 656120; Thermo Fisher Scientific, Inc.) at room
temperature for 1 h. The blot was incubated with horse radish
peroxidase (HRP) substrate (cat no. 34075; Thermo Fisher
Scientific, Inc.) and imaged using the ChemiDoc touch imaging
system (Bio-Rad Laboratories, Inc.). The HBV capsid DNA was
transferred to nylon membranes and hybridized with
digoxigenin-labeled DNA probes according to the standard procedure
of southern blotting.
Southern and northern blotting
The HBV capsid DNAs were hybridized with DNA probes
and detected by alkaline phosphatase-conjugated anti-digoxigenin
antibody (23). For RNA detection,
5 µg tRNAs were resolved on a 1.5% agarose gel containing 0.66 M
formaldehyde, transferred onto nylon membranes and detected with
HBV RNA probes (22).
Western blotting
Cells were washed once with Tris-buffered saline
(TBS) and lysed with radioimmunoprecipitation assay lysis buffer
(cat no. 89,900; Thermo Fisher Scientific, Inc.) supplemented with
a phosphatase and protease inhibitor cocktail (cat no. 78440;
Thermo Fisher Scientific, Inc.). Total protein concentrations in
the lysate were measured using a bicinchoninic acid protein assay
kit (cat no. 23225; Thermo Fisher Scientific, Inc.). Cell lysates
were mixed with 4× SDS loading buffer (cat no. NP0007; Thermo
Fisher Scientific, Inc.) and 10 mM DTT, and were boiled for 5 min
at 95°C. A total of 5 µg of protein was loaded onto each lane. The
samples were subjected to 4–12% SDS-PAGE (cat no. NP0322; Thermo
Fisher Scientific, Inc.) and were then transferred to
nitrocellulose membranes (cat no. IB23001; Thermo Fisher
Scientific, Inc.). The membranes were blocked with 10% nonfat milk
(cat no. 9999; Cell Signaling Technology, Inc., Danvers, MA, USA)
in TBS-0.1% Tween 20 for at least 30 min and were then incubated
with the indicated primary antibodies. The primary antibodies used
were as follows: β-actin (cat. no. A3854; Sigma-Aldrich; Merck
KGaA), HBV core (cat. no. B0586; Dako; Agilent Technologies, Inc.,
Santa Clara, CA, USA), AKT (cat. no. 9272; Cell Signaling
Technology, Inc., Danvers, MA, USA), p-AKT (cat. no. 9275; Cell
Signaling Technology, Inc.), HBsAg (cat. no. 30R-AH018; Fitzgerald
Industries International, North Acton, MA, USA), and HBx (kindly
provided by Roche Innovation Centre Shanghai, Shanghai, China). The
membrane was incubated with 1:1,000 diluted primary antibody at 4°C
with gentle shaking overnight. The secondary antibodies, including
anti-rabbit antibody (cat no. 656120) and anti-mouse antibody (cat
no. 32430) were purchased from Thermo Fisher Scientific, Inc.
Following washing three times with TBS, the membrane was incubated
with 1:2,000 diluted secondary antibody at room temperature for 1
h. The blot was incubated with HRP substrate (cat no. 34075; Thermo
Fisher Scientific, Inc.) and imaged using the ChemiDoc touch
imaging system (Bio-Rad Laboratories, Inc.).
Immunofluorescence
HepAD38 cells (8×104) were seeded into
each well of chamber slides (cat. no. 177445; Thermo Fisher
Scientific, Inc.). Following treatment with the inhibitors for
three days, cells were fixed with 4% paraformaldehyde at room
temperature for 20 min. The samples were incubated for 10 min with
PBS containing either 0.5% Triton X-100 at room temperature for 10
min, and then were washed three times with PBS for 5 min. Then,
samples were incubated with HBsAg antibody (1:100; cat. no.
18–0023; Invitrogen; Thermo Fisher Scientific, Inc.) in PBST buffer
supplemented with 5% normal serum for 2 h at room temperature, and
washed three times with PBST buffer for 5 min. Samples were then
incubated with goat anti-mouse antibody (1:400; cat. no. A28175;
Invitrogen; Thermo Fisher Scientific, Inc.) at room temperature for
30 min in the dark and washed thrice with PBST buffer for 5 min.
Samples were incubated with 100 nM DAPI in PBS buffer at room
temperature for 5 min, and then cells were washed three times with
PBS for 5 min in the dark. The cells were visualized with a Zeiss
confocal microscope (Zeiss GmbH, Jena, Germany) at 400×
magnification.
Chemiluminescence immunoassay
(CLIA)
HBsAg and HBeAg in the culture medium and cell
lysate were tested using Autobio CLIA kits (Autobio Co., Ltd.,
Zhengzhou, China; cat. nos. CL0310-2 and CL0310-2 for HBsAg and
HBeAg, respectively), according to the manufacturer's protocol.
Statistical analysis
HBV DNA/RNA and HBsAg levels were quantified by PCR
and CLIA assay respectively. Data represent the mean ± standard
deviation of experiments performed in triplicate, and are presented
as absolute copy numbers or relative to control (cells treated with
DMSO). One-way analysis of variance was used with Dunn's multiple
comparisons test (GraphPad Prism version 7.03 software; GraphPad
Software, Inc., La Jolla, CA, USA) to compare the different groups
in the present study. P<0.05 was considered to indicate a
statistically significant difference.
Results
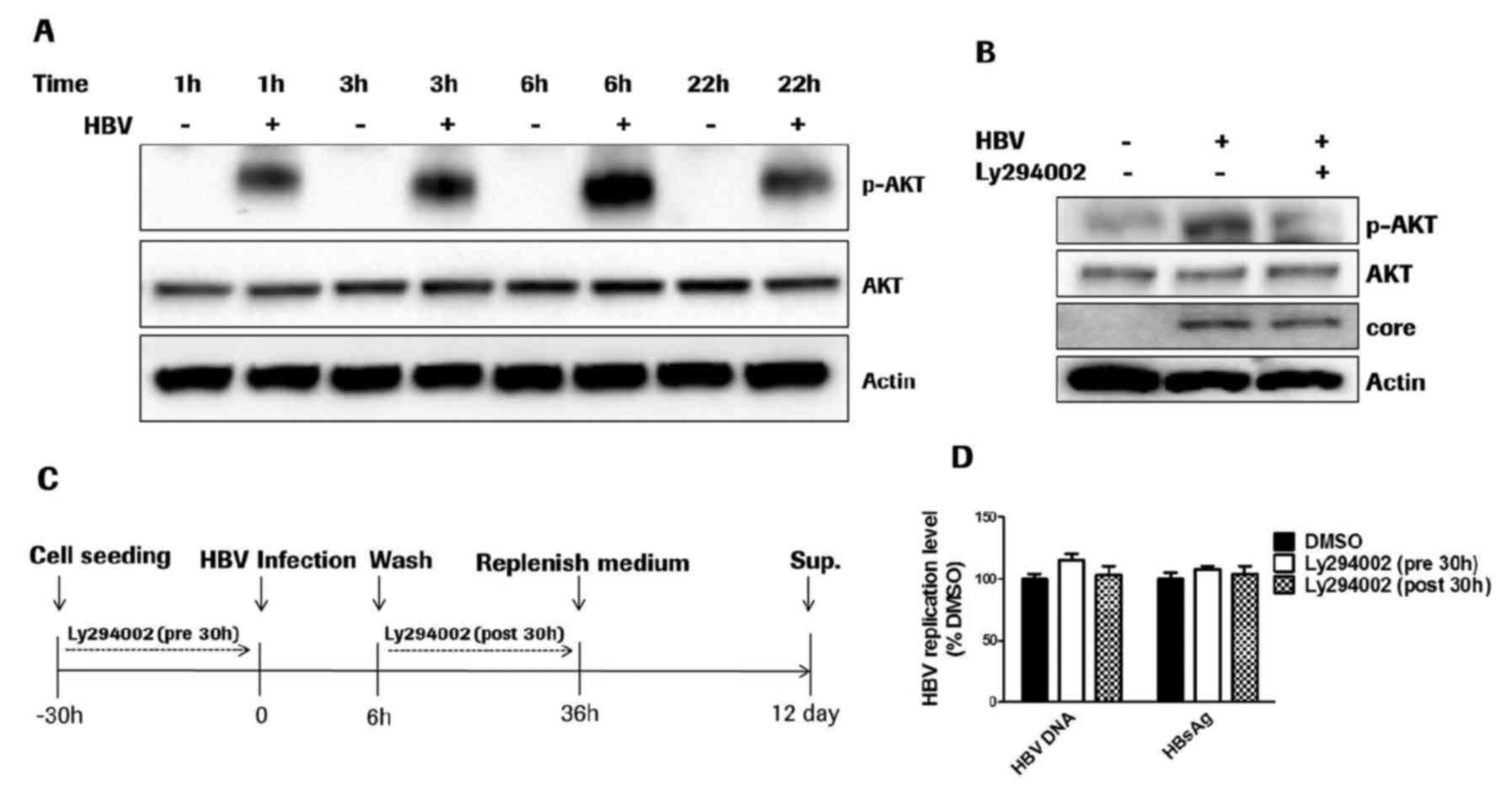
HBV entry process activates the AKT
pathway
In order to evaluate the AKT phosphorylation induced
by the HBV entry process, differentiated HepaRG cells were utilized
for HBV infection, and cell lysates were collected at different
time points post-HBV infection. HBV cellular entry transiently
activated AKT and resulted in AKT phosphorylation at threonine 308
as early as 1 h post-HBV infection (Fig. 1A). AKT phosphorylation peaked at 6
h post-HBV infection and gradually decreased at 22 h
post-infection.
Furthermore, AKT activation was transiently
inhibited by the PI3K inhibitor Ly294002 at 10 µM, at which
concentration the AKT phosphorylation was markedly inhibited. HBV
entry activated AKT at 36 h post-viral infection in primary human
hepatocytes, as hypothesized (Fig.
1B). Inhibition of AKT phosphorylation exerted no effect on HBV
cellular entry, as the HBV core protein level in the cell lysate
was not affected at 36 h post-HBV infection (Fig. 1B). The results demonstrated that
although the HBV cellular entry process was able transiently
activate the AKT pathway, AKT phosphorylation was not essential for
HBV entry compared with a variety of viruses (11–13).
The HBV infection and compound treatment procedure is illustrated
in Fig. 1C. The PI3K inhibitor
Ly294002 was applied for 30 h prior to or following HBV infection.
Quantification of HBV DNA and HBsAg levels at 12 days
post-infection additionally indicated that transient inhibition of
AKT phosphorylation with 10 µM Ly294002 had no effect on the HBV
entry process (Fig. 1D).
Prolonged treatment with PI3K-AKT-mTOR
pathway inhibitors promotes HBV replication
First of all, the effect of PI3K-AKT-mTOR inhibitors
was evaluated in HBV plasmid transfected HepG2 and Huh7 cells. All
three inhibitors, including Ly294002, AKTi and rapamycin, promoted
HBV DNA levels in the supernatant following 5 days of treatment
(Fig. 2A). In HBV infected dHepaRG
cells, prolonged treatment with PI3K-AKT inhibitors increased the
extracellular HBV DNAs in addition to the intracellular HBV RNAs
(Fig. 2B and C). The prolonged
effect of Ly294002 on HBV replication was further confirmed in
HepG2.2.15 cells. The PI3K inhibitor Ly294002 dose-dependently
increased the levels of HBV capsids and capsid DNAs in the cell
lysates (Fig. 2D). These results
were consistent with previous studies (13,15),
and demonstrated that the PI3K-AKT-mTOR pathway negatively
regulates HBV replication.
 | Figure 2.PI3K-AKT inhibitors promote HBV
replication. (A) pHBV1.3 plasmid-transfected HepG2 and Huh7 cells
were treated with PI3K-AKT inhibitors for 5 days. HBV DNA in the
supernatants was detected by quantitative polymerase chain reaction
analysis. (B) In HBV-infected dHepaRG cells, the prolonged presence
of PI3K-AKT inhibitors between days 5 and 12 increased the HBV DNA
level. (C) HBV RNAs were promoted in dHepaRG cells treated with
PI3K-AKT inhibitors. (D) In HepG2.2.15 cells, the intracellular
capsids were detected with agarose gel electrophoresis of viral
particles. Capsid DNA was detected by Southern blotting. Treatment
with Ly294002 for 5 days increased HBV capsids and capsid DNAs in a
dose-dependent manner. (E) In enticavir- and tetracycline-treated
HepAD38 cells, HBx expression was detected by western blotting. (F)
HBV RNA transcription in HepAD38 cells was evaluated via northern
blotting. The HBx gene was introduced into HepAD38 cells via
lentiviral transduction. The cells were treated with PI3K-AKT-mTOR
inhibitors for 5 days. 18S and 28S ribosomal RNAs were used as the
loading control. **P<0.01, ***P<0.001 vs. DMSO. PI3K,
phosphatidylinositol 3-kinase; AKT, RAC-α serine/threonine-protein
kinase; HBV, hepatitis B virus; DMSO, dimethyl sulfoxide; AKTi,
AKT1/2 inhibitor; GE, genome equivalents. |
In addition, HepAD38 cells were treated without
tetracycline for 8 days to accumulate intracellular covalently
closed circular (ccc)DNAs. The cells were treated with 200 nM
entecavir and tetracycline for 5 days to inhibit reverse
transcription and transcription from integrated HBV DNA,
respectively. Transcripts from episomal cccDNA were not affected by
treatment with entecavir and tetracycline. Lentiviruses were
utilized to introduce HBx into HepAD38 cells. The effect of the
PI3K-AKT-mTOR signaling pathway inhibitors on HBV transcription was
evaluated. In entecavir- and tetracycline-treated HepAD38 cells,
HBx expression was undetectable by western blotting (Fig. 2E). The PI3K-AKT-mTOR pathway
inhibitors promoted HBV 3.5 kb pgRNA transcription in
HBx-expressing cells, and exerted no effect on pgRNA transcription
in HBx negative cells. The enhanced transcription was specific to
3.5 kb mRNAs, as it exerted no impact on the transcription of 2.4
kb and 2.1 kb S mRNAs (Fig. 2F).
The results demonstrated that the PI3K-AKT-mTOR pathway inhibitors
promoted HBV 3.5 kb pgRNA transcription in an HBx-dependent
manner.
Ly294002 inhibits small HBsAg
secretion in a PI3K-AKT pathway-independent manner
HepG2.2.15 cells were treated with PI3K-AKT-mTOR
inhibitors for 4 days. Ly294002 was observed to be the only
inhibitor which was able to decrease HBsAg levels in the cell
culture medium (Fig. 3A). It was
subsequently identified that Ly294002 was the only compound
increasing intracellular HBsAg in HepG2.2.15 cells (data not
shown). The results indicated that Ly294002 may block HBsAg
secretion processes.
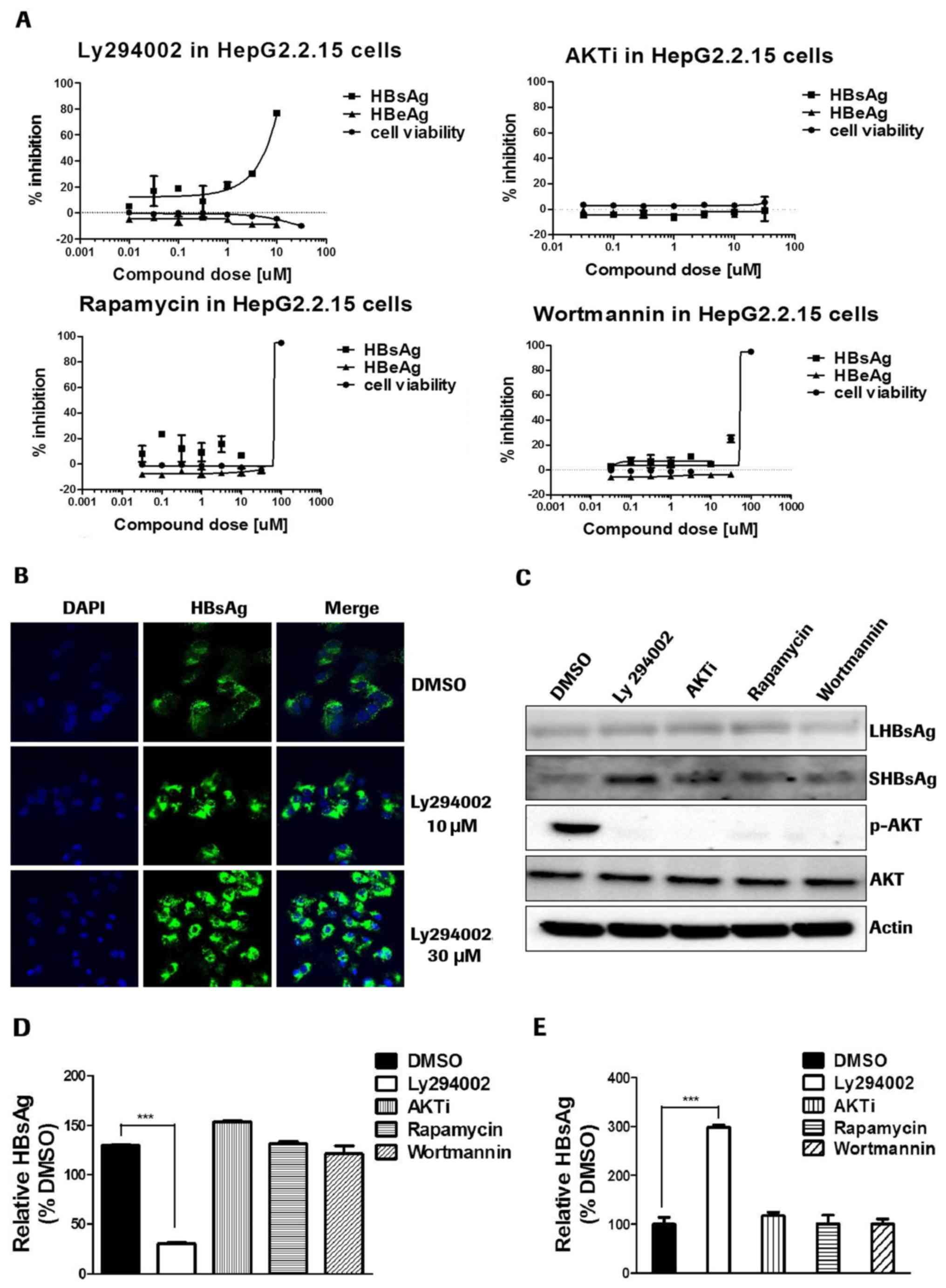 | Figure 3.Ly294002 inhibits small HBsAg
secretion in a PI3K-AKT-mTOR pathway-independent manner. (A) For
HepG2.2.15 cells, HBsAg and HBeAg were detected via
chemiluminescence immunoassays. Cell viability was detected by Cell
Counting Kit-8 assay. (B) HepAD38 cells were treated with Ly294002
for 4 days. The cells were immunostained with the antibody to HBsAg
(green color) and counterstained with DAPI (blue color). The images
were acquired on a confocal microscope at 400× magnification. (C)
HepG2.2.15 cells were treated with inhibitors for 4 days. HBsAg,
AKT and β-actin were detected via western blotting. (D) In SHBsAg
plasmid-transfected HepG2 cells, treatment with Ly294002 decreased
extracellular SHBsAg levels. (E) In SHBsAg plasmid-transfected
HepG2 cells, Ly294002 promoted intracellular SHBsAg levels.
***P<0.001. Ag, antigen; PI3K, phosphatidylinositol 3-kinase;
AKT, RAC-α serine/threonine-protein kinase; mTOR,
serine/threonine-protein kinase mTOR; HB, hepatitis B; DMSO,
dimethyl sulfoxide; p, phosphorylated; LHB, large surface envelope
protein; SHB, small surface envelope protein; AKTi, AKT1/2
inhibitor. |
This HBsAg secretion inhibition capability of the
PI3K inhibitor Ly294002 was further confirmed by immunofluorescence
staining of HBsAg in HepAD38 cells (Fig. 3B). The results also indicated that
Ly294002 blocked HBsAg secretion and increased the intracellular
HBsAg level in a dose dependent manner. Western blot detection of
intracellular HBsAg demonstrated that all of the four inhibitors
intensively inhibited AKT phosphorylation. However, only Ly294002
blocked small HBsAg (SHBs) secretion, and exerted no effect on
large HBsAg (Fig. 3C). In SHB
plasmid-transfected HepG2 cells, following treatment with Ly294002,
AKTi, rapamycin and wortmannin, only Ly294002 inhibited the
secretion of sHBsAg (Fig. 3D and
E). These results further demonstrated that the PI3K inhibitor
Ly294002 may specifically inhibit the secretion of small HBsAg in a
PI3K-AKT-mTOR pathway-independent manner.
Discussion
In the present study, the regulatory effect of the
PI3K-AKT-mTOR pathway in HBV infection and replication was
investigated using different in vitro cell culture systems.
The role of this pathway in hepatocellular carcinoma (HCC) was not
evaluated. In addition, certain cell lines used in the present
study, including HepG2, HepG2.2.15 and HepAD38, are actually
hepatoblastoma cells and are not unsuitable for HCC study. The role
of AKT activation in the HBV entry process was evaluated in a HBV
natural infection model. The results indicated that AKT
phosphorylation at threonine 308 was markedly induced by HBV entry
as early as 1 h post-HBV inoculation. Blocking AKT activation
during the first 30 h of the HBV entry process via treatment with
Ly294002 did not affect HBV entry, as short-term treatment with AKT
inhibitors exerted no effect on HBV core levels at 30 h
post-infection, nor on HBV DNA production at day 12. These results
indicated that the HBV entry process activated the AKT pathway,
demonstrated by AKT phosphorylation in HBV natural infection.
However, inhibition of AKT phosphorylation via short-term treatment
with AKT inhibitors was unable to inhibit HBV entry, suggesting
that AKT activation induced by HBV infection is not essential for
the viral entry process.
The PI3K-AKT-mTOR signaling pathway has been
observed to negatively regulate HBV replication in the HepG2 cell
line (13,15). The present study used HBV natural
infection to study the role of this multifunctional pathway in the
HBV entry, transcription and HBsAg secretion processes. The results
demonstrated that the PI3K-AKT inhibitors strongly increased HBV
DNA levels by enhancing HBV 3.5 kb pgRNA transcription in an
HBx-dependent manner. As the HBeAg in the cell culture medium
remained unchanged in HepAD38 cells (data not shown), the
dramatically increased 3.5 kb RNAs were almost pgRNAs, not the 3.5
kb pcRNAs which encode HBeAg. This result was consistent with
previous studies (13,15), which indicated that the
PI3K-AKT-mTOR pathway was able to negatively regulate HBV
replication and balance cell survival. However, these inhibitors
only promoted pgRNA, and not S mRNA transcription, which suggested
that the transcriptional enhancement was specific to pgRNAs.
To the best of our knowledge, the present study was
the first to utilize a HBV natural infection model to study the
role of the PI3K-AKT-mTOR pathway in HBV infection and replication.
In HBV-infected primary human hepatocytes (Fig. 1D), Ly294002 was added at 30 h prior
to or following HBV inoculation to inhibit the AKT phosphorylation
induced by HBV. The short-term existence of the PI3K inhibitor
Ly294002 exerted no influence on the HBV entry process. This result
indicated that, although HBV infection may trigger AKT activation,
the AKT activation induced by HBV infection is not essential for
HBV entry. In the HBV replication and infection models (Fig. 2A-C), to evaluate the influence of
PI3K-AKT-mTOR inhibitors on the long-term replication of HBV,
including HBV transcription, reverse transcription and expression,
these inhibitors were applied for at least 5 days. The results
suggested that the inhibitors promoted HBV replication in different
HBV replicating systems. Further investigation may be required to
identify whether off-target effects of PI3K-AKT-mTOR inhibitors
contribute to the regulation of HBV replication.
In addition, the PI3K inhibitor Ly294002 blocked
small HBsAg secretion in a PI3K-AKT independent manner, and thus
decreased the HBsAg level in the cell culture medium. The other
PI3K-AKT pathway inhibitors, AKTi, rapamycin and wortmannin, had no
effect on HBsAg secretion. Decreased HBsAg levels in patient serum
may be of benefit in reversing HBV-specific T-cell dysfunction and
pDC impairment. As mentioned previously, low HBsAg levels in
patient serum confer an increased HBsAg clearance potential
(7). Ly294002 may be used as a
tool for the development of drugs which are able to decrease the
HBsAg level in patient serum. Combination treatment with HBsAg
inhibitors and immune-modulators may improve anti-HBV therapy
(24).
Acknowledgements
K. Xiang is supported by the education assistance
program of Roche Innovation Center Shanghai (RICS). The authors
would like to thank RICS for providing reagents and materials for
the present study.
References
|
1
|
Yu X and Mertz JE: Promoters for synthesis
of the pre-C and pregenomic mRNAs of human hepatitis B virus are
genetically distinct and differentially regulated. J Virol.
70:8719–8726. 1996.PubMed/NCBI
|
|
2
|
Raney AK and McLachlan A: Characterization
of the hepatitis B virus large surface antigen promoter Sp1 binding
site. Virology. 208:399–404. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gearhart TL and Bouchard MJ: The hepatitis
B virus X protein modulates hepatocyte proliferation pathways to
stimulate viral replication. J Virol. 84:2675–2686. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benhenda S, Cougot D, Buendia MA and
Neuveut C: Hepatitis B virus X protein molecular functions and its
role in virus life cycle and pathogenesis. Adv Cancer Res.
103:75–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Woltman AM, Op den Brouw ML, Biesta PJ,
Shi CC and Janssen HL: Hepatitis B virus lacks immune activating
capacity, but actively inhibits plasmacytoid dendritic cell
function. PLoS One. 6:e153242011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schurich A, Khanna P, Lopes AR, Han KJ,
Peppa D, Micco L, Nebbia G, Kennedy PT, Geretti AM, Dusheiko G and
Maini MK: Role of the coinhibitory receptor cytotoxic T lymphocyte
antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B
virus infection. Hepatology. 53:1494–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lok AS and McMahon BJ: Chronic hepatitis
B: Update 2009. Hepatology. 50:661–662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
European Association For The Study Of The
Liver, . EASL clinical practice guidelines: Management of chronic
hepatitis B virus infection. J Hepatol. 57:167–185. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cooray S: The pivotal role of
phosphatidylinositol 3-kinase-Akt signal transduction in virus
survival. J Gen Virol. 85:1065–1076. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buchkovich NJ, Yu Y, Zampieri CA and
Alwine JC: The TORrid affairs of viruses: Effects of mammalian DNA
viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol.
6:266–275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Z, Tian Y, Machida K, Lai MM, Luo G,
Foung SK and Ou JH: Transient activation of the PI3K-AKT pathway by
hepatitis C virus to enhance viral entry. J Biol Chem.
287:41922–41930. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Soares JA, Leite FG, Andrade LG, Torres
AA, De Sousa LP, Barcelos LS, Teixeira MM, Ferreira PC, Kroon EG,
Souto-Padrón T and Bonjardim CA: Activation of the PI3K/Akt pathway
early during vaccinia and cowpox virus infections is required for
both host survival and viral replication. J Virol. 83:6883–6899.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo H, Zhou T, Jiang D, Cuconati A, Xiao
GH, Block TM and Guo JT: Regulation of hepatitis B virus
replication by the phosphatidylinositol 3-kinase-akt signal
transduction pathway. J Virol. 81:10072–10080. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu H, Xu J, Zhou L, Yun X, Chen L, Wang
S, Sun L, Wen Y and Gu J: Hepatitis B virus large surface antigen
promotes liver carcinogenesis by activating the Src/PI3K/Akt
pathway. Cancer Res. 71:7547–7557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rawat S and Bouchard MJ: The hepatitis B
virus (HBV) HBx protein activates AKT to simultaneously regulate
HBV replication and hepatocyte survival. J Virol. 89:999–1012.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YI, Kang-Park S, Do SI and Lee YI: The
hepatitis B virus-X protein activates a phosphatidylinositol
3-kinase-dependent survival signaling cascade. J Biol Chem.
276:16969–16977. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shih WL, Kuo ML, Chuang SE, Cheng AL and
Doong SL: Hepatitis B virus X protein activates a survival
signaling by linking SRC to phosphatidylinositol 3-kinase. J Biol
Chem. 278:31807–31813. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou Q, Lui VW and Yeo W: Targeting the
PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol.
7:1149–1167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sarbassov DD, Ali SM, Sengupta S, Sheen
JH, Hsu PP, Bagley AF, Markhard AL and Sabatini DM: Prolonged
rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell.
22:159–168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cerec V, Glaise D, Garnier D, Morosan S,
Turlin B, Drenou B, Gripon P, Kremsdorf D, Guguen-Guillouzo C and
Corlu A: Transdifferentiation of hepatocyte-like cells from the
human hepatoma HepaRG cell line through bipotent progenitor.
Hepatology. 45:957–967. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gripon P, Rumin S, Urban S, Le Seyec J,
Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C and
Guguen-Guillouzo C: Infection of a human hepatoma cell line by
hepatitis B virus. Proc Natl Acad Sci USA. 99:pp. 15655–15660.
2002; View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang S, Guo JT, Wu JZ and Yang G:
Identification and characterization of multiple TRIM proteins that
inhibit hepatitis B virus transcription. PLoS One. 8:e700012013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi L, Li S, Shen F, Li H, Qian S, Lee DH,
Wu JZ and Yang W: Characterization of nucleosome positioning in
hepadnaviral covalently closed circular DNA minichromosomes. J
Virol. 86:10059–10069. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lanford RE, Guerra B, Chavez D, Giavedoni
L, Hodara VL, Brasky KM, Fosdick A, Frey CR, Zheng J, Wolfgang G,
et al: GS-9620, an oral agonist of Toll-like receptor-7, induces
prolonged suppression of hepatitis B virus in chronically infected
chimpanzees. Gastroenterology. 144:1508–1517. 2013. View Article : Google Scholar : PubMed/NCBI
|