Introduction
Non-traumatic intracerebral hemorrhage (ICH) is the
second most common type of stroke and is associated with high
mortality and high morbidity; 50–70% of survivors suffer from
paralysis, aphasia and severe disability (1,2).
Previous studies have demonstrated that brain damage following ICH
is due not only to hematoma mass effect and its direct damage to
the surrounding brain tissues, but also to secondary brain injury.
In addition, apoptosis and degeneration of neurons post-ICH are
important factors in secondary brain injury (3,4).
Following ICH, there is an increase in the local concentration of
glutamate, which overstimulates N-methyl-D-aspartate receptors
(NMDARs) in the brain (5); this
leads to an increase in intracellular Ca2+ and neuron
death, suggesting that NMDARs serve an important role in neuronal
apoptosis post-ICH.
Pannexins were discovered in vertebrates by Panchin
et al in 2000 (6), and
there are three subtypes: Pannexin-1, Pannexin-2 and Pannexin-3.
Pannexins exhibit sequence homology to the invertebrate family of
gap junction proteins called innexins (6). Pannexin-1 is a non-selective ion
channel that is widely distributed in various tissues and is
involved in several important physiological and pathophysiological
functions. Pannexin-l may be part of the postsynaptic channel
complex and may regulate postsynaptic activity through the
formation of hemichannels (7).
Pannexin-l directly mediates the release of ATP, adjusts the
extracellular regenerative currents in neurons, and serves an
important role in signal transduction in glial cells (8,9).
Furthermore, Pannexin-1 channels can be activated by ischemia, as
indicated by a previous report that demonstrated that during
ischemic stroke, the Pannexin-1 hemichannel opening in neurons
increases after hypoxic-ischemic stress injury (10). Another study revealed that the
addition of a Pannexin-1 inhibitor to hippocampal slice cultures
significantly reduced the activation of caspase-3 and neuronal cell
death (11). In addition,
Pannexin-1 channels have been revealed to be activated by NMDARs
(12,13), which further suggests an important
role for Pannexin-1 in neuronal apoptosis following ICH. However,
little is currently known regarding the role of Pannexin-1
post-ICH. The present study aimed to explore the expression and the
role of Pannexin-1 post-ICH, which, to the best of our knowledge,
has not previously been reported.
Materials and methods
Animals and ethics
All applicable international, national, and/or
institutional guidelines for the care and use of animals were
followed. Animal experimental protocols, including all use, care
and operative procedures, were approved by the Institutional Animal
Care and Use Committee of Soochow University (Suzhou, China) and
complied with the 8th version of the Guide for the Care and Use of
Laboratory Animals by the National Institutes of Health (2012).
Rat ICH model
A total of 146 male Sprague-Dawley rats (weight,
250–300 g) were purchased from the Animal Center of Soochow
University (Suzhou, China) and were raised on a 12-h dark-light
cycle with free access to food and water. They were anesthetized
with an intraperitoneal injection of pentobarbital (45 mg/kg;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany). Core
temperature was maintained at 37°C using a feedback-controlled
heating pad. Rats were positioned in a stereotactic frame (David
Kopf Instruments, Tujunga, CA, USA) and a cranial burr hole (1 mm)
was drilled on the right coronal suture, 3.5 mm lateral to midline.
A 30-gauge needle was introduced through the burr hole into the
caudate nucleus, 3.5 mm lateral to midline and 0.2 mm anterior to
the bregma, and to a depth of 5.5 mm below the surface of the
skull. A microinjector was used to infuse 1 µl buffered saline
containing 0.23 units type VII collagenase (Merck Millipore) over 5
min, to break up the basement of vessels and cause internal
bleeding. The needle was kept in place for an additional 5 min
post-injection to avoid reflux. Sham controls only had an
intracerebral needle insertion. Following injection, the needle was
removed, the burr hole was filled with bone wax and the skin
incision was closed with sutures (14,15).
Animals were then re-anesthetized as above and perfused for 5 min
through the left cardiac ventricle with 0.9% NaCl solution, until
effluent from the right atrium was clear, the brain was removed and
a coronal tissue slice (3 mm) was cut 4 mm from the frontal pole.
Two tissue samples, the ipsilateral and the contralateral cortex,
were obtained from each brain.
Experimental design
In experiment 1 (Fig.
1A), 42 of the 54 rats survived and were assigned randomly into
7 groups (n=6/group): 1 sham group (control) and 6 post-ICH groups
(6, 12, 24, 48 and 72 h, and 7 days). The animals in the post-ICH
groups were subjected to experimental induction of ICH on day 0 and
were sacrificed via the aforementioned procedure, after 6, 12, 24,
48, 72 h and 7 days, respectively.
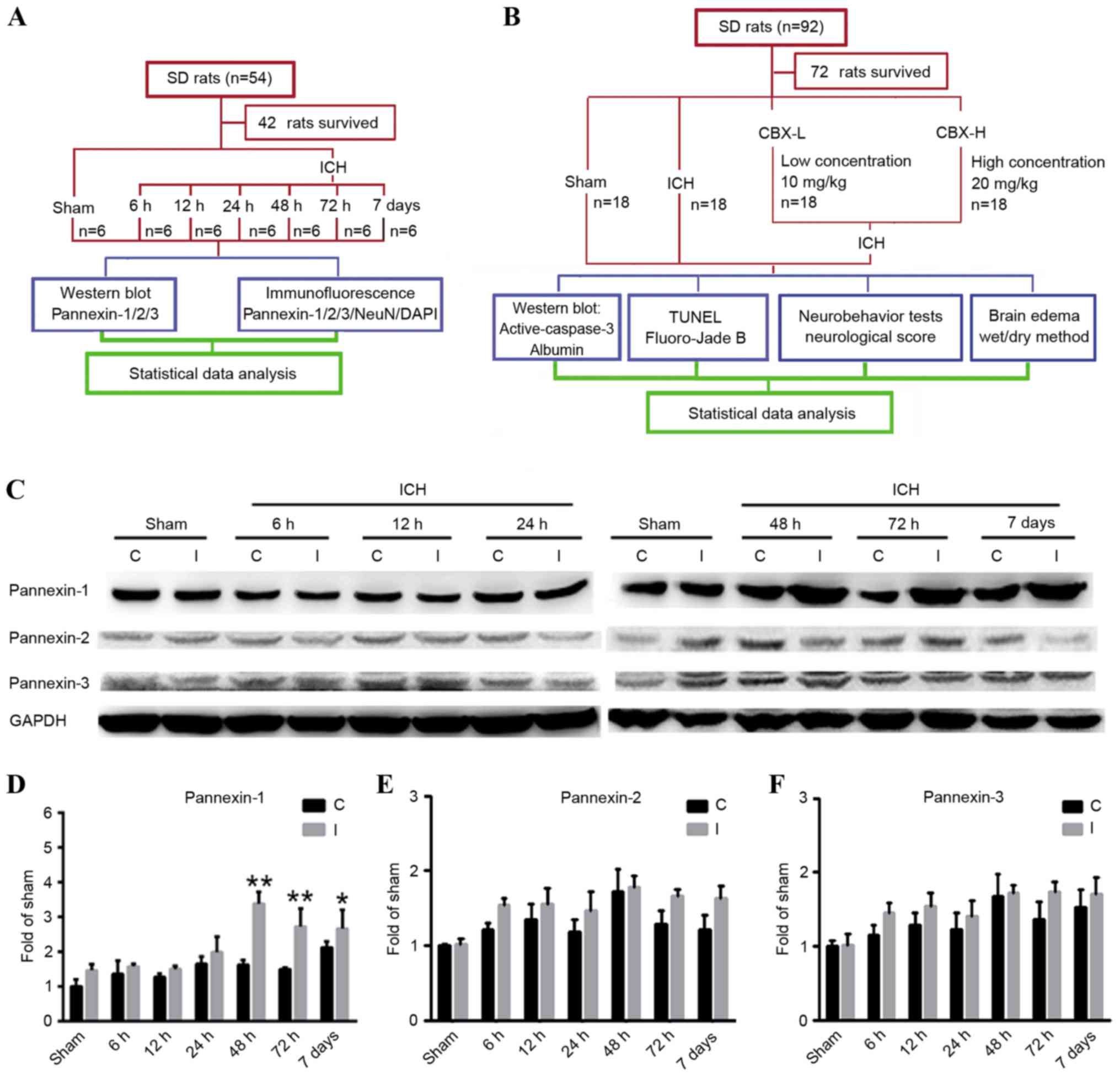 | Figure 1.Experimental designs and time course
of pannexin protein expression detection in rat brain tissues
following ICH. (A) Experiment 1 was designed to detect changes in
the levels of protein expression of Pannexin-1, Pannexin-2 and
Pannexin-3 in the brain tissues of rats following ICH. (B)
Experiment 2 was designed to investigate the role of Pannexin-1 in
neuronal apoptosis and degeneration in rats post-ICH. (C) Western
blot analyses and quantitative analyses of the relative protein
expression levels of (D) Pannexin-1, (E) Pannexin-2 and (F)
Pannexin-3 in brain tissues post-ICH; the mean band intensity of
the sham group was normalized to 1.0. Data are presented as the
mean ± standard error of the mean; *P<0.05, **P<0.01 vs. sham
group; n=6/group. C, contralateral; CBX, carbenoxolone; I,
ipsilateral; ICH, intracerebral hemorrhage; NeuN, Neuronal nuclei;
SD, Sprague-Dawley; TUNEL, terminal
deoxynucleotidlyl-transferase-mediated dUTP nick end labeling. |
In experiment 2 (Fig.
1B), 72 of the 92 rats survived and were assigned randomly into
4 groups (n=18/group): Sham group; ICH group; ICH + low
concentration carbenoxolone (CBX-L; cat. no. A8389; 10 mg/kg;
ApexBio Technology, Houstoun, TX, USA) group; and ICH + high
concentration CBX (CBX-H; 20 mg/kg) group. The concentration of CBX
treatment used was according to a previous study (16). A CBX stock solution was diluted to
50 mg/ml in sterile saline; the required concentration for each
group was then prepared prior to intraperitoneal injection, which
occurred 1 h post-ICH induction. A total of 6 rats were selected
randomly from each group and sacrificed using the aforementioned
procedure, 48 h post-ICH. An additional 6 rats were used to
evaluate brain edema and the remaining rats underwent nerve
function score tests 48 h following ICH.
Western blot analysis
Brain tissue samples from the ipsilateral and
contralateral cortex were mechanically lysed in lysis buffer
containing 20 mmol Tris (pH 7.6), 0.2% SDS, 1% Triton X-100, 1%
deoxycholate, 1 mmol phenylmethylsulfonyl fluoride and 0.11 U/ml
aprotinin (Merck Millipore). The lysates were centrifuged at 12,000
× g for 20 min at 4°C, and the protein concentration was estimated
using the Bradford method. Samples (60 µg) were separated by 10%
SDS-PAGE and electrotransferred onto a nitrocellulose membrane
(Merck Millipore). The membranes were blocked with 5% non-fat milk
for 1 h at room temperature and then incubated with primary
antibodies against Pannexin-1 (cat. no. ab124131; Abcam, Cambridge,
MA, USA), Pannexin-2 (cat. no. sc-51384; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), Pannexin-3 (cat. no. ab98093; Abcam),
active-caspase-3 (cat. no. ab2302; Abcam) or albumin (cat. no.
ab207327; Abcam) at dilutions of 1:500 in 5% bovine serum albumin
(BSA; Beyotime Institute of Biotechnology, Haimen, China) in TBS +
0.1% Tween-20 overnight at 4°C. GAPDH (1:6,000; Merck Millipore)
was used as a loading control. The membranes were washed 3 times
for 5 min each in TBS + 0.1% Tween-20 and then incubated with the
appropriate horseradish peroxidase-conjugated secondary antibodies
(cat. no. SC-2004; 1:5,000 in 5% BSA; Santa Cruz Biotechnology,
Inc.) for 2 h at room temperature. Finally, protein bands were
visualized using enhanced chemiluminescence (ECL; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and were exposed to X-ray film.
Relative changes in protein expression were estimated from the mean
pixel density using UN-SCAN-IT, normalized to β-actin, and
calculated as target protein expression/β-actin expression
ratios.
Immunofluorescence microscopy
Following collection, brain tissue samples from the
ipsilateral cortex were embedded in Tissue-Tek Optimal Cutting
Temperature Embedding Compound (Sakura Finetek Japan Co., Ltd.,
Tokyo, Japan). Sections 4 µm thick were sliced and blocked with 5%
normal fetal bovine serum in PBS containing 0.1% Triton X-100 for 2
h at room temperature. Following this, sections (4 µm) were then
incubated with primary antibodies against Pannexin-1, Pannexin-2 or
Pannexin-3 and Neuronal nuclei (NeuN; cat. no. ab104224; Abcam)
overnight at 4°C. Following this, sections were washed three times
with PBS for 45 min and were immunolabeled with the appropriate
secondary antibodies (Alexa Fluor 488, cat. no. A21206 and Alexa
Fluor 555, cat. no. A31570; 1:200; Invitrogen, Thermo Fisher
Scientific, Inc.) for 1 h at room temperature. The slides were
washed with PBS again three times for 45 min prior to
counterstaining with DAPI to stain the nuclei, for 2 min. A total
of 3 random sections from each rat were examined and imaged using a
fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Relative fluorescence intensity was analyzed using ImageJ software
version 2 (National Institutes of Health, Bethesda, MA, USA).
Neurobehavioral testing and
neurological scoring
Three behavioral activity examinations were
performed 48 h post-ICH on 24 rats that were separated into 4
groups (n=6/group: Sham; ICH; ICH + CBX-L; and ICH + CBX-H), to
record appetite, activity and neurological deficits using the
scoring system reported previously (17). The standard of behavior and
activity scores are shown in Table
I.
 | Table I.Behavior and activity scores. |
Table I.
Behavior and activity scores.
| Category | Behavior | Score |
|---|
| Appetite | Finished meal | 0 |
|
| Left meal
unfinished | 1 |
|
| Scarcely ate | 2 |
| Activity | Walked and reached
at least three corners of the cage | 0 |
|
| Walked with some
stimulations | 1 |
|
| Almost always lying
down | 2 |
| Deficits | No deficits | 0 |
|
| Unstable walk | 1 |
|
| Impossible to
walk | 2 |
Brain edema and blood-brain barrier
(BBB) integrity
Brain water content was evaluated using the wet/dry
weight method as previously described (18), and BBB integrity was measured by
evaluating albumin levels in the ipsilateral cortex, detected via
western blotting. To assess brain water content, 24 rats were
separated into 4 groups (n=6/group): Sham; ICH; ICH + CBX-L; and
ICH + CBX-H. Rats were anesthetized with an intraperitoneal
injection of 45 mg/kg pentobarbital and decapitated 48 h post-ICH.
Brains were removed, the olfactory bulbs, cerebella and brain stems
were discarded, and the contralateral and ipsilateral hemispheres
were separated. Each sample was weighed to obtain the wet weight
and then dried in an oven at 100°C for 24 h to obtain the dry
weight. Water content was expressed as a percentage of the wet
weight: (wet weight - dry weight)/wet weight ×100%.
Terminal
deoxynucleotidlyl-transferase-mediated dUTP nick end labeling
(TUNEL)/NeuN immunofluorescence double labeling
As described previously (18), cell apoptosis in brain tissues was
detected by TUNEL staining using the In Situ Cell Death
Detection kit (Roche Diagnostics, Indianapolis, IN, USA), according
to the manufacturer's protocol. To further identify neuronal
apoptosis in the brain, TUNEL-stained slides were incubated with
anti-NeuN primary antibody (cat. no. ab104224, 1:200; Abcam)
overnight, and then incubated with secondary donkey anti-mouse
immunoglobulin G (Alexa Fluor 555; cat. no. A31570; 1:200;
Molecular Probes; Thermo Fisher Scientific, Inc.) at room
temperature, visualized by a fluorescence microscope (Olympus
Corporation). A total of 3 sections per rat were examined and
imaged in parallel for counting TUNEL-positive cells.
Fluoro-Jade B staining
Frozen brain (the ipsilateral cortex) sections were
stained with Fluoro-Jade B as previously described (19). Briefly, brain sections were
incubated in 100% ethanol for 3 min, 70% ethanol for 1 min and then
washed with deionized water. Sections were incubated for 10 min in
0.06% potassium permanganate followed by 60 min in Fluoro-Jade B
solution (Histo-Chem, Inc., Jefferson, AR, USA). Subsequently, the
sections were washed 3 times with PBS and dried overnight at room
temperature. Following drying, samples were cleared with xylene, a
coverslip was added using DPX Mountant (Electron Microscopy
Sciences, Hatfield, PA, USA) and visualized using a fluorescence
microscope (Olympus Corporation). A total of 3 sections per rat
were examined and imaged in parallel for counting Fluoro-Jade
B-positive cells using ImageJ software, version 2 (National
Institutes of Health).
Statistical analysis
All data are presented as the mean ± standard error
of the mean. GraphPad Prism 5 software (GraphPad Software, Inc., La
Jolla, CA, USA) was used for all statistical analysis. All data
were analyzed using one-way analysis of variance. The significance
of differences among experimental groups was determined by Fisher's
least significant difference post-test and P<0.05 was considered
to indicate a statistically significant difference.
Results
Expression of pannexin proteins in rat
brain tissues at various time points following ICH
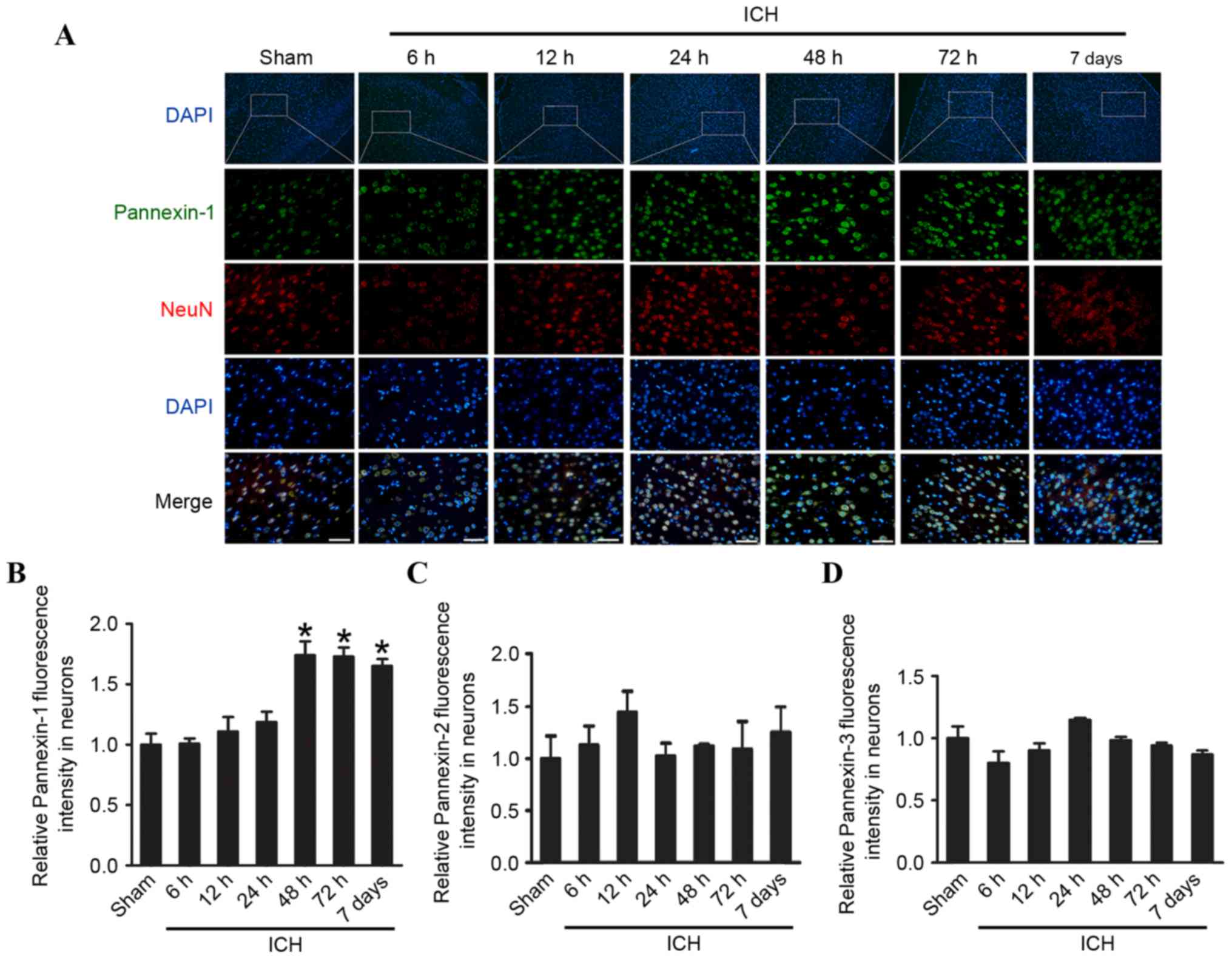
Pannexin-1 was expressed in the ipsilateral and
contralateral cortices in the sham and ICH groups at each of the
six time points (Fig. 1C and D).
However, compared with the sham group, the expression levels of
Pannexin-1 were significantly higher at 48 and 72 h (P<0.01), as
well as at 7 days (P<0.05) following ICH; the expression of
Pannexin-1 peaked at 48 h post-ICH. Although Pannexin-2 and
Pannexin-3 were also expressed in ICH groups at each time point and
in the sham group (Fig. 1C), there
was no significant difference between the two groups (P>0.05;
Fig. 1E and F). Immunofluorescence
analysis confirmed that the expression of Pannexin-1 protein in the
rat brain neurons gradually increased over time and peaked 48 h
post-ICH (Fig. 2). Conversely, the
expression levels of Pannexin-2 and Pannexin-3 at each time point
revealed no significant differences between ICH and sham groups
(Fig. 1C-F).
CBX treatment improves cognitive
function in rats post-ICH
Following ICH, rats treated with CBX demonstrated
significantly improved activity and appetite compared with the
ICH-only group. The total combined score of the ICH-only group was
significantly higher than that of sham group (P<0.01). However,
neurological scores of the CBX-L and CBX-H treatment groups were
markedly lower than the ICH group (P<0.05 and P<0.01,
respectively). As shown in Table
II, the mean neurological scores were 0.61 (sham group), 3.01
(ICH-only group), 2.11 (ICH + CBX-L group) and 1.87 (ICH + CBX-H
group).
| Table II.Clinical behavior scores in each
group. |
Table II.
Clinical behavior scores in each
group.
| Group | Mean score |
|---|
| Sham (n=18) | 0.61 |
| ICH (n=18) | 3.01a |
| ICH + CBX (low,
n=18) | 2.11b |
| ICH + CBX (high,
n=18) | 1.87c |
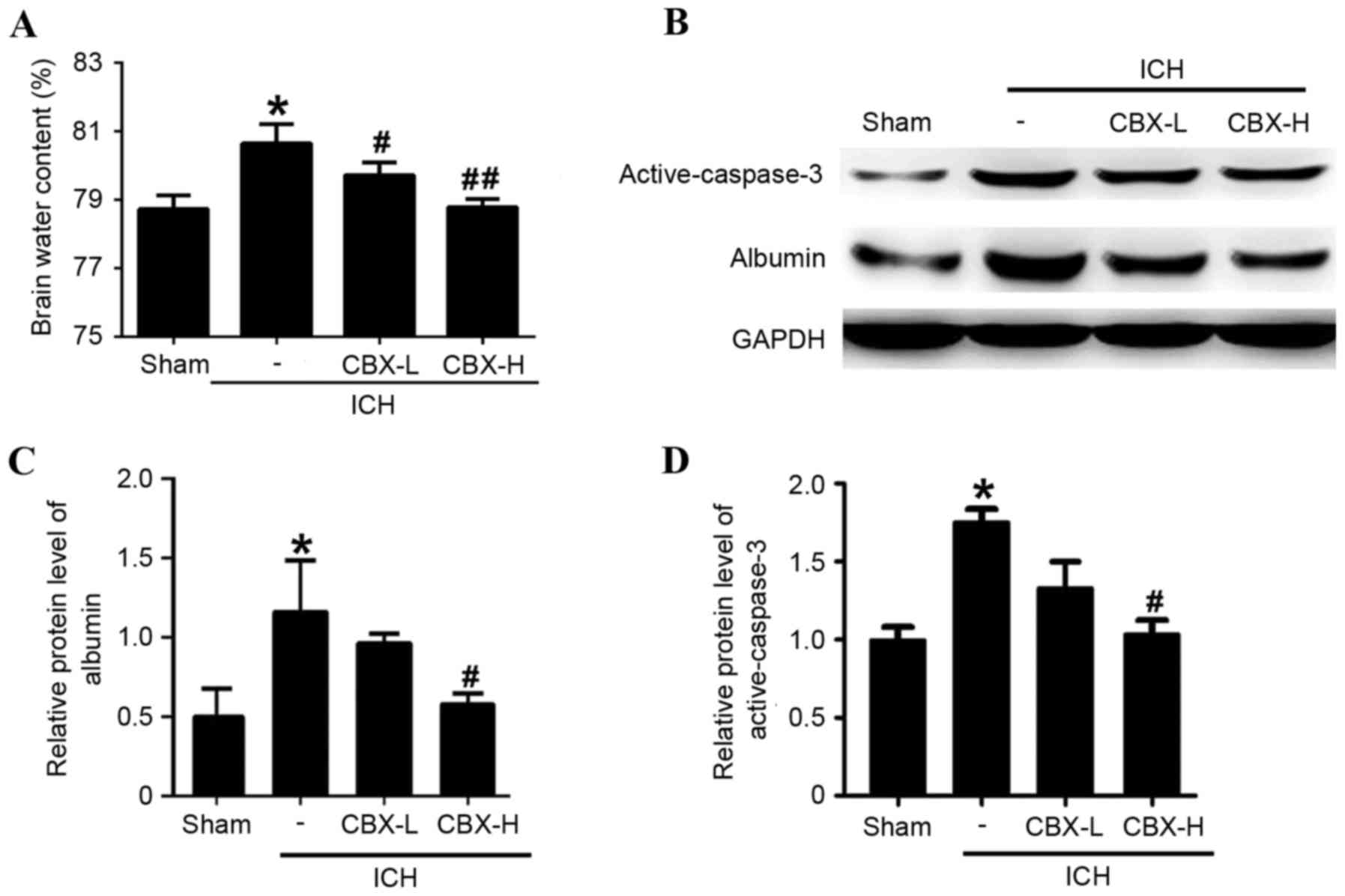
CBX treatment reduces brain edema and
BBB injury post-ICH
Brain water content was calculated using the wet/dry
weight method. Compared with the sham group, the brain water
content was significantly higher in the ICH-only group (P<0.05;
Fig. 3A). However, brain water
content of the ICH + CBX-L and ICH + CBX-H groups was lower than in
the ICH-only group (P<0.05 and P<0.01, respectively; Fig. 3A). To evaluate the effects of CBX
treatment on BBB integrity following ICH, the protein expression
levels of albumin were detected by western blot analysis (Fig. 3B). The results suggested that CBX
treatment could reduce the leakage of albumin content in brain
tissue which indicated that CBX treatment may attenuate BBB injury
after ICH (Fig. 3B and C). In
addition, CBX treatment also reduced ICH-induced increase in the
level of active-caspase3, which indicated that CBX treatment may
attenuate brain cell apoptosis following ICH (Fig. 3B and D).
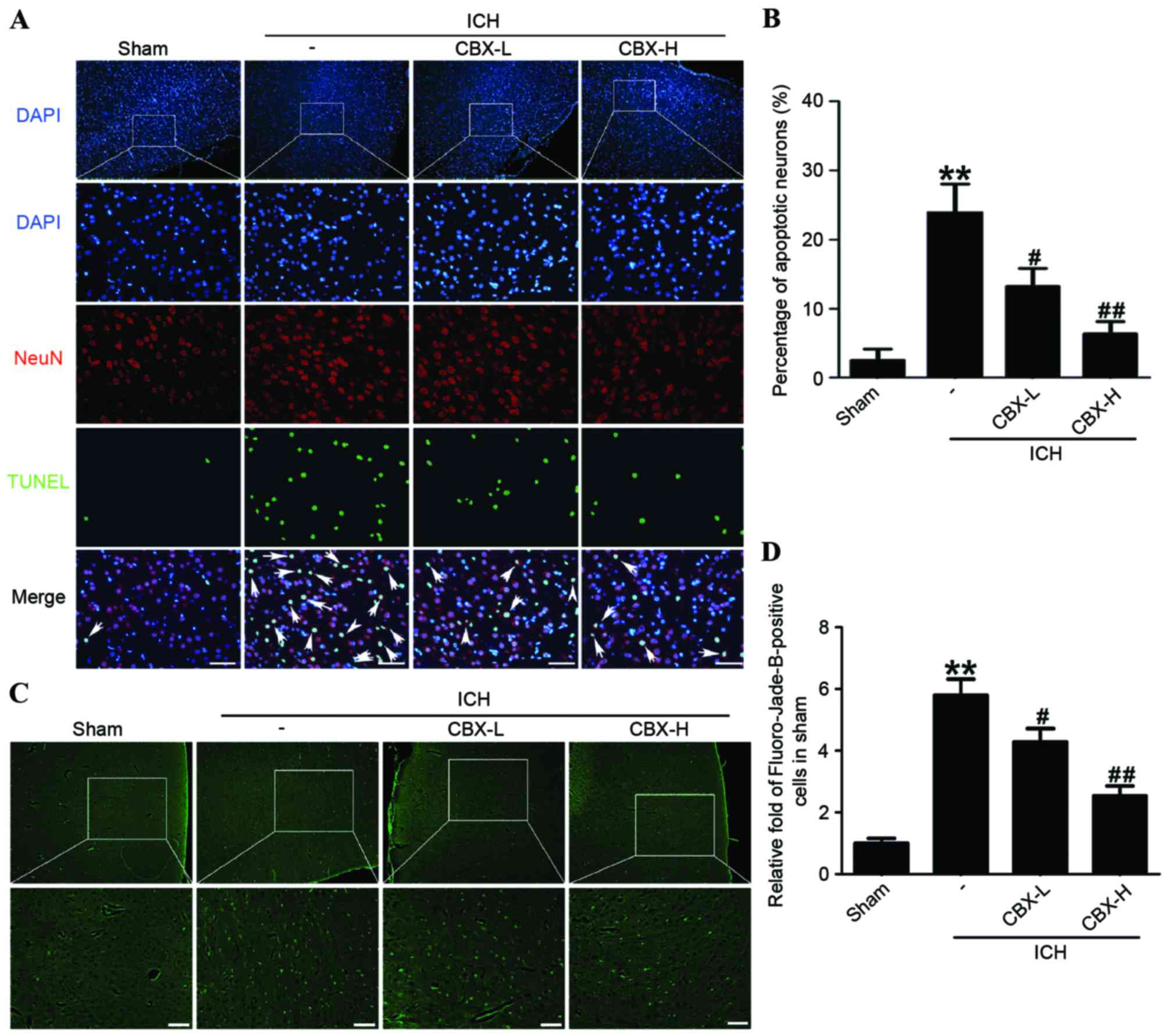
CBX treatment inhibits neuronal
apoptosis and neuronal degeneration in rat brain tissues following
ICH
Neuronal apoptosis was detected by TUNEL staining in
each of the four treatment groups. The results indicated that the
rate of apoptosis in neurons was significantly higher in the
ICH-only group compared with the sham group (P<0.01), whereas
the number of apoptotic neurons in the CBX-L and CBX-H treatment
groups was significantly lower than in the ICH-only group
(P<0.05 and P<0.01, respectively; Fig. 4A and B). Results of Fluoro-Jade B
assay revealed that the rate of neuronal degeneration was markedly
increased in the ICH-only group compared with the sham group
(P<0.01). However, the rates of neuronal degeneration in the
CBX-L or CBX-H groups were significantly lower than that in the
ICH-only group (P<0.05 and P<0.01, respectively; Fig. 4C and D). Western blot analysis
confirmed that the expression levels of active-caspase-3 protein
were significantly higher in the ICH-only group compared with the
sham group (P<0.05; Fig. 3B and
D). However, the expression levels of active-caspase-3 were
significantly lower in the ICH + CBX-H treatment group compared
with the ICH-only group (P<0.05). There was no significant
difference in the expression of active-caspase-3 between the CBX-L
treatment group and the ICH-only group (P>0.05; Fig. 3B and D).
Discussion
Pannexin-1 is a member of the vertebrate pannexin
family of gap junction proteins, which was identified after the
connexins and exhibits sequence homology with the invertebrate
innexins (6). Pannexin-1 is able
to form functional gap junctions when expressed in paired
Xenopus oocytes, and forms unconjugated hemichannels in
cultured neurons and glial cells. The hemichannels are typically
closed under resting conditions and permit Ca2+ and ATP
flux when they are open (7).
Numerous studies have reported that the pannexins may form
hemichannels in the cell membrane and mediate the release of ATP,
the transmission of intercellular Ca2+ waves, the
regulation of blood flow, immune responses and other physiological
functions (8,20). Pannexins are also involved in
inflammation, cancer, cerebral ischemia, epilepsy and other
pathologies (8,20,21).
ICH is a devastating disease that is associated with
high mortality, and there are no effective treatments to reduce
mortality and to improve the outcome for survivors (22). Nerve injury after ICH is complex.
Initial damage is due to the mechanical force induced by formation
of the hematoma. Subsequent hematoma expansion, edema and
inflammation cause further damage to brain tissues (23). Excitatory amino acid toxicity is a
major factor in secondary brain injury following ICH, and glutamate
is the primary excitatory neurotransmitter (24). Following ICH, high concentrations
of glutamate cause excessive activation of NMDARs, which leads to a
large increase in Ca2+ influx, thus triggering a series
of neurotoxic cascades (25,26).
Animal experiments have revealed that the glutamate concentration
sharply increases in tissues surrounding a hematoma within 3 h
following ICH and peaks at 12 h post-ICH (25). However, as the volume of the
hematoma decreased due to absorption 48–72 h post-ICH, the
glutamate concentration also significantly decreased, although it
remained higher than the sham group.
Pannexin-1 is one of the primary downstream proteins
of NMDAR. A previous study revealed that, following repeated or
prolonged stimulation, postsynaptic NMDARs induced a secondary
inward current (9). A Pannexin-l
specific inhibitory peptide was able to block the inward current,
indicating that NMDARs can activate Pannexin-1 hemichannels.
Pannexin-1 hemichannels open in response to depolarization, hypoxia
and mechanical stress. When open, they permit a large efflux of
ATP, which can bind to the P2Y subclass of purinergic receptors
present on vascular endothelial cells and red blood cells. The
binding of ATP leads to the release of more ATP, diffusion of
intracellular Ca2+ and the release of nitric oxide
directly into the vascular smooth muscle cells, which promotes
vasodilation and increases blood flow (27). The present study demonstrated that
the expression of Pannexin-1 increased over time following ICH,
peaking at 48 h post-ICH; the delay in expression relative to the
peak time point of increased glutamate concentration (12 h
post-ICH) is probably because Pannexin-1 is downstream of NMDAR
signaling.
CBX is a traditional anti-ulcer drug, but it also
exhibits anti-inflammatory effects by stimulating the adrenal
glands or enhancing endogenous corticosteroids (28). CBX is a broad-spectrum gap junction
inhibitor that permeates the BBB (29). CBX binds directly to gap junction
channels, which results in a conformational change in the gap
junction and the closing of hemichannels (30). A previous study revealed that CBX
effectively reduces the depolarization-induced pannexin hemichannel
current in Xenopus oocyte assays, and the effect was
concentration-dependent (20).
Another study compared the effects of compounds known to inhibit
cation and anion channels, and demonstrated that CBX was the most
effective inhibitor of Pannexin-1 hemichannels in mammalian cell
lines (31). The suppressive
effect of CBX was rapid and readily reversible. These results
suggested that CBX may act directly on Pannexin-1 hemichannels. To
confirm this finding, further studies are necessary in the
future.
Inhibition of gap junctions may serve a
neuroprotective role, which has been demonstrated in several in
vitro and in vivo models of nerve injury. One study
demonstrated that CBX significantly reduced the activation of
caspase-3 and decreased neuronal death in hippocampal slice
cultures after oxygen-glucose deprivation for 60 min (11). Using an intrauterine
hypoxic-ischemic model, it was revealed that treatment with CBX
significantly reduced newborn cub mortality and improved long-term
developmental defects (11).
However, there have been almost no studies on the effects of gap
junction inhibitors on secondary brain injury following ICH. One
study demonstrated that intravenous and intraperitoneal injections
of CBX significantly reduced seizure severity in rats that were
genetically susceptible to epilepsy; the effect was dose-dependent
within the range of 5–30 mg/kg CBX (16). Therefore, CBX may effectively
provide neuroprotection and merits further study. The present
study, to the best of our knowledge, is the first to investigate
the effect of gap junction inhibition on secondary brain injury,
and suggested that inhibition of Pannexin-1 may be a target for the
treatment of ICH.
In conclusion, the present study confirmed that
Pannexin-1 expression was increased following ICH. Furthermore, the
elevated expression of Pannexin-1 served an important role in
cognitive dysfunction post-ICH. In addition, CBX inhibition of
Pannexin-1 effectively reduced ICH-induced brain edema and improved
cognitive function. CBX also significantly reduced the expression
levels of active-caspase-3 and decreased neuronal apoptosis and
degeneration. Results of the present study indicated that CBX may
have a protective role in brain damage following ICH.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81371279, 81422013
and 81471196), Jiangsu Province's Outstanding Medical Academic
Leader program (grant no. LJ201139), the Scientific Department of
Jiangsu Province (grant no. BL2014045), the Suzhou Government
(grant nos. LCZX201301, SZS201413 and SYS201332), and A Project
Funded by the Priority Academic Program Development of Jiangsu
Higher Education Institutions.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Broderick JP, Brott T, Tomsick T, Miller R
and Huster G: Intracerebral hemorrhage more than twice as common as
subarachnoid hemorrhage. J Neurosurg. 78:188–191. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang LF, Yang J, Hong Z, Yuan GG, Zhou
BF, Zhao LC, Huang YN, Chen J and Wu YF; Collaborative Group of
China Multicenter Study of Cardiovascular Epidemiology, :
Proportion of different subtypes of stroke in China. Stroke.
34:2091–2096. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qureshi AI, Ling GS, Khan J, Suri MF,
Miskolczi L, Guterman LR and Hopkins LN: Quantitative analysis of
injured, necrotic and apoptotic cells in a new experimental model
of intracerebral hemorrhage. Crit Care Med. 29:152–157. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rincon F and Mayer SA: Novel therapies for
intracerebral hemorrhage. Curr Opin Crit Care. 10:94–100. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hossain MI, Kamaruddin MA and Cheng HC:
Aberrant regulation and function of Src family tyrosine kinases:
Their potential contributions to glutamate-induced neurotoxicity.
Clin Exp Pharmacol Physiol. 39:684–691. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Panchin Y, Kelmanson I, Matz M, Lukyanov
K, Usman N and Lukyanov S: A ubiquitous family of putative gap
junction molecules. Curr Biol. 10:R473–R474. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zoidl G, Petrasch-Parwez E, Ray A, Meier
C, Bunse S, Habbes HW, Dahl G and Dermietzel R: Localization of the
pannexin1 protein at postsynaptic sites in the cerebral cortex and
hippocampus. Neuroscience. 146:9–16. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Iglesias R, Dahl G, Qiu F, Spray DC and
Scemes E: Pannexin 1: The molecular substrate of astrocyte
‘hemichannels’. J Neurosci. 29:7092–7097. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thompson RJ, Jackson MF, Olah ME, Rungta
RL, Hines DJ, Beazely MA, MacDonald JF and MacVicar BA: Activation
of pannexin-1 hemichannels augments aberrant bursting in the
hippocampus. Science. 322:1555–1559. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang L, Deng T, Sun Y, Liu K, Yang Y and
Zheng X: Role for nitric oxide in permeability of hippocampal
neuronal hemichannels during oxygen glucose deprivation. J Neurosci
Res. 86:2281–2291. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
de Pina-Benabou MH, Szostak V, Kyrozis A,
Rempe D, Uziel D, Urban-Maldonado M, Benabou S, Spray DC, Federoff
HJ, Stanton PK and Rozental R: Blockade of gap junctions in vivo
provides neuroprotection after perinatal global ischemia. Stroke.
36:2232–2237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Madry C, Haglerød C and Attwell D: The
role of pannexin hemichannels in the anoxic depolarization of
hippocampal pyramidal cells. Brain. 133:3755–3763. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weilinger NL, Tang PL and Thompson RJ:
Anoxia-induced NMDA receptor activation opens pannexin channels via
Src family kinases. J Neurosci. 32:12579–12588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liew HK, Pang CY, Hsu CW, Wang MJ, Li TY,
Peng HF, Kuo JS and Wang JY: Systemic administration of urocortin
after intracerebral hemorrhage reduces neurological deficits and
neuroinflammation in rats. J Neuroinflammation. 9:132012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okauchi M, Hua Y, Keep RF, Morgenstern LB,
Schallert T and Xi G: Deferoxamine treatment for intracerebral
hemorrhage in aged rats: Therapeutic time window and optimal
duration. Stroke. 41:375–382. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gareri P, Condorelli D, Belluardo N, Russo
E, Loiacono A, Barresi V, Trovato-Salinaro A, Mirone MB, Ferreri IG
and De Sarro G: Anticonvulsant effects of carbenoxolone in
genetically epilepsy prone rats (GEPRs). Neuropharmacology.
47:1205–1216. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen G, Li Q, Feng D, Hu T, Fang Q and
Wang Z: Expression of NR2B in different brain regions and effect of
NR2B antagonism on learning deficits after experimental
subarachnoid hemorrhage. Neuroscience. 231:136–144. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y, Gao A, Xu X, Dang B, You W, Li H,
Yu Z and Chen G: The neuroprotection of lysosomotropic agents in
experimental subarachnoid hemorrhage probably involving the
apoptosis pathway triggering by cathepsins via chelating
intralysosomal iron. Mol Neurobiol. 52:64–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Friedrich V, Flores R and Sehba FA: Cell
death starts early after subarachnoid hemorrhage. Neurosci Lett.
512:6–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bruzzone R, Hormuzdi SG, Barbe MT, Herb A
and Monyer H: Pannexins, a family of gap junction proteins
expressed in brain. Proc Natl Acad Sci USA. 100:pp. 13644–13649.
2003; View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chekeni FB, Elliott MR, Sandilos JK, Walk
SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW,
Salvesen GS, et al: Pannexin 1 channels mediate ‘find-me’ signal
release and membrane permeability during apoptosis. Nature.
467:863–867. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Balami JS and Buchan AM: Complications of
intracerebral haemorrhage. Lancet Neurol. 11:101–118. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xi G, Hua Y, Bhasin RR, Ennis SR, Keep RF
and Hoff JT: Mechanisms of edema formation after intracerebral
hemorrhage: Effects of extravasated red blood cells on blood flow
and blood-brain barrier integrity. Stroke. 32:2932–2938. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Castillo J, Dávalos A, Naveiro J and Noya
M: Neuroexcitatory amino acids and their relation to infarct size
and neurological deficit in ischemic stroke. Stroke. 27:1060–1065.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qureshi AI, Ali Z, Suri MF, Shuaib A,
Baker G, Todd K, Guterman LR and Hopkins LN: Extracellular
glutamate and other amino acids in experimental intracerebral
hemorrhage: An in vivo microdialysis study. Crit Care Med.
31:1482–1489. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mailly F, Marin P, Israël M, Glowinski J
and Prémont J: Increase in external glutamate and NMDA receptor
activation contribute to H2O2-induced
neuronal apoptosis. J Neurochem. 73:1181–1188. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thompson RJ, Zhou N and MacVicar BA:
Ischemia opens neuronal gap junction hemichannels. Science.
312:924–927. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Turpie AG and Thomson TJ: Carbenoxolone
sodium in the treatment of gastric ulcer with special reference to
side-effects. Gut. 6:591–594. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Traub RD, Whittington MA, Buhl EH, LeBeau
FE, Bibbig A, Boyd S, Cross H and Baldeweg T: A possible role for
gap junctions in generation of very fast EEG oscillations preceding
the onset of and perhaps initiating, seizures. Epilepsia.
42:153–170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Carlen PL, Skinner F, Zhang L, Naus C,
Kushnir M and Perez VJ: The role of gap junctions in seizures.
Brain Res Brain Res Rev. 32:235–241. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma W, Hui H, Pelegrin P and Surprenant A:
Pharmacological characterization of pannexin-1 currents expressed
in mammalian cells. J Pharmacol Exp Ther. 328:409–418. 2009.
View Article : Google Scholar : PubMed/NCBI
|