Introduction
Lung cancer is the second most prevalent and the
most lethal malignancy in both men and women throughout the world
(1). The majority of which are
non-small cell lung cancer (NSCLC) related to tobacco-driven
carcinogenesis (2). NSCLC
originates from respiratory epithelial cells (3), the early stage of which can be
treated by curative intent surgery, but NSCLC is always
un-diagnosable until advanced stage IIIB or IV because of its
nonspecific early symptoms, or relapse after surgery because of its
unlimited proliferation and metastasis (4). Generally, NSCLC cells could migrate
to regional lymph nodes through lymphatic spreading and to distant
organs through hematogenous spreading, most commonly to the liver,
bone marrow, adrenal glands and the brain (1). Therefore, effective therapeutic
approaches to control the proliferation and metastasis of NSCLC are
expected to reduce mortality of this disease.
Tubeimoside-1 (TBMS1) is an active ingredient from
the tuber of Bolbostemma paniculatum (Maxim) Franquet
(5), the traditional Chinese herb
with detoxification and detumescent properties (6). Prior studies have been identified
that TBMS1 exerted potent antitumor activity with low toxicity to
non-tumor cells (7). It can arrest
the cell cycle at G2/M phase to inhibit proliferation (6,8,9), as
well as induce the release of cytochrome c via increasing
mitochondrial dysfunction and endoplasmic reticulum stress to
enhance the apoptosis in various cancer cells (10–13).
In lung cancer, TBMS1 exerts its cytotoxicity by increasing the Bax
to Bcl-2 ratio and triggering mitochondrial-related apoptotic
pathway (14,15). However, neither the contribution of
TBMS1 to NSCLC metastasis nor the internal mechanism has been
substantiated.
miRNAs are evolutionarily conserved non-coding RNAs
capable of negatively regulating gene expression by binding to the
3′-untranslated region (3′-UTR) on target mRNAs (16). miR-126-5p, as known as miR-126*, is
a 5′ part of the miR-126 transcript that located in the epidermal
growth factor-like domain 7 (EGFL7) gene (17). miR-126 is involved in multiple
processes of cellular activities via suppressing target genes such
as vascular endothelial growth factor (VEGF), PI3K, EGFL7, CRK,
ADAM9, IRS-1, SOX-2 and SLC7A5 (16). Previous studies have found that
miR-126-5p was significantly downregulated in patients with
coronary artery disease, overexpression of miR-126-5p promoted
endothelial proliferation, increased pro-angiogenic endothelial
cell behavior and limited atherosclerosis (18–20).
Apart from that, the expression of miR-126-5p is notably reduced in
alcohol-related hepatocellular carcinoma, breast carcinoma and
prostate cancer (21,22). Cho and colleagues showed that
miR-126-5p was significantly downregulated using miRNA microarrays,
which was the most differentially expressed miRNA in lung cancer
(23). Accordingly, we speculated
that TBMS1 might repress the progression of NSCLC through
regulating the expression of miR-126-5p.
Therefore, this study observed the status of
survival, migration and invasion in TBMS1-treated NCI-H1299 cells
with miR-126-5p inhibitor transfection. Then we detected miR-126-5p
targeted VEGF-A-related pathway through overexpression of VEGF-A
and VEGFR-2 in TBMS1-treated NCI-H1299 cells. The results showed
that the cytostatic and anti-metastatic effects of TBMS1 was
mediated by overexpressing miR-126-5p caused inactivation of
VEGF-A/VEGFR-2/ERK pathway.
Materials and methods
Cell culture
Human non small cell lung cancer NCI-H1299 cell line
was purchased from Shanghai Institutes for Biological Sciences,
Chinese Academy of Sciences (Shanghai, China). Cells were grown in
Roswell Park Memorial Institute-1640 (RPMI-1640; Gibco, Grand
Island, NY, USA) supplemented with 10% fetal bovine serum (FBS;
HyClone, Logan, UT, USA) and streptomycin/penicillin (100 U/ml) at
37°C in a humidified atmosphere containing 5% CO2.
Drug treatment
TBMS1 (Shanghai PureOne Biotechnology Co., Shanghai,
China) was completely dissolved in ddH2O. NCI-H1299
cells were exposed to TBMS1 of an ascending concentration range (0,
2.5, 5, 10, 25, 50 µM) for 48 h followed by CCK-8 assay to find the
optimum concentration. Then NCI-H1299 cells with or without
transfection were exposed to 10 µmol/l TBMS1 for 48 h for further
experiments. The untreated NCI-H1299 cells with or without
transfection were experimented in parallel as control. For the
detection of ERK pathway, NCI-H1299 cells were administrated with
10 µmol/l TBMS1 and tert-butylhydroquinone (TBHQ; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 48 h. The TBMS1
treatment alone and untreated NCI-H1299 cells were employed as
positive control and negative control.
Patients
We collected tumor tissues from 14 patients who
underwent thoracoscopic lobectomy surgery for non-small cell lung
cancer between May 2013 and January 2016 at the Third Affiliated
Hospital of Qiqihar Medical University (Heilongjiang, China), and
14 paraneoplastic lung tissue samples (>5 cm away from tumors)
were taken as healthy control. All tissue specimens were obtained
with permission from the Medical Ethics Committee of The Third
Affiliated Hospital of Qiqihar Medical University. The median age
of all patients was 66.57 years (range, 43–78 years). None of the
patients received chemotherapy, radiotherapy or immunotherapy
before surgery. Each patient was agreed to participate in our study
and signed an informed consent. All tissue specimens were obtained
with permission from the Medical Ethics Committee of Qiqihar
Medical University.
Cell Counting Kit-8 (CCK-8) assay
NCI-H1299 cells were seeded in 96-well plates with a
density of 1×104/well and incubated to 80% confluence,
followed by incubating with indicated concentrations of TBMS1 for
48 h, with five replicates for each testing point including the
negative control and blank wells. Thereafter, the cell growth was
measured by Enhanced Cell Counting Kit-8 (Beyotime Institute of
Biotechnology, Haimen, China) completely following the
manufacturer's directions. Optical density (OD) values were
evaluated at 450 nm by a microplate reader (BioTek Instruments,
Inc., Winooski, VT, USA).
Plasmids, miR-126-5p inhibitor and
transfection
The plasmids pcDNA3.1-VEGF-A and pcDNA3.1-VEGFR-2
harboring VEGF-A-coding sequences and VEGFR-2-coding sequences,
along with miR-126-5p inhibitor were all constructed by Vipotion
Co., Ltd. (Guangzhou, China) and were transfected into NCI-H1299
cells respectively using the Lipofectamine 2000 reagent
(Invitrogen, Carlsbad, CA, USA) strictly following the
manufacturer's instructions. Cells were harvested after 24 h of
transfection for RT-qPCR detection and 48 h of transfection for
western blot detection.
Hoechst staining assay
NCI-H1299 cells in each group were grown onto
coverslips in 12-well plates at a density of 5×104/well.
After growing to 80% confluences, cells were administrated with 10
µmol/l TBMS1 for 48 h followed by Hoechst Staining assay strictly
according to the protocol of Hoechst Staining kit (Beyotime
Institute of Biotechnology). Then all coverslips were mounted
inversely onto slides with anti-fluorescein quencher addition
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) and observed under a fluorescence microscope (Olympus,
Tokyo, Japan) and photographed.
Flow cytometric analysis of cell
apoptosis
Cell apoptosis analysis was performed with a Annexin
V-FITC/PI apoptosis detection kit (Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China) strictly following the manufacturer's
instructions. In brief, Cells in each group was resuspended in 500
µl binding buffer containing 5 µl Annexin V-FITC and 5 µl PI. Then
the mixture was incubated for 15 min at room temperature in the
dark. Cell apoptosis was detected through flow cytometry and
calculated by CellQuest software.
Wound healing assay
Cells were planted in 6-well plates and grown to 80%
confluence. A artificial wound was gently created with a 200 µl
pipette tip on the confluent cell monolayer in each group. The
detached cells were removed by FBS-free RPMI-1640 medium.
Thereafter, cells were cultured in FBS free PMI-1640 medium
supplemented with indicated drug for 48 h. The migrating cells were
imaged using an inverted microscope. The migration rate was the
ratio of the migrated distance to the initial distance.
Matrigel-based invasion assay
The upper chambers of 24-well Transwell system
(Corning, Tewksbury, MA, USA) were pre-coated with matrigel (BD
Biosciences, San Jose, CA, USA). Cells were resuspended in FBS-free
RPMI-1640 medium with indicated drug and plated in the upper
chamber of the transwell system with a density of
2×104/well. RPMI-1640 (800 µl) plus 30% FBS was added
into the lower chamber. Cells were incubated in the Transwell
system for 48 h, then the non-invading cells at the upper-surface
of the membrane were removed by cotton swabs, and the invading
cells at the under-surface of the membrane were fixed in 4%
paraformaldehyde for 20 min and stained with crystal violet for 5
min. The number of invading cells at each group was counted in five
randomly selected fields with a blinded manner under an inverted
microscope.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA in each sample was extracted by an RNA
extraction kit (Tiangen Biotech Co., Ltd., Beijing, China)
completely following the manufacturer instructions and
reverse-transcribed into cDNA. RT-qPCR was carried out by
Stratagene Mx3000P (Stratagene; Agilent Technologies, Inc., Santa
Clara, CA, USA) using Bestar® SybrGreen qPCR MasterMix
(DBI® Bioscience, Ludwigshafen, Germany) with the
following protocol: Initial denaturation at 95°C for 2 min, 40
cycles consisting of 94°C for 20 sec, 58°C for 20 sec, and 72°C for
20 sec. The following primers were used: miR-126-5p,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCGCGTA-3′ (sense) and
5′-ACACTCCAGCTGGGCATTATTACTTTTGGTA-3′ (antisense); VEGFR-A,
5′-AGGGCAGAATCATCACGAAGT-3′ (sense) and 5′-AGGGTCTCGATTGGAT-GGCA-3′
(antisense); VEGFR-2, 5′-ATAGAAGGTGCCCAGGAAAAG-3′ (sense) and
5′-GTCTTCAGTTCCCCTCCATTG-3′ (antisense); MEK1,
5′-GGGCTTCTA-TGGTGCGTTCTA-3′ (sense) and
5′-CCCACGGGAGTTGACTAGGAT-3′ (antisense); ERK,
5′-TCTGGAGCAGTATTACGACCC-3′ (sense) and 5′-CTGGCTGGAATCTAGCAGTCT-3′
(antisense); β-actin, 5′-ATCGTGCGTGACA-TTAAGGAGAAG-3′ (sense) and
5′-AGGAAGGAAGGCTGGAAGAGTG-3′ (antisense). Relative mRNA expressions
were obtained by 2−∆∆CT method. β-actin was served as an
internal control.
Western blot
Cells in each group were lysed with NP-40 lysate
(Beyotime Institute of Biotechnology) including 1%
phenylmethanesulfonyl fluoride (PMSF). The concentration of total
proteins was quantitated by a bicinchoninic acid (BCA) protein
assay kit (Beyotime Institute of Biotechnology). Then equal amounts
(20 µg) of different proteins were loaded and separated using
SDS-polyacrylamide gel electrophoresis (PAGE) followed by
electrotransferred onto polyvinylidene fluoride (PVDF) membranes
(Millipore, Bedford, MA, USA). After being blocked with 5% non-fat
milk at 4°C overnight, the membranes were probed with specific
primary antibodies against VEGFR-2, p-VEGFR-2 (Tyr1175), ERK1/2,
p-ERK1 (pT202/pY204) + p-ERK2 (pT185/pY187) (all 1:1,500 diluted);
and VEGF-A, MEK1, p-MEK1 (pS298) (all 1:1,000 diluted; Abcam,
Cambridge, MA, USA Abcam) at 4°C overnight. The membranes were then
incubated with the secondary goat anti-rabbit IgG-HRP antibody
(1:20,000 diluted; Wuhan Boster Biological Technology, Ltd., Wuhan,
China) at 37°C for 40 min. The unbound antibodies in each step were
washed with TBST for three times. The target bands were visualized
by an enhanced chemiluminescence (ECL; Millipore), and the protein
intensities were detected by Gel-Pro Analyzer software (Media
Cybernetics, Inc., Bethesda, MD, USA). GAPDH was used as an
internal control.
Statistical analysis
Statistical analysis was performed with GraphPad
Prism 5.0 software. All data are presented as mean ± standard
deviation (SD). Differences between groups in RT-PCR detection of
clinical tissues were calculated with unpaired Student's t-test.
Other differences comparison between groups were analyzed with
one-way analysis of variance (ANOVA). P<0.05 were considered to
indicate a statistically significant difference.
Results
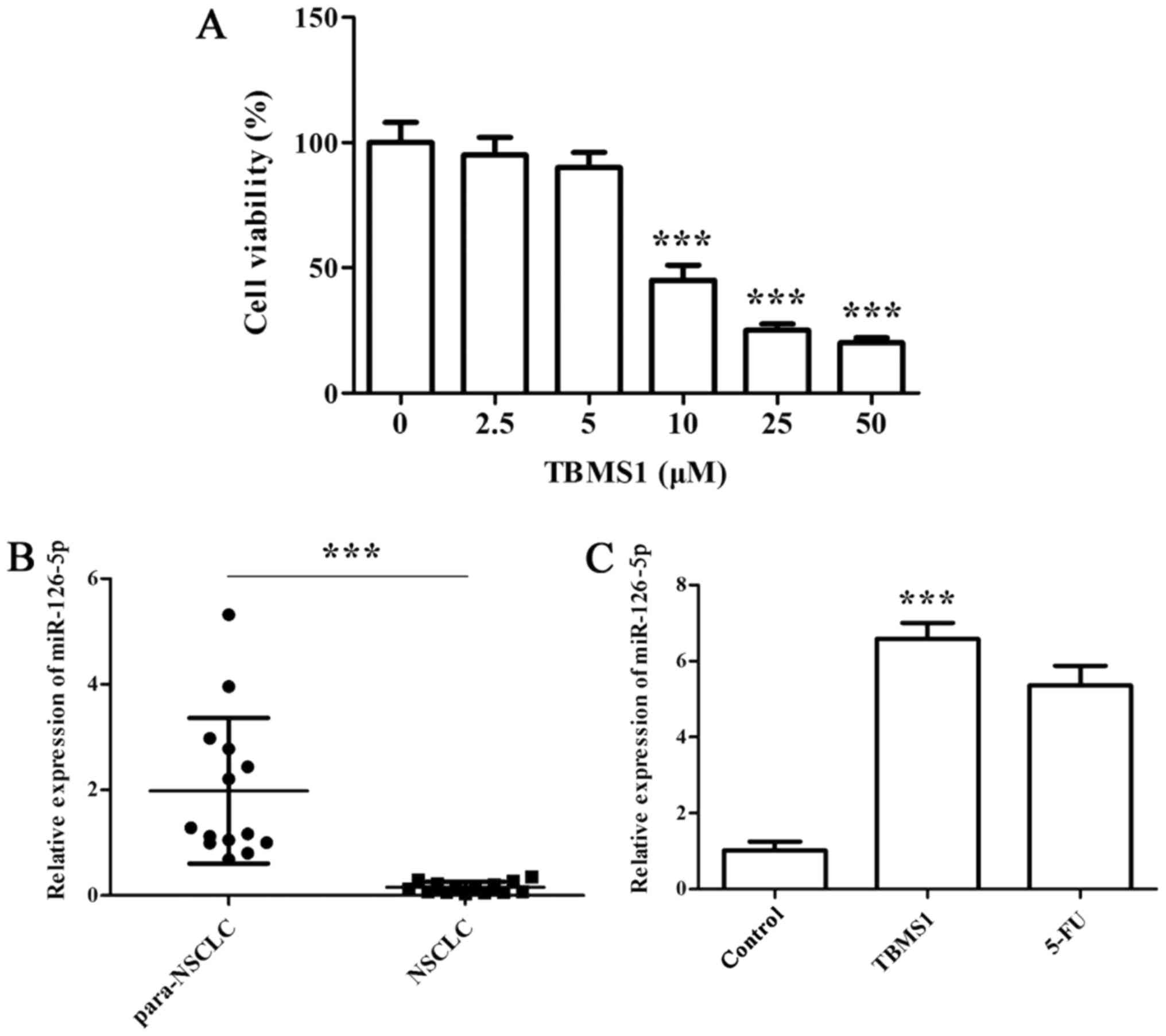
10 µM TBMS1 could significantly reduce
cell growth and upregulate miR-126-5p expression
We first employed CCK-8 assay to determine the
effect of TBMS1 on NCI-H1299 cell growth. The results showed that
cell viability exhibited dose-dependent inhibitions after TBMS1
treatment for 48 h, and the inhibitory effect was sharply increased
at 10 µM TBMS1 treatment, so we chose 10 µM TBMS1 for subsequent
experiments (Fig. 1A;
P<0.001).
Next, we conducted RT-qPCR to detect the expression
of miR-126-5p in both NSCLC pathological specimens and
TBMS1-treated NCI-H1299 cells. We found that miR-126-5p was notably
downregulated in NSCLC tissues compared with normal adjacent
tissues (Fig. 1B; P<0.001).
Furthermore, the significantly elevated miR-126-5p level was
observed in NCI-H1299 cells upon TBMS1 treatment for 48 h compared
with control (Fig. 1C;
P<0.001).
The above results demonstrated that 10 µM TBMS1
could significantly reduce cell growth and upregulate miR-126-5p
expression.
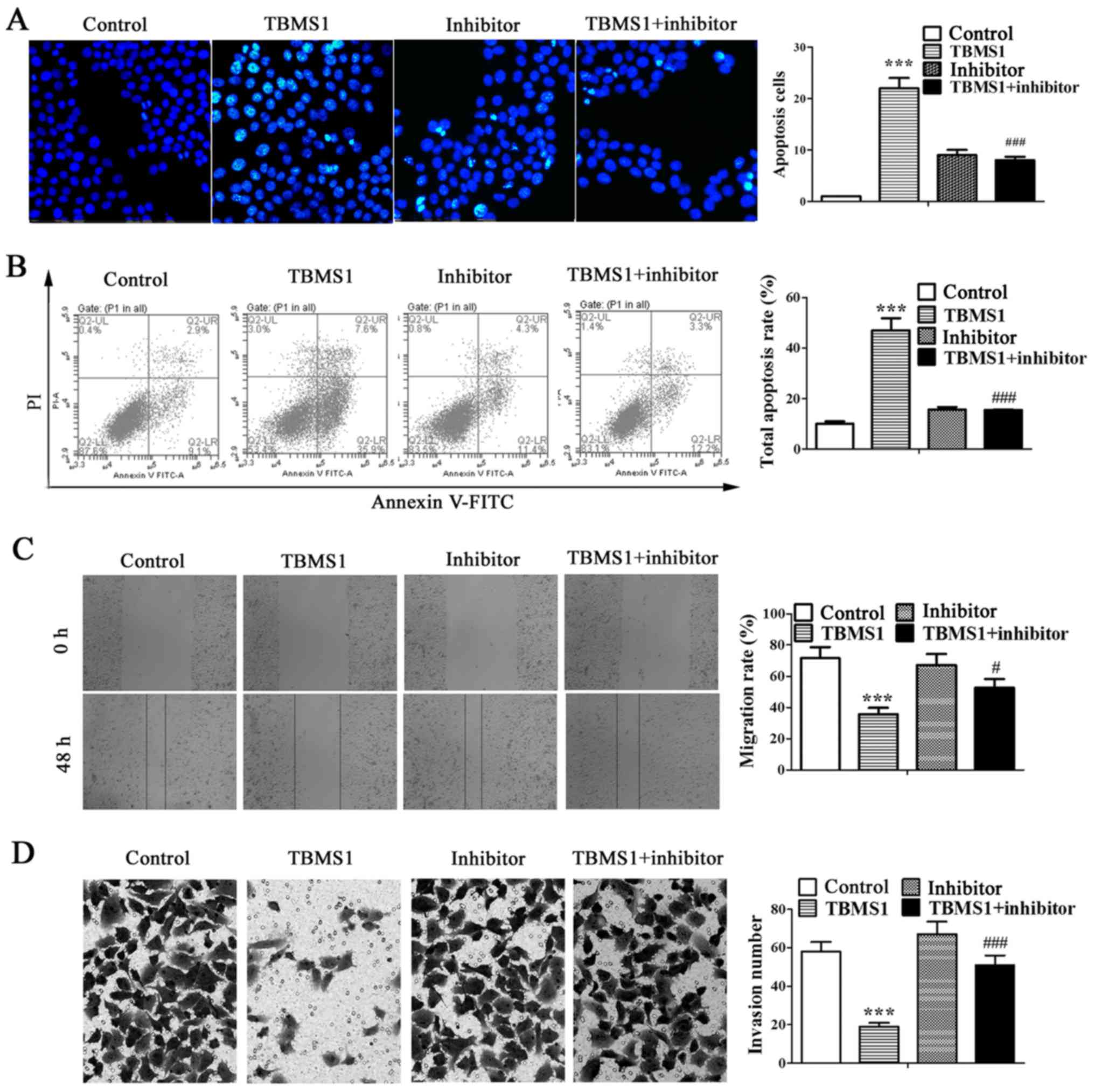
TBMS1 stimulates the apoptosis and
inhibits the metastasis of NCI-H1299 cells by promoting miR-126-5p
expression
To identify whether miR-126-5p is involved in the
antitumor effects of TBMS1 in NSCLC cells, the miR-126-5p inhibitor
was transfected into NCI-H1299 cells. Then Hoechst staining and
flow cytometry were employed to investigate cell apoptosis, and
wound healing and matrigel-based invasion assays were used to
evaluate the migration and invasion of NCI-H1299 cells. The results
showed that substantial nuclear condensation and apoptotic bodies
were appeared in TBMS1-treated NCI-H1299 cells, which was reversed
after inhibiting miR-126-5p (Fig.
2A; P<0.001). Similarly, the total apoptosis rate of
NCI-H1299 cells was increased significantly after TBMS1 treatment
for 48 h compared with control, but was decline after inhibiting
miR-126-5p (Fig. 2B; P<0.001).
Besides, the wound healing rate of TBMS1 treated NCI-H1299 cells
was much lower than control at the point of 48 h after wound
creation (P<0.001), but was notably elevated after inhibiting
miR-126-5p (Fig. 2C; P<0.05).
Simultaneously, the number of invading cells was 19.10±2.40 after
TBMS1 treatment for 48 h, which was obviously decreased as compared
to control cells (58.16±5.03), but the effect of TBMS1 was
diminished by inhibiting miR-126-5p (Fig. 2D; P<0.001). The above results
suggested that TBMS1 stimulates the apoptosis and inhibits the
metastasis of NCI-H1299 cells by promoting miR-126-5p
expression.
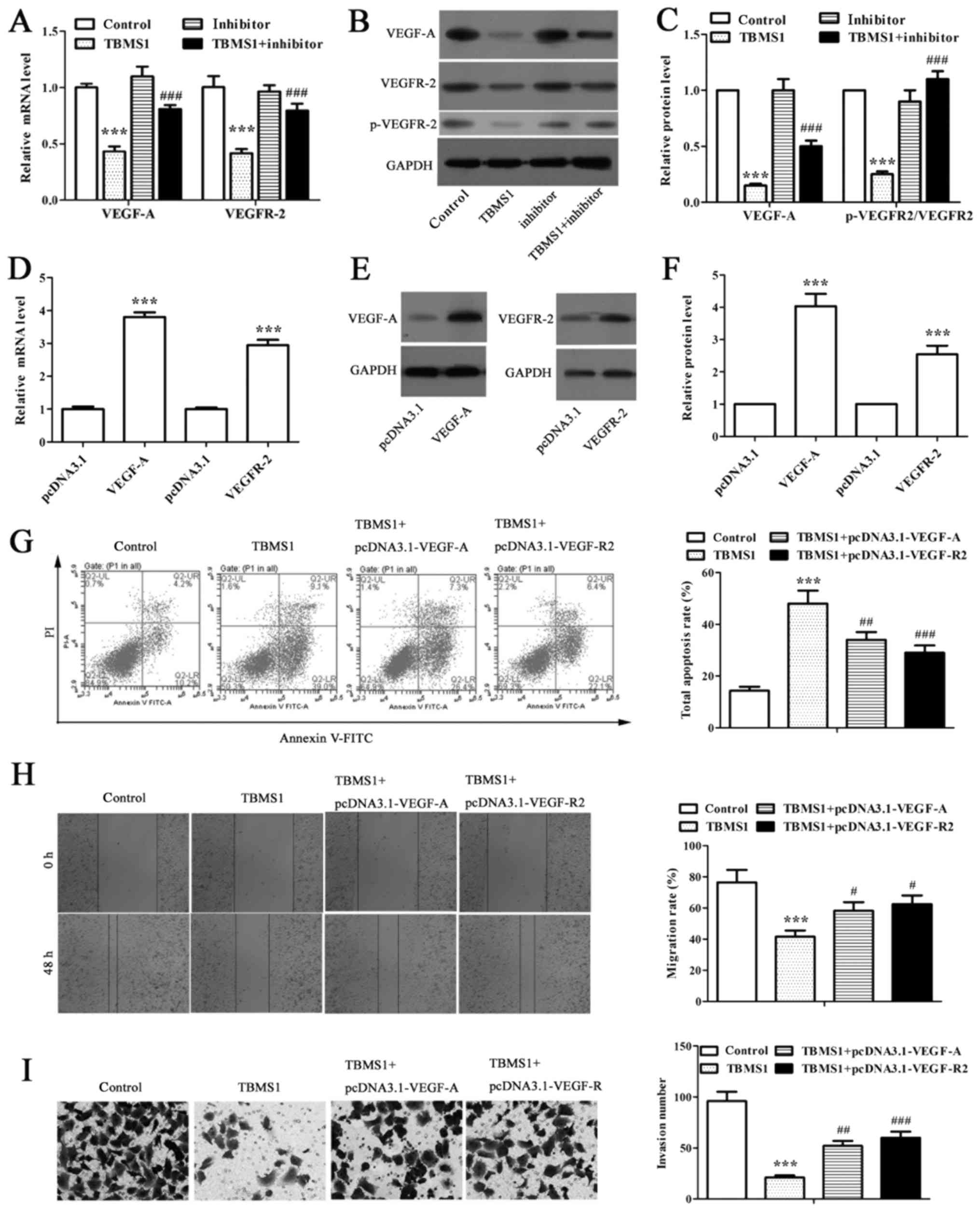
TBMS1 increased miR-126-5p
downregulates the expressions of VEGF-A and VEGFR-2 to enhance the
apoptosis and reduce the metastasis of NCI-H1299 cells
RT-qPCR and western blot analysis were performed to
detect the expression of VEGF-A and VEGFR-2 in miR-126-5p inhibitor
transfected NCI-H1299 cells under TBMS1 treatment. We found that
TBMS1 coordinately downregulated VEGF-A and VEGFR-2 expressions at
both mRNA and protein levels in NCI-H1299 cells compared with
control, as well as the significantly suppressed phosphorylation of
VEGFR-2 (Fig. 3A-C; P<0.001).
While inhibiting miR-126-5p remarkably reversed above effects
(Fig. 3A-C; P<0.001). As VEGF-A
is a target for miR-126-5p, the results implied a link between
VEGF-A/VEGFR-2 axis and TBMS1 induced pro-apoptotic and
anti-metastatic effects.
We then overexpressed VEGF-A and VEGFR-2
respectively in NCI-H1299 cells and conducted flow cytometry, wound
healing and matrigel-based invasion assays to explore the
apoptosis, migration and invasion of NCI-H1299 cells. NCI-H1299
cells transfected with pcDNA3.1-VEGF-A and pcDNA3.1-VEGFR-2
displayed 3.80-fold and 2.94-fold higher mRNA levels along with
4.03-fold and 2.54-fold higher protein levels than those with
respective pcDNA3.1 vectors transfected (Fig. 3D-F; P<0.001), indicating
successfully overexpressed VEGF-A and VEGFR-2 in NCI-H1299 cells.
Further study revealed that the apoptosis rates were decreased
significantly in VEGF-A-overexpressing and VEGFR-2-overexpressing
NCI-H1299 cells respectively than in non-transfected cells
following the same TBMS1 treatment (Fig. 3G; P<0.01). As expected, the
migration rates were notably elevated to 1.4- and 1.5-fold after 48
h of TBMS1 treatment in VEGF-A-overexpressing and
VEGFR-2-overexpressing NCI-H1299 cells respectively compared with
non-transfected TBMS1-treated cells (Fig. 3H; P<0.05). Meanwhile,
overexpressing VEGF-A and VEGFR-2 stimulated the invasion of
TBMS1-treated NCI-H1299 cells (Fig.
3I; P<0.01). Thus, the above results indicated that
TBMS1-driven miR-126-5p enhanced the apoptosis and reduced the
metastasis of NCI-H1299 cells through downregulating VEGF-A/VEGFR-2
axis.
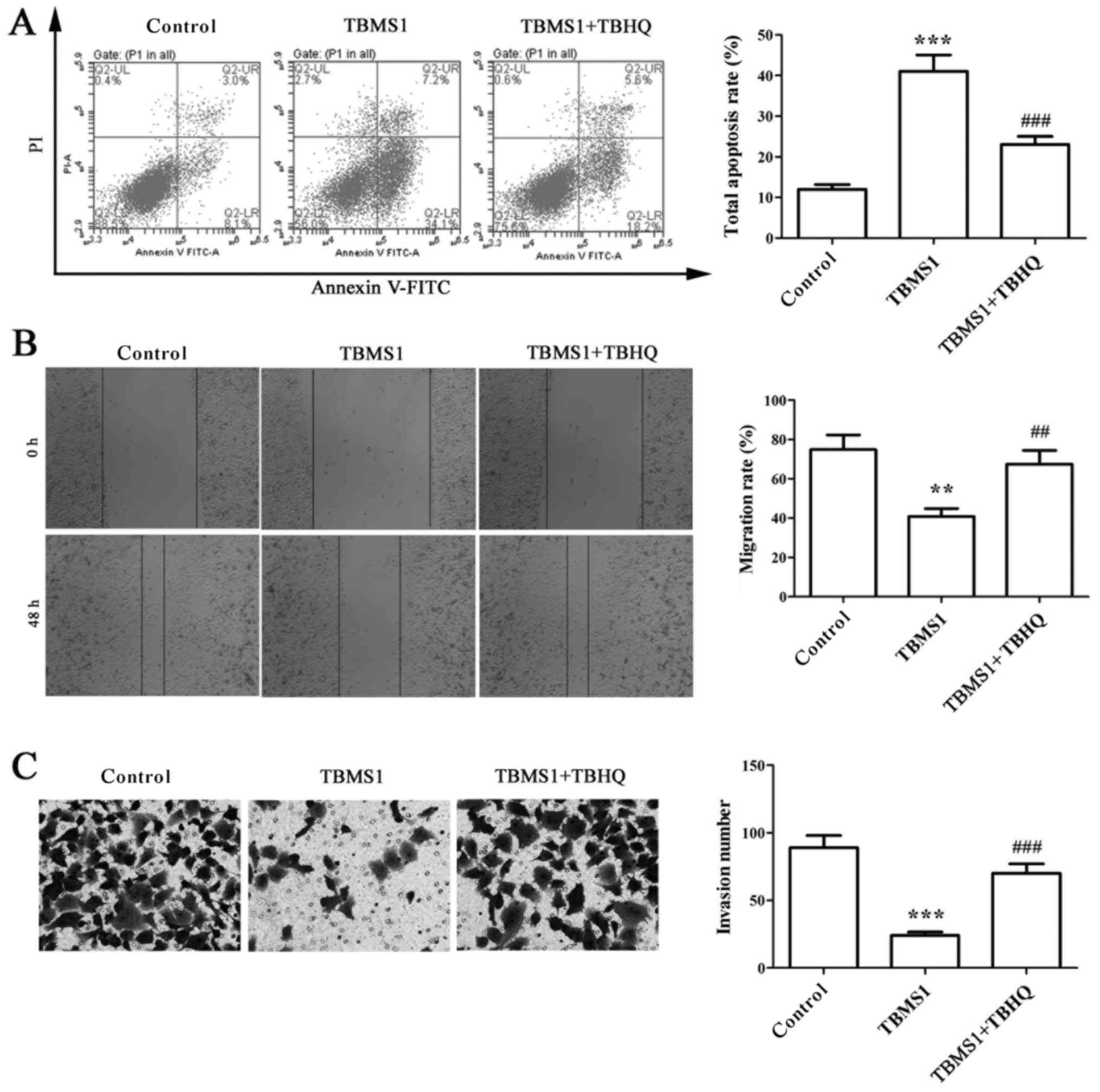
TBMS1 inactivates ERK pathway to
enhance the apoptosis and reduce the metastasis of NCI-H1299
cells
TBHQ is a phenolic antioxidant functioning as an
activator of ERK, and it requires the help from upstream signaling
kinase MAPK/ERK kinase (MEK). To deduce changes in ERK1/2 pathway
as a function of TBMS1, NCI-H1299 cells were exposed to 10 µmol/l
TBMS1 for 48 h combined with TBHQ, then flow cytometry, wound
healing and matrigel-based invasion assays were used to detect the
apoptosis, migration and invasion of NCI-H1299 cells. The results
showed that the elevated apoptosis rate of NCI-H1299 cells with
TBMS1 treatment was sharply declined after combination with TBHQ
(Fig. 4A; P<0.001). In
addition, the migration rate and invasion number in TBMS1-treated
NCI-H1299 cells upon addition of TBHQ were increased significantly
compared with TBMS1 treatment alone (Fig. 4B and C; P<0.01). These results
indicated that TBMS1 could inactivate ERK pathway to enhance the
apoptosis and reduce the metastasis of NCI-H1299 cells.
The pro-apoptotic and anti-metastatic
effects of TBMS1 in NCI-H1299 cells are mediated by overexpressing
miR-126-5p induced inactivation of VEGF-A/VEGFR-2/ERK pathway
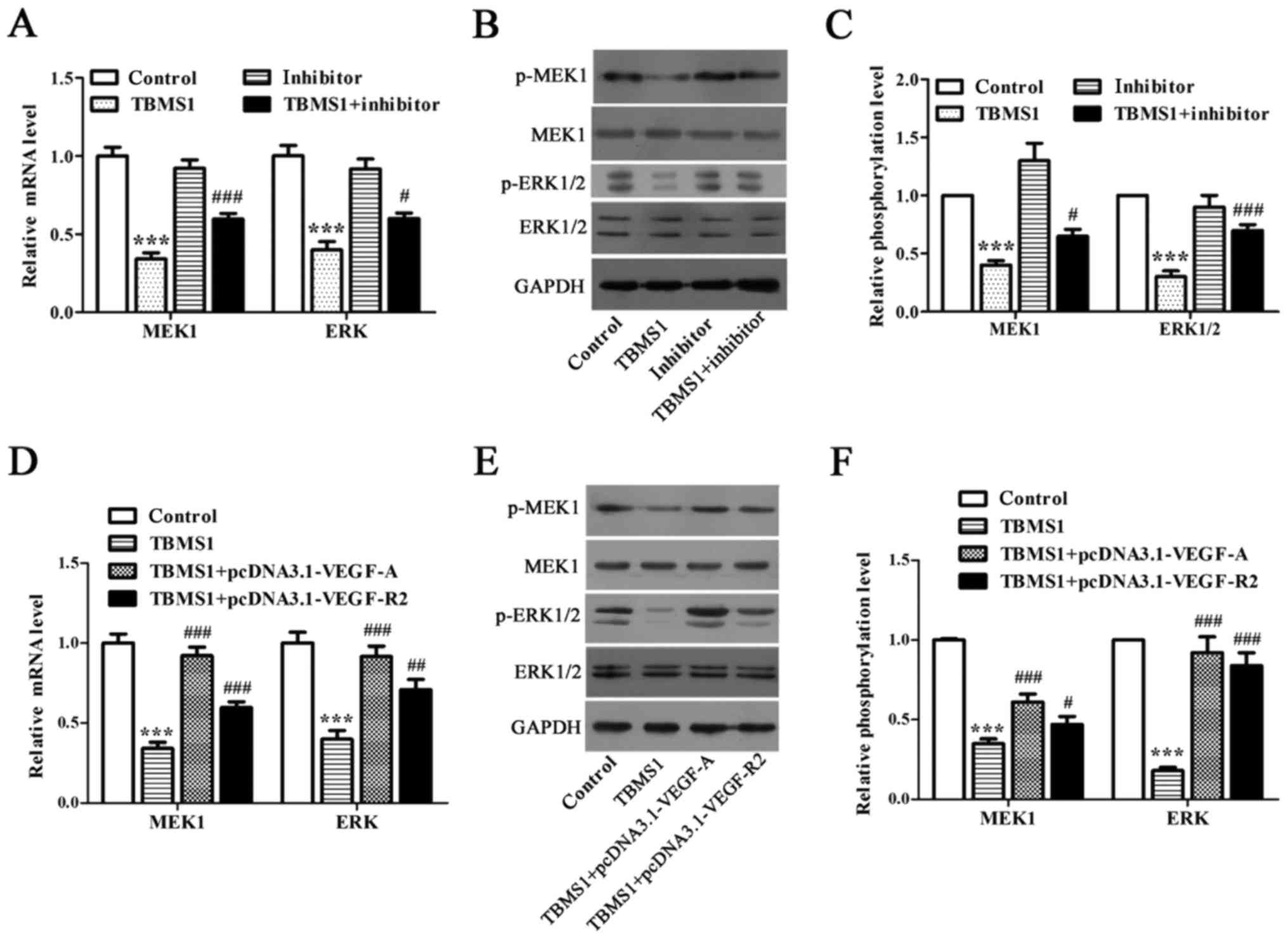
We employed RT-qPCR and Western blot to address
whether ERK1/2 pathway is the downstream mediator of miR-126-5p
downregulated VEGF-A/VEGFR-2. We discovered that both mRNA and
phosphorylation levels of MEK1 and ERK were declined significantly
in NCI-H1299 cells upon TBMS1 treatment for 48 h in comparison with
control (Fig. 5A-C; P<0.001),
while inhibiting miR-126-5p prevented this decline (Fig. 5A-C; P<0.05). Next, we detected
that overexpressing VEGF-A or VEGFR-2 in TBMS1-treated NCI-H1299
cells caused the elevated mRNA and phosphorylation levels of both
MEK1 and ERK (Fig. 5D-F;
P<0.05). Taken together, we concluded that the pro-apoptotic and
anti-metastatic effects of TBMS1 in NCI-H1299 cells are mediated by
overexpressing miR-126-5p induced inactivation of
VEGF-A/VEGFR-2/ERK axis.
Discussion
Our study demonstrate that TBMS1 boosts the
apoptosis and inhibits the migration and invasion of NSCLC cells by
overexpressing miR-126-5p induced inactivation of
VEGF-A/VEGFR-2/ERK pathway, highlighting a therapeutic pathway of
TBMS1.
Clinical researches proved that the reduced
miR-126-5p is the most differentially expressed miRNA in lung
cancers (24). In addition, Felli
et al revealed that overexpression of miR-126-5p actuated
the reduction of proliferation, invasion, propagation and
chemotaxis in melanoma cells (25). In the present study, we detected
the significantly downregulated miR-126-5p in NSCLC tumor tissues,
but the expression of miR-126-5p was raised much higher after TBMS1
administration in NCI-H1299 cells, we therefore speculate that
TBMS1 may upregulate miR-126-5p to induce antitumor effects in
NSCLC cells. Then we found that the pro-apoptotic and
anti-metastatic effects of TBMS1 were notably abrogated by
inhibition of miR-126-5p in NCI-H1299 cells, which results proved
our conjecture.
Except for angiogenesis, VEGF-A-stimulated response
also contains the invasion, migration, and survival of cancer
(26–28), which is largely attributed to
VEGFR-2 for its potent tyrosine kinase activity (29). VEGF-A binding to VEGFR-2 induces
dimerization and transautophosphorylation of cytoplasmic tyrosine
residues to trigger diverse cellular responses (29,30).
Overexpression of VEGF-A has been implicated in most human tumors,
including NSCLC, and is associated with increased tumor recurrence,
metastasis, and poorer prognosis (31–36).
It is a well-established fact that VEGF-A is a direct miR-126-5p
target, and miR-126-downregulated VEGF could inhibit the
proliferation of lung cancer cells (37). Accordingly, we found that TBMS1
downregulated VEGF-A through miR-126-5p, and overexpressing VEGF-A
prevented TBMS1-mediated, miR-126-5p-dependent antitumor effects in
NCI-H1299 cells, overexpressing VEGFR-2 yielded similar results,
indicating that VEGF-A/VEGFR-2 are the downstream pathway of TBMS1
induced overexpression of miR-126-5p.
ERK1/2 pathway is an important member of MAPKs
pathway capable of promoting growth, invasion, angiogenesis and
metastasis by increasing the secretion of MMP-9 and the
phosphorylation of ATF-2 (38–40).
Liu et al identified that TBMS1 sensitizes cisplatin in
cisplatin-resistant human ovarian cancer cells through
downregulation of ERK and upregulation of p38 signaling pathways
(41). In NSCLC, we discovered
that inhibiting ERK pathway in TBMS1 treated NCI-H1299 cells could
resulted in the pro-apoptotic and anti-metastatic effects, but
above inactivation of ERK pathway was prevented upon inhibiting
miR-126-5p, confirming that ERK pathway is also the downstream
pathway of TBMS1-mediated, miR-126-5p-dependent antitumor
effects.
VEGF-A/VEGFR-2/MEK1/ERK1/2 signaling pathway
converge on the nucleus leading to DNA replication, cellular
proliferation and migration. In vitro study demonstrated
that overexpression of VEGF-A augmented cell proliferation and
migration through this pathway, thereby inducing a more invasive
tumor phenotype (42,43). The phosphorylation of VEGFR2 at
Tyr1175 induces PIP2 hydrolysis by activating PLCγ to trigger ERK
pathway. Silencing VEGFR-2 blocked the phosphorylation of ERK1/2 in
lung cancer cells (44). Santos
et al found that the intracellular VEGFR-2 inhibitor
promoted apoptosis of acute myeloid leukemia cell lines through
inhibiting ERK1/2 pathway (45).
We found the reduced phosphorylation level of VEGFR2 under TBMS1
treatment, and further discovered that the inactivation of ERK
pathway induced by TBMS1 was prevented when VEGF-A or VEGFR-2 was
overexpressed in NCI-H1299 cells, indicating that
VEGF-A/VEGFR-2/MEK1/ERK1/2 axis act as a direct mediator of
TBMS1-mediated, miR-126-5p-dependent antitumor effects.
In conclusion, our present study gradually
recognized and confirmed that TBMS1 could increase the expression
of miR-126-5p, then miR-126-5p targeted VEGF-A inactivated the
VEGF-A/VEGFR-2/ERK1/2 pathway to boost apoptosis and suppress
migration and invasion in NCI-H1299 cells. Our data explained the
mechanism of TBMS1-mediated antitumor effects in NCI-H1299 cells.
TBMS1 may become a promising candidate for NSCLC therapy.
Glossary
Abbreviations
Abbreviations:
|
TBMS1
|
tubeimoside-1
|
|
VEGF-A
|
vascular endothelial growth
factor-A
|
|
VEGF-2
|
vascular endothelial growth factor
receptor-2
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
NSCLC
|
non-small cell lung cancer
|
References
|
1
|
Wald O, Shapira OM and Izhar U:
CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic
roles and therapeutic potential. Theranostics. 3:26–33. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kumarakulasinghe NB, van Zanwijk N and Soo
RA: Molecular targeted therapy in the treatment of advanced stage
non-small cell lung cancer (NSCLC). Respirology. 20:370–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Travis WD, Brambilla E, Noguchi M,
Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ,
Van Schil PE, et al: International association for the study of
lung cancer/american thoracic society/european respiratory society
international multidisciplinary classification of lung
adenocarcinoma. J Thorac Oncol. 6:244–285. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fennell DA, Summers Y, Cadranel J, Benepal
T, Christoph DC, Lal R, Das M, Maxwell F, Visseren-Grul C and Ferry
D: Cisplatin in the modern era: The backbone of first-line
chemotherapy for non-small cell lung cancer. Cancer Treat Rev.
44:42–50. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gu Y, Körbel C, Scheuer C, Nenicu A,
Menger MD and Laschke MW: Tubeimoside-1 suppresses tumor
angiogenesis by stimulation of proteasomal VEGFR2 and Tie2
degradation in a non-small cell lung cancer xenograft model.
Oncotarget. 7:5258–5272. 2016.PubMed/NCBI
|
|
6
|
Yin Y, Chen W, Tang C, Ding H, Jang J,
Weng M, Cai Y and Zou G: NF-κB, JNK and p53 pathways are involved
in tubeimoside-1-induced apoptosis in HepG2 cells with oxidative
stress and G2/M cell cycle arrest. Food Chem Toxicol. 49:3046–3054.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu L, Ma R, Wang Y and Nishino H: Potent
anti-tumor activity and low toxicity of tubeimoside 1 isolated from
Bolbostemma paniculatum. Planta Med. 60:204–208. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartoli Klugman F, Decorti G, Candussio L,
Mallardi F, Grill V, Zweyer M and Baldini L: Effect of ketotifen on
adriamycin toxicity: Role of histamine. Cancer Lett. 39:145–152.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu Y, Wang G, Chen Q, Lin T, Zeng Z, Luo
Q, Liu J and Sun C: Intrinsic apoptotic pathway and G2/M cell cycle
arrest involved in tubeimoside I-induced EC109 cell death. Chin J
Cancer Res. 25:312–321. 2013.PubMed/NCBI
|
|
10
|
Jia G, Wang Q, Wang R, Deng D, Xue L, Shao
N, Zhang Y, Xia X, Zhi F and Yang Y: Tubeimoside-1 induces glioma
apoptosis through regulation of Bax/Bcl-2 and the ROS/Cytochrome
C/Caspase-3 pathway. Onco Targets Ther. 8:303–311. 2015.PubMed/NCBI
|
|
11
|
Xu Y, Chiu JF, He QY and Chen F:
Tubeimoside-1 exerts cytotoxicity in HeLa cells through
mitochondrial dysfunction and endoplasmic reticulum stress
pathways. J Proteome Res. 8:1585–1593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang P, Yu C, Liu XQ, Ding YB, Wang YX
and He JL: Cytotoxicity of tubeimoside I in human choriocarcinoma
JEG-3 cells by induction of cytochrome c release and apoptosis via
the mitochondrial-related signaling pathway. Int J Mol Med.
28:579–587. 2011.PubMed/NCBI
|
|
13
|
Wang F, Ma RD and Yu LJ: Role of
mitochondria in tubeimoside I-mediated apoptosis in human cervical
carcinoma HeLa cell line. Zhongguo Zhong Yao Za Zhi. 30:1935–1939.
2005.(In Chinese). PubMed/NCBI
|
|
14
|
Lin Y, Xie G, Xia J, Su D, Liu J, Jiang F
and Xu Y: TBMS1 exerts its cytotoxicity in NCI-H460 lung cancer
cells through nucleolar stress-induced p53/MDM2-dependent
mechanism, a quantitative proteomics study. Biochim Biophys Acta.
1864:204–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Xu X and He P: Tubeimoside-1
inhibits proliferation and induces apoptosis by increasing the Bax
to Bcl-2 ratio and decreasing COX-2 expression in lung cancer A549
cells. Mol Med Rep. 4:25–29. 2011.PubMed/NCBI
|
|
16
|
Ebrahimi F, Gopalan V, Smith RA and Lam
AK: miR-126 in human cancers: Clinical roles and current
perspectives. Exp Mol Pathol. 96:98–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meister J and Schmidt MH: miR-126 and
miR-126*: New players in cancer. ScientificWorldJournal.
10:2090–2100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schober A, Nazari-Jahantigh M, Wei Y,
Bidzhekov K, Gremse F, Grommes J, Megens RT, Heyll K, Noels H,
Hristov M, et al: MicroRNA-126-5p promotes endothelial
proliferation and limits atherosclerosis by suppressing Dlk1. Nat
Med. 20:368–376. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Esser JS, Saretzki E, Pankratz F, Engert
B, Grundmann S, Bode C, Moser M and Zhou Q: Bone morphogenetic
protein 4 regulates microRNAs miR-494 and miR-126-5p in control of
endothelial cell function in angiogenesis. Thromb Haemost.
117:734–749. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li HY, Zhao X, Liu YZ, Meng Z, Wang D,
Yang F and Shi QW: Plasma microRNA-126-5p is associated with the
complexity and severity of coronary artery disease in patients with
stable angina pectoris. Cell Physiol Biochem. 39:837–846. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ladeiro Y, Couchy G, Balabaud C,
Bioulac-Sage P, Pelletier L, Rebouissou S and Zucman-Rossi J:
MicroRNA profiling in hepatocellular tumors is associated with
clinical features and oncogene/tumor suppressor gene mutations.
Hepatology. 47:1955–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang J, Du YY, Lin YF, Chen YT, Yang L,
Wang HJ and Ma D: The cell growth suppressor, mir-126, targets
IRS-1. Biochem Biophys Res Commun. 377:136–140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cho WC, Chow AS and Au JS: Restoration of
tumour suppressor hsa-miR-145 inhibits cancer cell growth in lung
adenocarcinoma patients with epidermal growth factor receptor
mutation. Eur J Cancer. 45:2197–2206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yanaihara N, Caplen N, Bowman E, Seike M,
Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, et
al: Unique microRNA molecular profiles in lung cancer diagnosis and
prognosis. Cancer Cell. 9:189–198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Felli N, Felicetti F, Lustri AM, Errico
MC, Bottero L, Cannistraci A, De Feo A, Petrini M, Pedini F,
Biffoni M, et al: miR-126&126* restored expressions play a
tumor suppressor role by directly regulating ADAM9 and MMP7 in
melanoma. PLoS One. 8:e568242013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dvorak HF: Vascular permeability
factor/vascular endothelial growth factor: A critical cytokine in
tumor angiogenesis and a potential target for diagnosis and
therapy. J Clin Oncol. 20:4368–4380. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Millauer B, Wizigmann-Voos S, Schnürch H,
Martinez R, Møller NP, Risau W and Ullrich A: High affinity VEGF
binding and developmental expression suggest Flk-1 as a major
regulator of vasculogenesis and angiogenesis. Cell. 72:835–846.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zeng H, Dvorak HF and Mukhopadhyay D:
Vascular permeability factor (VPF)/vascular endothelial growth
factor (VEGF) peceptor-1 down-modulates VPF/VEGF
receptor-2-mediated endothelial cell proliferation, but not
migration, through phosphatidylinositol 3-kinase-dependent
pathways. J Biol Chem. 276:26969–26979. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roskoski R Jr: Vascular endothelial growth
factor (VEGF) signaling in tumor progression. Crit Rev Oncol
Hematol. 62:179–213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koch S and Claesson-Welsh L: Signal
transduction by vascular endothelial growth factor receptors. Cold
Spring Harb Perspect Med. 2:a0065022012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen P, Zhu J, Liu DY, Li HY, Xu N and Hou
M: Over-expression of survivin and VEGF in small-cell lung cancer
may predict the poorer prognosis. Med Oncol. 31:7752014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ferrara N: The role of vascular
endothelial growth factor in pathological angiogenesis. Breast
Cancer Res Treat. 36:127–137. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mattern J, Koomägi R and Volm M:
Association of vascular endothelial growth factor expression with
intratumoral microvessel density and tumour cell proliferation in
human epidermoid lung carcinoma. Br J Cancer. 73:931–934. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brown LF, Berse B, Jackman RW, Tognazzi K,
Manseau EJ, Dvorak HF and Senger DR: Increased expression of
vascular permeability factor (vascular endothelial growth factor)
and its receptors in kidney and bladder carcinomas. Am J Pathol.
143:1255–1262. 1993.PubMed/NCBI
|
|
35
|
Brown LF, Berse B, Jackman RW, Tognazzi K,
Guidi AJ, Dvorak HF, Senger DR, Connolly JL and Schnitt SJ:
Expression of vascular permeability factor (vascular endothelial
growth factor) and its receptors in breast cancer. Hum Pathol.
26:86–91. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Seto T, Higashiyama M, Funai H, Imamura F,
Uematsu K, Seki N, Eguchi K, Yamanaka T and Ichinose Y: Prognostic
value of expression of vascular endothelial growth factor and its
flt-1 and KDR receptors in stage I non-small-cell lung cancer. Lung
Cancer. 53:91–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu B, Peng XC, Zheng XL, Wang J and Qin
YW: miR-126 restoration down-regulate VEGF and inhibit the growth
of lung cancer cell lines in vitro and in vivo. Lung Cancer.
66:169–175. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fearnley GW, Odell AF, Latham AM, Mughal
NA, Bruns AF, Burgoyne NJ, Homer-Vanniasinkam S, Zachary IC,
Hollstein MC, Wheatcroft SB and Ponnambalam S: VEGF-A isoforms
differentially regulate ATF-2-dependent VCAM-1 gene expression and
endothelial-leukocyte interactions. Mol Biol Cell. 25:2509–2521.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shankar S, Ganapathy S, Hingorani SR and
Srivastava RK: EGCG inhibits growth, invasion, angiogenesis and
metastasis of pancreatic cancer. Front Biosci. 13:440–452. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaneshiro T, Morioka T, Inamine M, Kinjo
T, Arakaki J, Chiba I, Sunagawa N, Suzui M and Yoshimi N:
Anthraquinone derivative emodin inhibits tumor-associated
angiogenesis through inhibition of extracellular signal-regulated
kinase 1/2 phosphorylation. Eur J Pharmacol. 553:46–53. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu HZ, Yu C, Yang Z, He JL, Chen WJ, Yin
J, Li WM, Liu HT and Wang YX: Tubeimoside I sensitizes cisplatin in
cisplatin-resistant human ovarian cancer cells (A2780/DDP) through
down-regulation of ERK and up-regulation of p38 signaling pathways.
Mol Med Rep. 4:985–992. 2011.PubMed/NCBI
|
|
42
|
Dias S, Hattori K, Zhu Z, Heissig B, Choy
M, Lane W, Wu Y, Chadburn A, Hyjek E, Gill M, et al: Autocrine
stimulation of VEGFR-2 activates human leukemic cell growth and
migration. J Clin Invest. 106:511–521. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tian Y, Xie Q, Tian Y, Liu Y, Huang Z, Fan
C, Hou B, Sun D, Yao K and Chen T: Radioactive ¹25I seed
inhibits the cell growth, migration, and invasion of nasopharyngeal
carcinoma by triggering DNA damage and inactivating VEGF-A/ERK
signaling. PLoS One. 8:e740382013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu Y, Qiao Y, Hu C, Liu L, Zhou L, Liu B,
Chen H and Jiang X: VEGFR2 inhibition by RNA interference affects
cell proliferation, migration, invasion, and response to radiation
in Calu-1 cells. Clin Transl Oncol. 18:212–219. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Santos SC and Dias S: Internal and
external autocrine VEGF/KDR loops regulate survival of subsets of
acute leukemia through distinct signaling pathways. Blood.
103:3883–3889. 2004. View Article : Google Scholar : PubMed/NCBI
|