Introduction
Vascular calcification is an important medical issue
due to the close association between the degree of vascular
calcification and the severity of various clinical diseases,
including atherosclerosis (1),
diabetes mellitus (2) and chronic
kidney disease (3). In vessel
calcification, vascular smooth muscle cells (VSMCs) serve an
integral mediatory role by undergoing differentiation into
osteoblast-like cells and generating matrix vesicles, serving as
nidi for calcium-phosphate deposition in the vessel wall (4). During this process, upregulation of
bone-associated genes, such as core binding factor α1 (CBFA1)
(5), and downregulation of smooth
muscle cell-associated marker genes, such as smooth muscle 22α
(SM22α) (6), can be observed.
Oxidized low-density lipoproteins (Ox-LDL) are
crucial in the progression of atherosclerosis, and promote
osteogenic differentiation and calcification of VSMCs (7). In addition, the catabolic process of
autophagy, which can occur as an adaptive response to cell stress,
has been demonstrated to limit VSMC calcification (8). Accumulating evidence has demonstrated
that the enhancement of autophagy by different types of medicinal
molecules may serve as a potential therapeutic approach for
Ox-LDL-induced pathological states (9,10).
Numerous polysaccharide-containing Chinese herbal
medicines are capable of exerting a wide variety of pharmacological
effects, including cardioprotection (11,12).
One such polysaccharide has previously been extracted from Radix
Aconitum carmichaelii Praeparata (also known as Fuzi), and
is designated as polysaccharide from Fuzi (FPS) (13). Previously, it was demonstrated that
FPS pretreatment protected hepatocytes against ischemia-reperfusion
injury through its potent anti-oxidative effects and necrosis
attenuation (14). Furthermore, it
was demonstrated that FPS increased autophagic activity to protect
against starvation-induced cytotoxicity in H9c2 cells (15). Therefore, it can be hypothesized
that FPS may also have protective effects against calcification in
human VSMCs during Ox-LDL-induced stress, via autophagy
upregulation.
Materials and methods
Cell culture
All cell culture reagents were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Written informed
consent was obtained from patients and approval was received from
the Ethics Committee of the First Affiliated Hospital of Sun
Yat-sen University (Guangzhou, China). The work described in the
present study was conducted in accordance with The Code of Ethics
of the World Medical Association (www.wma.net/).
VSMCs were isolated from human femoral arteries (4 patients,
45.4±4.6 years old, 2 male and 2 female, recruited from Guangzhou,
China, from June 2016 to May 2017) using the explant method
described previously (16). VSMCs
were maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum at 37°C in 5%
CO2. Both DMEM and fetal serum were purchased from
Sigma-Aldrich (Merck KGaA). Cells between passages 3 and 6 were
used in this study. To induce calcification, growth medium was
replaced with osteogenic DMEM, which was supplemented with 10 mM
β-glycerophosphate, which was purchased from Sigma-Aldrich (Merck
KGaA).
Ox-LDL preparations
LDLs (1.019–1.063 g/ml) were separated from human
plasma by sequential density gradient ultracentrifugation (145,000
× g at 10°C for 20 h). The LDL fraction was dialyzed against PBS
(pH 7.4) at 4°C for 24 h. Ox-LDL was prepared by incubation of LDL
with 5 mM copper sulfate (Sigma-Aldrich, Merck KGaA) at 37°C for 3
h, and was subsequently sterilized by passaging through a 0.22-µm
filter. Cells were treated with Ox-LDL (10, 50 or 100 µg/ml) for 14
days (37°C, treated once every two days).
Mineralization assay
Mineral deposition in cultured VSMCs was assessed by
Alizarin Red S staining as previously described (17). Calcified VSMCs (2×106)
were fixed in 4% formaldehyde for 10 min at room temperature and
exposed to 2% Alizarin Red S (pH 4.2) for 5 min at room
temperature. Subsequently, cells were washed with deionized water
to remove excess dye and imaged using an inverted phase contrast
microscope. For the quantification of Alizarin Red S staining, 10%
formic acid was used to elute the Alizarin Red S dye, and the
absorbance at 405 nm was determined with a microplate reader and
normalized to the protein content. For quantification of calcium
content, cells were washed with PBS and decalcified with 0.6 M
hydrogen chloride for 24 h. The calcium content of the cultures was
determined colorimetrically using the O-cresolphthalein complexone
method and normalized to protein content as previously described
(18).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cultured VSMCs using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), and was reverse transcribed using an avian
myeloblastosis virus Reverse Transcriptase (Roche Diagnostics GmbH,
Mannheim, Germany) according to the manufacturer's protocol. The
thermocycling conditions were 37°C for 1 h and then 94°C for 10
min. RT-qPCR was performed using a SYBR®-Green PCR
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.) in
a StepOne Real Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The primers used for RT-qPCR were as follows:
CBFA1 forward, 5′-CCGTGGCCTTCAAGGTTGT-3′ and reverse,
5′-TTCATAACAGCGGGAGGCATTTC-3′; SM22α forward,
5′-TTGGATCCGACATGGCCAACAAG-3′ and reverse,
5′-GCCCTAAGCTTTCTAACTGATGATCT-3′; and β-actin forward,
5′-CCAGCTCACCATGGATGATG-3′ and reverse, 5′-GAGCCGTTGTCGACGACG-3′.
Values were normalized to those of β-actin. The results were
calculated using the comparative threshold cycle (Cq) method
(19).
Treatments
In all assays human VSMCs were cultured in
osteogenic medium at 37°C with 5% CO2 for 14 days.
Experiment 1 (Fig. 1) human VSMCs
were treated with Ox-LDL (0, 10, 50 and 100 µg/ml). In experiment 2
(Fig. 2) cells were treated with 0
or 50 µg/ml Ox-LDL and different concentrations of FPS (0, 10, 100
or 1,000 µg/ml). Experiment 3 (Fig.
3) was performed with 0 or 50 µg/ml Ox-LDL and 1,000 µg/ml FPS.
In experiment 4 (Fig. 4) human
VSMCs with 0 or 50 µg/ml Ox-LDL, 1,000 µg/ml FPS and 5 mM 3MA.
 | Figure 3.Human VSMCs were cultured in
osteogenic medium with or without 50 µg/ml Ox-LDL and 1,000 µg/ml
FPS for 14 days. Representative images of (A) LC3-I, LC3-II, P62
and GAPDH proteins and (B) quantification of the relative
LC3-II/LC3-I ratio and relative P62 protein expression levels, as
measured by western blotting. (C) Representative images of GFP-LC3
dots after the indicated treatments. Blue arrows represent VSMCs
expressing GFP-LC3 dots. Data are expressed as the mean ± standard
error (n=4). Group 1, human VSMCs were cultured in osteogenic
medium without Ox-LDL or FPS. In group 2 human VSMCs were cultured
in osteogenic medium with 50 µg/ml Ox-LDL without FPS. *P<0.05
vs. group 1; &P<0.05 vs. group 2. VSMCs, vascular
smooth muscle cells; Ox-LDL, oxidized low-density lipoproteins;
FPS, polysaccharide from Fuzi; GFP, green fluorescent protein; LC3,
microtubule-associated protein 1A/1B light chain 3; OD, optical
density. |
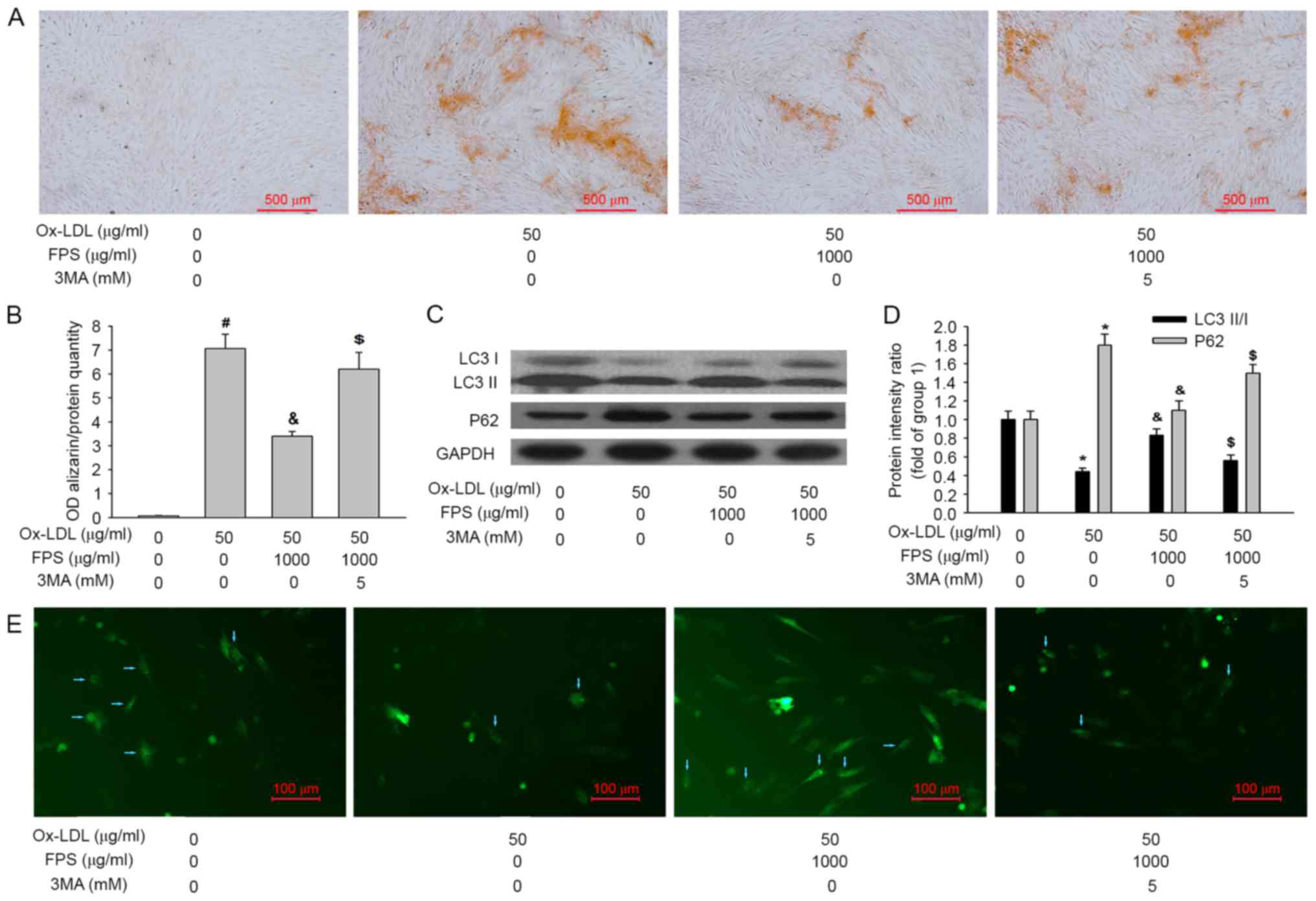 | Figure 4.Human VSMCs were cultured in
osteogenic medium with or without 50 µg/ml Ox-LDL, 1,000 µg/ml FPS
and 5 mM 3MA for 14 days. Representative images of VSMCs stained
with (A) Alizarin Red S to assess mineralization and (B)
quantification of it. Representative images of (C) LC3-I, LC3-II,
P62 and GAPDH proteins and (D) quantification of the relative
LC3-II/LC3-I ratio and relative P62 protein expression levels, as
assessed by western blotting. (E) Representative images of GFP-LC3
dots after the indicated treatments. Blue arrows represent VSMCs
expressing GFP-LC3 dots. Data are expressed as the mean ± standard
error (n=4). In group 1 human VSMCs were cultured in osteogenic
medium without Ox-LDL or FPS. Group 2, human VSMCs were cultured in
osteogenic medium with 50 µg/ml Ox-LDL without FPS. In group 3
human VSMCs were cultured in osteogenic medium with 50 µg/ml Ox-LDL
and 1,000 µg/ml FPS. #P<0.001 vs. group 1; *P<0.05
vs. group 1; &P<0.05 vs. group 2;
$P<0.05 vs. group 3. VSMCs, vascular smooth muscle
cells; Ox-LDL, oxidized low-density lipoproteins; FPS,
polysaccharide from Fuzi; GFP, green fluorescent protein; LC3,
microtubule-associated protein 1A/1B light chain 3; OD, optical
density; 3MA, 3-methyladenine. |
Western blot analysis
Cells (1×107) were plated in 100-mm
diameter Petri dishes and were treated as indicated above following
70–80% confluency. Following treatment, cells were harvested and
resuspended in ice-cold cell lysis buffer (Sigma-Aldrich, Merck
KGaA), and the homogenate was centrifuged at 10,000 × g for 15 min
at 4°C. The total supernatant protein concentration was measured
using a bicinchoninic acid protein assay kit. Total protein (50 µg)
from each sample was separated by 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to a
polyvinylidene difluoride membrane. The membrane was blocked with
5% fat-free dry milk in TBS with Tween-20 for 1.5 h at room
temperature, followed by incubation with primary antibodies against
microtubule-associated protein 1A/1B light chain 3 (LC3B),
nucleoporin P62 and GAPDH (cat. nos. 2775, 5114 and 5014; 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA) overnight with
gentle agitation at 4°C. The next day, the membranes were washed
with PBS and subsequently incubated with horseradish
peroxidase-conjugated secondary antibodies (cat. no. 7075; 1:1,000;
Cell Signaling Technology, Danvers, MA, USA) for 1.5 h at room
temperature. Following three washes with TBS with Tween-20,
membranes were developed using an enhanced chemiluminescence kit
(Applygen Technologies, Inc., Beijing, China) and exposed to X-ray
films. ImageJ software (1.48u version; National Institutes of
Health, Bethesda, MD, USA) was used to quantify the protein
expression levels.
Cell transfections and green
fluorescent protein (GFP)-LC3 dot-per-cell quantification
Cells were transfected with pEGFP-LC3 plasmids (cat.
no. 24920; Addgene, Cambridge, MA, USA) using Lipofectamine™ LTX
(cat. no. 11668019; Thermo Fisher Scientific, Inc.) reagent as
previously described (20); a
minimum of 40% transfection efficiency was achieved and
GFP-LC3-transfected cells were visualized using a fluorescence
microscope.
Statistical analysis
All assays were performed ≥4 times and the values
were expressed as the mean ± standard error. Statistical analysis
of between-group differences was performed by one-way analysis of
variance with a Bonferroni post hoc test with using SPSS 16.0
software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
Ox-LDL increases human VSMC
calcification in a concentration-dependent manner
Human VSMCs were treated with 10, 50 or 100 µg/ml
Ox-LDL for 14 days to determine the effect of Ox-LDL on vascular
calcification. Alizarin Red S staining was used to assess
mineralization, which is positively associated with VSMC
mineralization. As illustrated in Fig.
1A and B, Ox-LDL increased human VSMC calcification in a
concentration-dependent manner, particularly in the 50 and 100
µg/ml Ox-LDL treatment groups. Additionally, RT-qPCR revealed that
Ox-LDL increased CBFA1 mRNA expression in a
concentration-dependent manner, whereas it decreased SM22α
mRNA expression (Fig. 1C), further
confirming that Ox-LDL increased human VSMC calcification. For the
subsequent experiments, 50 µg/ml Ox-LDL treatment for 14 days was
selected as this induced VSMC calcification and increased Alizarin
Red S staining along with increasing CBFA1 mRNA expression and
decreasing SM22α mRNA expression.
FPS treatment attenuates
Ox-LDL-induced human VSMC calcification in a
concentration-dependent manner
Various concentrations of FPS were added to the
cells to determine its protective effect against vascular
calcification induced by Ox-LDL. As demonstrated in Fig. 2A, FPS treatment attenuated the
Ox-LDL-induced increase in Alizarin Red S staining in human VSMCs,
particularly in the 1,000 µg/ml FPS-treatment group (Fig. 2B). Furthermore, the CBFA1
and SM22α mRNA changes induced by Ox-LDL were significantly
attenuated by 1,000 µg/ml FPS (Fig.
2C), suggesting that FPS treatment protected against
Ox-LDL-induced calcification in human VSMCs.
FPS treatment alleviates the
Ox-LDL-induced downregulation of autophagy activity
It was also investigated whether autophagy was
upregulated or downregulated following 14 days of Ox-LDL treatment,
and whether FPS treatment affected autophagic activity. The protein
expression levels of LC3-I, LC3-II, P62 and GAPDH were measured by
western blotting. As presented in Fig.
3A and B, following 50 µg/ml Ox-LDL-treatment for 14 days, the
LC3-II/LC3-I ratio was decreased and P62 protein expression was
increased, indicating that autophagy was inhibited in the vascular
calcification progress. Notably, treatment with 1,000 µg/ml FPS
attenuated the aforementioned changes in LC3-II/LC3-I ratio and P62
protein intensity induced by Ox-LDL, suggesting that FPS treatment
protected against the Ox-LDL-induced downregulation of autophagic
activity. Additionally, no change in autophagic activity was
detected in native FPS-treated cells compared with the control
group of untreated cells. As demonstrated in Fig. 3C, GFP-LC3 puncta, as indicators of
autophagosomes, were also used to monitor autophagic activity. A
greater number of GFP-LC3 dots in each VSMC represents increased
autophagy. Compared with the control group, fewer GFP-LC3 dots were
detected in the Ox-LDL treatment group. When 1,000 µg/ml FPS was
added, the Ox-LDL-induced reduction in the number of GFP-LC3 dots
was attenuated. This further confirmed that FPS treatment
alleviated the Ox-LDL-induced downregulation of autophagy.
Autophagy inhibition by
3-methyladenine (3MA) decreases the protective effect of FPS
against VSMC calcification
Whether increased autophagy activity contributed to
FPS-mediated protection against vascular calcification was also
investigated. Since 3MA is commonly used as a pharmacological agent
for the inhibition of autophagy in vitro, VSMCs were treated
with 5 mM 3MA (Sigma-Aldrich, Merck KGaA) to inhibit autophagy, and
the effects on FPS-mediated protection against vascular
calcification were assessed. As presented in Fig. 4A and B, when 3MA was added, the
protective effect of FPS against VSMC calcification was also
inhibited. Additionally, 3MA treatment decreased the LC3-II/LC3-I
ratio and increased the P62 protein level, indicating that
autophagic activity was inhibited (Fig. 4C and D). Furthermore, a decrease in
the number of GFP-LC3 dots also demonstrated that autophagy was
inhibited (Fig. 4E). Therefore, it
may be concluded that inhibition of autophagy by 3MA decreased the
protective effect of FPS against VSMC calcification.
Discussion
Consistent with the observed calcification-inducing
effect of Ox-LDL reported previously (7,21),
the present study demonstrated that Ox-LDL increased human VSMC
calcification in a concentration-dependent manner, as determined by
Alizarin Red S staining and measurement of CBAF1 and
SM22α mRNA expression levels by RT-qPCR. In addition, FPS
treatment attenuated Ox-LDL-induced calcification of human VSMCs
and alleviated the Ox-LDL-induced downregulation of autophagy.
Furthermore, inhibition of autophagic activity by 3MA decreased the
protective effect of FPS against VSMC calcification. Therefore, to
the best of the authors' knowledge, the present study is the first
to demonstrate that FPS attenuates Ox-LDL-induced human VSMC
calcification, at least partially, via upregulation of
autophagy.
Vascular calcification is a pathological process
whereby extraskeletal calcium-phosphate crystals are deposited in
the vascular system (22). It is
highly prevalent, as presented by the Multi-Ethnic Study of
Atherosclerosis, in which the prevalence of coronary calcification
in individuals of Caucasian, African-American, Hispanic and Chinese
ethnicity was reported to be 70.4, 52.0, 56.6 and 59.2%,
respectively, for men, and 44.7, 37.0, 34.8 and 41.9%,
respectively, for women (23).
Vascular calcification is a key risk factor for numerous adverse
clinical outcomes and, during its development, a phenotypic change
in VSMCs serves a critical role (24,25).
VSMCs undergo a phenotypic change from a contractile to a synthetic
and osteochondrogenic phenotype, which is accompanied by
downregulation of contractile markers expression, such as
SM22α, and upregulation of osteochondrogenic markers
expression, such as CBFA1 (26).
Oxidative stress has been implicated in vascular
calcification, and has been demonstrated to promote VSMC
differentiation (27). Liu et
al (28) demonstrated that
hydrogen peroxide enhanced vascular calcification by inducing
osteoblastic differentiation of VSMCs. In our previous research, it
was demonstrated that Ox-LDL promotes the osteogenic
differentiation and calcification of human VSMCs (21,29).
In the present study, Ox-LDL treatment was illustrated to increase
human VSMC mineralization, as indicated by Alizarin Red S staining,
and significantly downregulated SM22α mRNA expression and
upregulated CBFA1 mRNA expression. These results confirmed
that Ox-LDL increased human VSMC calcification.
Autophagy is a dynamic and highly regulated process
of self-digestion, which is responsible for cell survival and
response to oxidative stress (30). It is demonstrated to limit VSMC
calcification by inhibiting matrix vesicle release (31). In an in vitro
phosphate-induced calcification model of VSMCs and an in
vivo model of chronic renal failure, it was reported that
valproic acid-induced autophagy could decrease calcification, and
that the inhibition of autophagy by 3MA could significantly promote
phosphate-induced matrix vesicle release (32). In addition, it was reported that
atorvastatin protected VSMCs from transforming growth factor
β1-stimulated calcification by inducing autophagy (33). Furthermore, a previous study
illustrated that ghrelin treatment attenuated the elevation of
calcium deposition in vascular calcification models in vivo
and in vitro. Simultaneously, protein levels of the
autophagy markers LC3 and beclin-1 were significantly upregulated
by ghrelin in the vascular calcification model, and inhibition of
autophagy by 3MA blocked the ameliorative effect of ghrelin on
vascular calcification (34). From
these results, it can be hypothesized that autophagy may be an
endogenous protective mechanism counteracting vascular
calcification, and that autophagy induction could be a therapeutic
strategy for vascular calcification.
P62 accumulates when autophagy is inhibited, and is
decreased when autophagy is induced; therefore, it can be used as a
marker to study autophagic flux (35). Furthermore, during autophagy, the
cytosolic form of LC3 (LC3-I) is conjugated to
phosphatidylethanolamine to form an LC3-phosphatidylethanolamine
conjugate (LC3-II), which is recruited to autophagosome membranes
(36). Thus, the LC3-II/I ratio
detected by western blot analysis is considered to be positively
correlated with autophagic activity (37). In the present study, it was
demonstrated that Ox-LDL treatment decreased the LC3-II/LC3-I ratio
and simultaneously increased the P62 protein levels, suggesting
that autophagic activity was inhibited under Ox-LDL stressor
activity.
Cell injury induced by Ox-LDL is a major
contributing factor to the pathogenesis of many cardiovascular
diseases, and the accompanying inhibition of autophagy can be
detected. For example, it was reported that in an Ox-LDL-induced
model of human umbilical vein endothelial cell injury, the number
of autophagosomes and the expression levels of autophagy-associated
proteins were decreased (38). In
an Ox-LDL-induced RAW264.7 cell injury model, autophagic activity
was also presented to be inhibited, as confirmed by
immunofluorescence, western blotting and monodansylcadaverine
staining (39). Taken together,
these results highlight that any treatment promoting autophagy may
be considered to have a therapeutic potential in the treatment of
Ox-LDL-associated injury.
FPS is a water-soluble polysaccharide that has
previously been isolated from Fuzi (13). The total sugar content of the crude
polysaccharide is determined to be 97.1%, with a purity of
>99.8% (13). Previously, it
was demonstrated that FPS protected against starvation-induced
cytotoxicity in H9c2 cells by increasing autophagy (15). Thus, in the present study, it was
aimed to investigate whether FPS treatment could attenuate
Ox-LDL-induced human VSMC calcification, and what role autophagy
serves in the protective effects of FPS. Different concentrations
of FPS were added to assess its effect on Ox-LDL-induced vascular
calcification. It was demonstrated that FPS treatment attenuated
the Ox-LDL-induced increase in Alizarin Red S staining in human
VSMCs, indicating the amelioration of vascular calcification.
Furthermore, the changes in CBFA1 and SM22α mRNA
levels induced by Ox-LDL were likewise reversed, further suggesting
that FPS serves a protective role against Ox-LDL-induced vascular
calcification.
Ox-LDL treatment decreased the LC3-II/LC3-I ratio
and simultaneously increased the P62 protein levels, suggesting
that autophagic activity was inhibited. Importantly, treatment with
1,000 µg/ml FPS could attenuate the aforementioned changes in the
LC3-II/LC3-I ratio and P62 protein levels caused by Ox-LDL,
suggesting that FPS treatment attenuated the Ox-LDL-induced
downregulation of autophagic activity. This was also confirmed by
the number of GFP-LC3 dots, which were used to monitor the number
of autophagosomes. Subsequently, VSMCs were treated with 3MA, a
commonly used pharmacological inhibitor of autophagy, and the
effects on FPS-mediated protection against vascular calcification
were observed. The addition of 3MA resulted in the inhibition of
FPS protection. The autophagy changes indicated by the LC3-II/LC3-I
ratio, P62 protein intensity and GFP-LC3 dots suggested that
autophagy inhibition by 3MA decreased the protective effect of FPS
against calcification.
In conclusion, the present study demonstrated for
the first time, to the best of the authors' knowledge that FPS
protects against Ox-LDL-induced vascular calcification in human
VSMCs, likely by activating autophagy activity. It is possible that
FPS can be developed into a novel type of medicine for vascular
calcification treatment.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Guangdong Province, China (grant nos.
2015A030310185 and 2015A030313582), the Science and Technology
Foundation of Guangdong Province, China (grant no. 2013B022000094)
and the Natural Science Fund of China (grant no. 81701378).
References
|
1
|
Albanese I, Daskalopoulou SS, Yu B, You Z,
Genest J, Alsheikh-Ali A and Schwertani AG: The Urotensin II system
and carotid atherosclerosis: A role in vascular calcification.
Front Pharmacol. 7:1492016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yahagi K, Kolodgie FD, Lutter C, Mori H,
Romero ME, Finn AV and Virmani R: Pathology of human coronary and
carotid artery atherosclerosis and vascular calcification in
diabetes mellitus. Arterioscler Thromb Vasc Biol. 37:191–204. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shigematsu T, Sonou T, Ohya M, Yokoyama K,
Yoshida H, Yokoo T, Okuda K, Masumoto AR, Iwashita Y, Iseki K, et
al: Preventive strategies for vascular calcification in patients
with chronic kidney disease. Contrib Nephrol. 189:169–177. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leopold JA: Vascular calcification:
Mechanisms of vascular smooth muscle cell calcification. Trends
Cardiovasc Med. 25:267–274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han X, Wang LY, Diao ZL and Liu WH:
Apelin: A novel inhibitor of vascular calcification in chronic
kidney disease. Atherosclerosis. 244:1–8. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang J, Chang JR, Duan XH, Yu YR and
Zhang BH: Erratum to: Thyroid hormone attenuates vascular
calcification induced by vitamin D3 plus nicotine in rats. Calcif
Tissue Int. 96:5802015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang Y, Xu Q, Peng H, Liu Z, Yang T, Yu Z,
Cheng G, Li X, Zhang G and Shi R: The role of vascular peroxidase 1
in ox-LDL-induced vascular smooth muscle cell calcification.
Atherosclerosis. 243:357–363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ogawa S: Pathological mechanism of
vascular calcification and new development in clinical strategy for
the therapy. Clin Calcium. 25:6332015.(In Japanese). PubMed/NCBI
|
|
9
|
Xue Z, Yuan W, Li J, Zhou H, Xu L, Weng J,
Li X, Zhang X, Wang Z and Yan J: Cyclophilin A mediates the
ox-LDL-induced activation and apoptosis of macrophages via
autophagy. Int J Cardiol. 230:142–148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang J, Yu J, Li D, Yu S, Ke J, Wang L,
Wang Y, Qiu Y, Gao X, Zhang J and Huang L: Store-operated calcium
entry-activated autophagy protects EPC proliferation via the
CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy. 13:82–98. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Krishnamurthy M, Selvaraju M and
Tamilarasan M: Turbinaria conoides (J. Agardh) sulfated
polysaccharide protects rat's heart against myocardial injury. Int
J Biol Macromol. 50:1275–1279. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Silva AK, Juenet M, Meddahi-Pelle A and
Letourneur D: Polysaccharide-based strategies for heart tissue
engineering. Carbohydr Polym. 116:267–277. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao C, Li M, Luo Y and Wu W: Isolation
and structural characterization of an immunostimulating
polysaccharide from fuzi, Aconitum carmichaeli. Carbohydr Res.
341:485–491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin S, Liu K, Wu W, Chen C, Wang Z and
Zhang X: Study on pretreatment of FPS-1 in rats with hepatic
ischemia-reperfusion injury. Am J Chin Med. 37:323–337. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liao LZ, Chen YL, Lu LH, Zhao YH, Guo HL
and Wu WK: Polysaccharide from Fuzi likely protects against
starvation-induced cytotoxicity in H9c2 cells by increasing
autophagy through activation of the AMPK/mTOR pathway. Am J Chin
Med. 41:353–367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patel JJ, Srivastava S and Siow RC:
Isolation, culture, and characterization of vascular smooth muscle
cells. Methods Mol Biol. 1430:91–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gregory CA, Gunn WG, Peister A and Prockop
DJ: An Alizarin red-based assay of mineralization by adherent cells
in culture: Comparison with cetylpyridinium chloride extraction.
Anal Biochem. 329:77–84. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhu D, Mackenzie NC, Shanahan CM, Shroff
RC, Farquharson C and MacRae VE: BMP-9 regulates the osteoblastic
differentiation and calcification of vascular smooth muscle cells
through an ALK1 mediated pathway. J Cell Mol Med. 19:165–174. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Z, Singh R and Aschner M: Methods
for the detection of autophagy in mammalian cells. Curr Protoc
Toxicol. 69:20.12.1–20.12.26. 2016. View
Article : Google Scholar
|
|
21
|
Song Y, Hou M, Li Z, Luo C, Ou JS, Yu H,
Yan J and Lu L: TLR4/NF-κB/Ceramide signaling contributes to
Ox-LDL-induced calcification of human vascular smooth muscle cells.
Eur J Pharmacol. 794:45–51. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gao J, Zhang K, Chen J, Wang MH, Wang J,
Liu P and Huang H: Roles of aldosterone in vascular calcification:
An update. Eur J Pharmacol. 786:186–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bild DE, Detrano R, Peterson D, Guerci A,
Liu K, Shahar E, Ouyang P, Jackson S and Saad MF: Ethnic
differences in coronary calcification: The Multi-Ethnic Study of
Atherosclerosis (MESA). Circulation. 111:1313–1320. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao YG, Meng FX, Li BW, Sheng YM, Liu MM,
Wang B, Li HW and Xiu RJ: Gelatinases promote calcification of
vascular smooth muscle cells by up-regulating bone morphogenetic
protein-2. Biochem Biophys Res Commun. 470:287–293. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen T, Mao H, Chen C, Wu L, Wang N, Zhao
X, Qian J and Xing C: The Role and Mechanism of α-Klotho in the
calcification of rat aortic vascular smooth muscle cells. Biomed
Res Int. 2015:1943622015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shanahan CM, Crouthamel MH, Kapustin A and
Giachelli CM: Arterial calcification in chronic kidney disease: Key
roles for calcium and phosphate. Circ Res. 109:697–711. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cui L, Li Z, Chang X, Cong G and Hao L:
Quercetin attenuates vascular calcification by inhibiting oxidative
stress and mitochondrial fission. Vascul Pharmacol. 88:21–29. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu H, Lu Q and Huang K: Selenium
suppressed hydrogen peroxide-induced vascular smooth muscle cells
calcification through inhibiting oxidative stress and ERK
activation. J Cell Biochem. 111:1556–1564. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liao L, Zhou Q, Song Y, Wu W, Yu H, Wang
S, Chen Y, Ye M and Lu L: Ceramide mediates Ox-LDL-induced human
vascular smooth muscle cell calcification via p38 mitogen-activated
protein kinase signaling. PLoS One. 8:e823792013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu L, Chen Y and Tooze SA: Autophagy
pathway: Cellular and molecular mechanisms. Autophagy. Sep
21–2017.(Epub ahead of print). View Article : Google Scholar
|
|
31
|
Shanahan CM: Autophagy and matrix
vesicles: New partners in vascular calcification. Kidney Int.
83:984–986. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dai XY, Zhao MM, Cai Y, Guan QC, Zhao Y,
Guan Y, Kong W, Zhu WG, Xu MJ and Wang X: Phosphate-induced
autophagy counteracts vascular calcification by reducing matrix
vesicle release. Kidney Int. 83:1042–1051. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu D, Cui W, Liu B, Hu H, Liu J, Xie R,
Yang X, Gu G, Zhang J and Zheng H: Atorvastatin protects vascular
smooth muscle cells from TGF-β1-stimulated calcification by
inducing autophagy via suppression of the β-catenin pathway. Cell
Physiol Biochem. 33:129–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu M, Liu L, Song C, Chen W and Gui S:
Ghrelin improves vascular autophagy in rats with vascular
calcification. Life Sci. 179:23–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
BenYounès A, Tajeddine N, Tailler M, Malik
SA, Shen S, Métivier D, Kepp O, Vitale I, Maiuri MC and Kroemer G:
A fluorescence-microscopic and cytofluorometric system for
monitoring the turnover of the autophagic substrate p62/SQSTM1.
Autophagy. 7:883–891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schläfli AM, Adams O, Galván JA, Gugger M,
Savic S, Bubendorf L, Schmid RA, Becker KF, Tschan MP, Langer R and
Berezowska S: Prognostic value of the autophagy markers LC3 and
p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget.
7:39544–39555. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tanida I, Ueno T and Uchiyama Y: A
super-ecliptic, pHluorin-mKate2, tandem fluorescent protein-tagged
human LC3 for the monitoring of mammalian autophagy. PLoS One.
9:e1106002014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Luo Y, Meng X, Zhou P, Lu S, Qin M, Xu X,
Sun G and Sun X: Elatoside C protects against ox-LDL-induced HUVECs
injury by FoxO1-mediated autophagy induction. Biochim Biophys Acta.
1863:1654–1665. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang BC, Zhang CW, Wang C, Pan DF, Xu TD
and Li DY: Luteolin attenuates foam cell formation and apoptosis in
Ox-LDL-stimulated macrophages by enhancing autophagy. Cell Physiol
Biochem. 39:2065–2076. 2016. View Article : Google Scholar : PubMed/NCBI
|
















