Introduction
The global prevalence of Alzheimer's disease (AD),
which is characterized by progressive deterioration in cognition
and behavior, particularly memory loss, places a considerable
burden on society (1). The
neuropathological hallmarks of AD include extracellular senile
plaques composed of β-amyloid (Aβ) deposits, intracellular
neurofibrillary tangles and cerebral atrophy (2). Pharmacological treatment of AD
currently primarily focus on cholinesterase inhibitors and
N-methyl-D-aspartic acid receptor antagonists. Unfortunately,
according to previous studies, treatment with these two classes
predominantly provided symptomatic benefits without counteracting
the progression of the disease (3). Therefore, investigating compounds
that target the underlying mechanisms of disease is of utmost
importance for the development of novel therapeutic agents against
AD (4).
The pathology of AD is complex and multifactorial;
aggregated Aβ elicits neurotoxicity and induces oxidative-stress
and inflammation in the brain of patients with AD (5). Emerging evidence has indicated that
oxidative stress is important in the mechanisms associated with
Aβ-induced neurotoxicity and cell loss, and is proposed as one of
the basic mechanisms that contributes to the process of AD
(6). In this regard, various
studies have focused on the use of antioxidants for the management
of AD (7). Additionally, A
previous study demonstrated that increased p38 mitogen-activated
protein kinase (MAPK) activity was associated with the
neuropathology of AD. For example, p38 MAPK and its upstream kinase
mitogen-activated protein kinase kinase 6 (MKK6) were activated in
AD brain tissue samples, as demonstrated by immunohistochemistry
(8). Activation of p38 MAPK
signaling was also reported in AD-relevant animal models (9). Furthermore, c-Jun N-terminal kinase
(JNK) MAPK activation was localized to amyloid deposits in AD
models and this activation was coincident with the age-dependent
increase in amyloid deposition, tau phosphorylation, and loss of
synaptophysin (10).
In previous years, N-acetyl cysteine (NAC) had been
extensively reported to exert neuroprotective effects on the
central nervous system and may be effective against neurological
conditions by rescuing severely compromised cells from an
unremitting burden of oxidative stress (11). However, the protective effects of
NAC on oxidative stress-induced cell death and the underlying
mechanisms are unclear in primary hippocampus neurons. Therefore,
in the present study, the protective effect of NAC against hydrogen
peroxide (H202)-mediated damage of
hippocampus neurons was investigated by measuring the cellular
viability and reactive oxygen species (ROS) levels. Furthermore,
the mechanisms underlying these neuroprotective effects were
investigated by targeting MAPK signal transduction.
Materials and methods
Approval
All experimental protocols were reviewed and
approved by the Ethical Committee of Wenzhou Medical University
(Wenzhou, China).
Primary rat hippocampus neurons
culture and treatments
Primary cultures of hippocampus neurons were
obtained and cultured according to previously described protocol
(12). Briefly, primary rat
hippocampus samples were prepared from Sprague-Dawley rat brains at
embryonic days 1–3, which were purchased from the Experimental
Animal Center of China Medical University (Beijing, China) and were
dissected in calcium- and magnesium-free Hank's balanced salt
solution (Beyotime Institute of Biotechnology, Haimen, China),
following incubation with a 0.25% trypsin solution for 30 min at
36°C in order to obtain primary hippocampus neuron cells. Cells
were maintained in Dulbecco's modified Eagle's medium (DMEM)/high
glucose, horse serum containing 10% fetal bovine serum, 1%
L-glutamine (3.6 mM), and 1% penicillin antibiotics and were grown
in a 5% CO2 atmosphere at 37°C. The primary rat
hippocampus cells were cultured on plates which were coated in the
fetal bovine serum (Beyotime Institute of Biotechnology) and were
cultured at 37°C in humidified air. Two-thirds of the growing
medium was changed every 2–3 days and the cells were subcultured
roughly once a week. On day 12 of culturing, the incubation media
were replaced with media with H2O2 (3, 30 and
300 µmol/l) to achieve oxidative stress injury. Following
incubation for 30 min, NAC was added to the media at concentrations
of 1, 10, 100 or 1,000 µmol/l.
Measurement of cytotoxicity by MTT
assay and light microscopy
Cell viability was measured using the MTT assay
(Beyotime Institute of Biotechnology), which is based on the
conversion of MTT to formazan crystals by mitochondrial
dehydrogenases (13). MTT is
absorbed by viable cells and then converted to formazan by the
enzyme, succinate dehydrogenase in the mitochondria. The quantity
of produced formazan thus correlates with the number of living
cells. Cells were seeded in 24-well polystyrene plates with
~3×103 cells per well. Plates were incubated at 37°C for
24 h to allow the cells to attach. After treatment with
H2O2 for 0.5 h at 37°C followed by NAC for 24
h, the same volume of medium was added to the control cultures.
Cell viability was determined using an MTT toxicity assay by adding
10 µl of 5 mg/ml MTT to each well. After 4 h of incubation at 37°C
in humid air, formazan crystals were solubilized in 200 ml dimethyl
sulfoxide. The optical density was measured at a wavelength of 570
nm with background correction at 655 nm using a Bio-Rad microplate
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The mean
averages of optical density from six replicate wells were used for
each experimental sample and the control sample. Cell viability was
calculated with a reference to the absorbance of control wells not
challenged with H2O2 (assumed as 100%
protection). Analyzed by light microscopy, viable cells displayed
normal nuclear size and dark brown granules, whereas toxic cells
exhibited condensed chromatin. The number of residual viable cells
was counted.
Measurement of glutathione (GSH) and
lipid peroxide
To assess the enzymatic activity of GSH-peroxidase
and lipid peroxide in primary hippocampus neuron culture after
H2O2 injury, the cultures were washed with
ice-cold phosphate-buffered saline (PBS) and then pooled and
homogenized in 0.1 mol/l PBS containing 0.05 mmol/l
ethylenediaminetetraacetic acid according to previous protocol
(14). GSH-peroxidase activity was
assessed using a GSH assay kit (Beyotime Institute of
Biotechnology) by quantifying the rate of oxidation of reduced GSH
to oxidized GSH. To investigate the effect of NAC on anti-oxidative
stress in the AD cell model, the biomarkers of oxidative stress,
including GSH and GSH disulfide (GSSG) were assessed. The level of
GSH activity by the means of GSH/GSSG ratio. GSSG was obtained by
determining the absorbance of 5-thio-2-nitrobenzoic acid produced
from the reaction of the reduced GSH with DTNB. The level of maleic
dialdehyde (MDA), a product of lipid peroxidation, was measured
using an MDA assay kit (Beyotime Institute of Biotechnology) based
on the thiobarbituric acid method (15).
Determination of intracellular ROS by
dichloro-dihydro-fluorescein diacetate (DCFH-DA)
The fluorescent probe DCFH-DA was used to monitor
intracellular accumulation of ROS (16). Hippocampus neurons were seeded in
collagen-coated 24-well plates at a density of 4×105
cells/ml and incubated for 72 h. Cells were incubated with 300
µmol/l H2O2, a mixture of 1, 10 or 100 µmol/l
NAC, or 300 µmol/l H2O2 alone at 37°C for 9
h. The cells were collected and washed with PBS three times.
DCFH-DA was diluted in fresh DMEM to a final concentration of 5 µm
and incubated with the cells for 30 min at 37°C. The chemicals were
then removed and the cells were washed three times with PBS.
Fluorescence emission was measured at excitation and emission
wavelengths of 485 and 520 nm, respectively using fluorescence
microplate. ROS production was expressed as a percentage of the
control sample.
Western blot analysis
The primary rat hippocampus neurons were homogenized
in protein extraction solution comprised of 20 mM Tris-HCl (pH
7.4), containing 1 mM NaF, 150 mM NaCl, 1% Triton X-100 and
freshly-added protease inhibitor cocktail (Roche Diagnostics,
Basel, Switzerland), and 100 µM phenylmethylsulfonyl fluoride
(Beyotime Institute of Biotechnology). The supernatant contained
total and membrane-enriched proteins. The Bicinchoninic Acid
protein determination method was employed for the concentration of
the proteins. Then, the proteins (30–50 ug) were separated by 8–12%
sodium dodecyl polyacrylamide gels at 80 V for 50 min followed by
120 V for 40 min and electrophoretically transferred to a
polyvinylidene fluoride membrane (PVDF) at 300 mA; the duration of
electrophoresis depended on the molecular weight of the proteins.
The PVDF membrane was blocked with freshly prepared Tris-buffered
saline with Tween-20 (0.1%) containing 5% non-fat dry milk for
30–60 min at room temperature with constant agitation.
Subsequently, the membrane was incubated with polyclonal rabbit
anti-phospho-p38 immunoglobulin G (IgG; 1:1,000; cat. no. 2729,
Cell Signaling Technology, Inc., Danvers, MA, USA), monoclonal
rabbit anti-phospho-JNK IgG (1:1,000; cat. no. 4671, Cell Signaling
Technology, Inc.), polyclonal rabbit anti-phospho-extracellular
regulated kinase (ERK; 1:1,000; cat. no. 4370, Cell Signaling
Technology, Inc.), polyclonal mouse anti-phospho-tau IgG (1:00;
cat. no. 9632, Cell Signaling Technology, Inc.), monoclonal rabbit
anti-p38 IgG (1:1,000; cat. no. 8690, Cell Signaling Technology,
Inc.), polyclonal rabbit anti-JNK IgG (1:1,000; cat. no. 5136, Cell
Signaling Technology, Inc.), polyclonal rabbit anti-ERK IgG
(1:1,000; cat. no. 8544, Cell Signaling Technology, Inc.),
polyclonal rabbit anti-tau IgG (1:500; cat. no. T7951,
Sigma-Aldrich; Merck KGaA) and monoclonal mouse anti-β-actin IgG
(1:1,000; cat. no. AA128, Beyotime Institute of Biotechnology)
overnight at 4°C. The membrane was then incubated with anti-rabbit
or anti-mouse horseradish peroxidase IgGs (1:1,000; A0208 or A0216,
respectively, Beyotime Institute of Biotechnology) for 1–2 h at
room temperature. Immunoreactive bands were visualized using an
Enhanced Chemiluminescent Western Blotting Substrate (cat. no.
32106, Pierce; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
and quantified using Quantity One software 3.0 (Image Lab, Bio-Rad
Laboratories, Inc.).
Statistical analysis
Data were expressed as the mean ± standard
deviation. Comparisons between different groups were performed by
one-way analysis of variance followed by least significant
difference post-hoc comparisons when appropriate. P<0.05 was
considered to indicate a statistically significant difference. All
analyses were performed using SPSS 16.0 (SPSS, Inc., Chicago, IL,
USA).
Results
Effects of NAC on cell viability in
H2O2-induced primary hippocampus neuron
injury
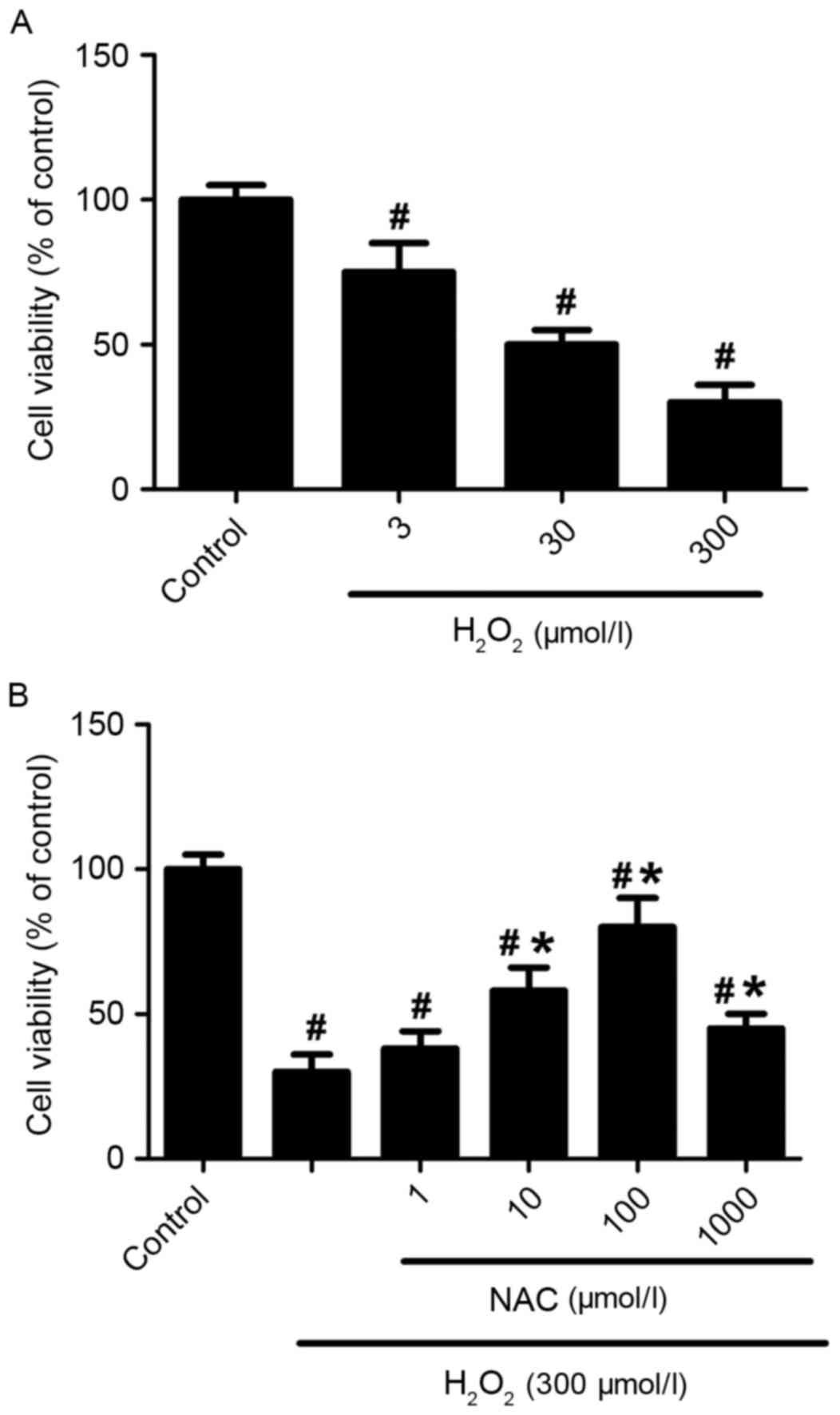
The viabilities of primary hippocampus neurons
exposed to different concentrations of H2O2
(3, 30 or 300 µmol/l) were detected after 24 h of
H2O2 incubation. The
H2O2 reduced cell viabilities in a
dose-dependent manner (P<0.05 vs. control group; Fig. 1A). The survival rate of the
hippocampus neurons was ~78% when the neurons were incubated with 3
µmol/l of H2O2 for 24 h. However, the
survival rate of neurons reduced to ~31% when treated with 300
µmol/l of H2O2 (P<0.01 vs. control group;
Fig. 1A). Exposure of cells to NAC
(1, 10, 100 or 1,000 µmol/l) significantly improved cell viability
(P<0.05 vs. control group: Fig.
1B), although treatment with 1 µmol/l NAC was not significantly
different when compared with H2O2 alone
(P>0.05; Fig. 1B). In addition,
100 µmol/l NAC almost completely saved neurons from 300 µmol/l
H2O2-induced cell deaths (82% survival rate
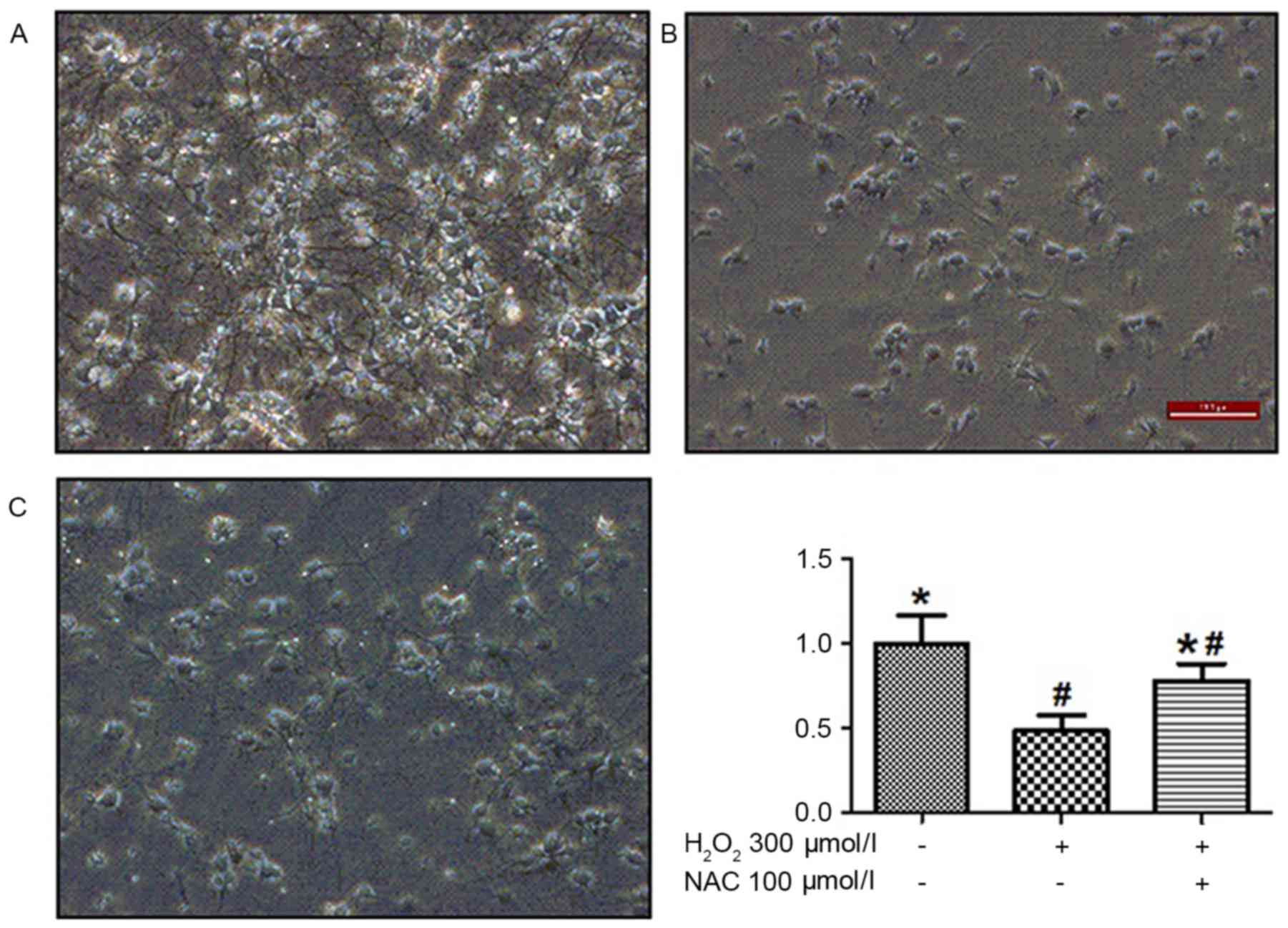
compared with the control group). Analysis under a light microscope
demonstrated that H2O2-induced neuron death
in 50% of cells and significantly reduced neurite length. The
pretreatment of cells with 100 µmol/l NAC tended to overcome these
detrimental effects of H2O2 incubation
(P<0.01; Fig. 2). Treatment
with 100 µmol/l NAC significantly reduced
H2O2-induced cell death (P<0.05; Fig. 2), indicating that NAC treatment
elicited a potent protection effect on
H2O2-induced cell viability.
Effects of NAC on MDA and GSH
activity
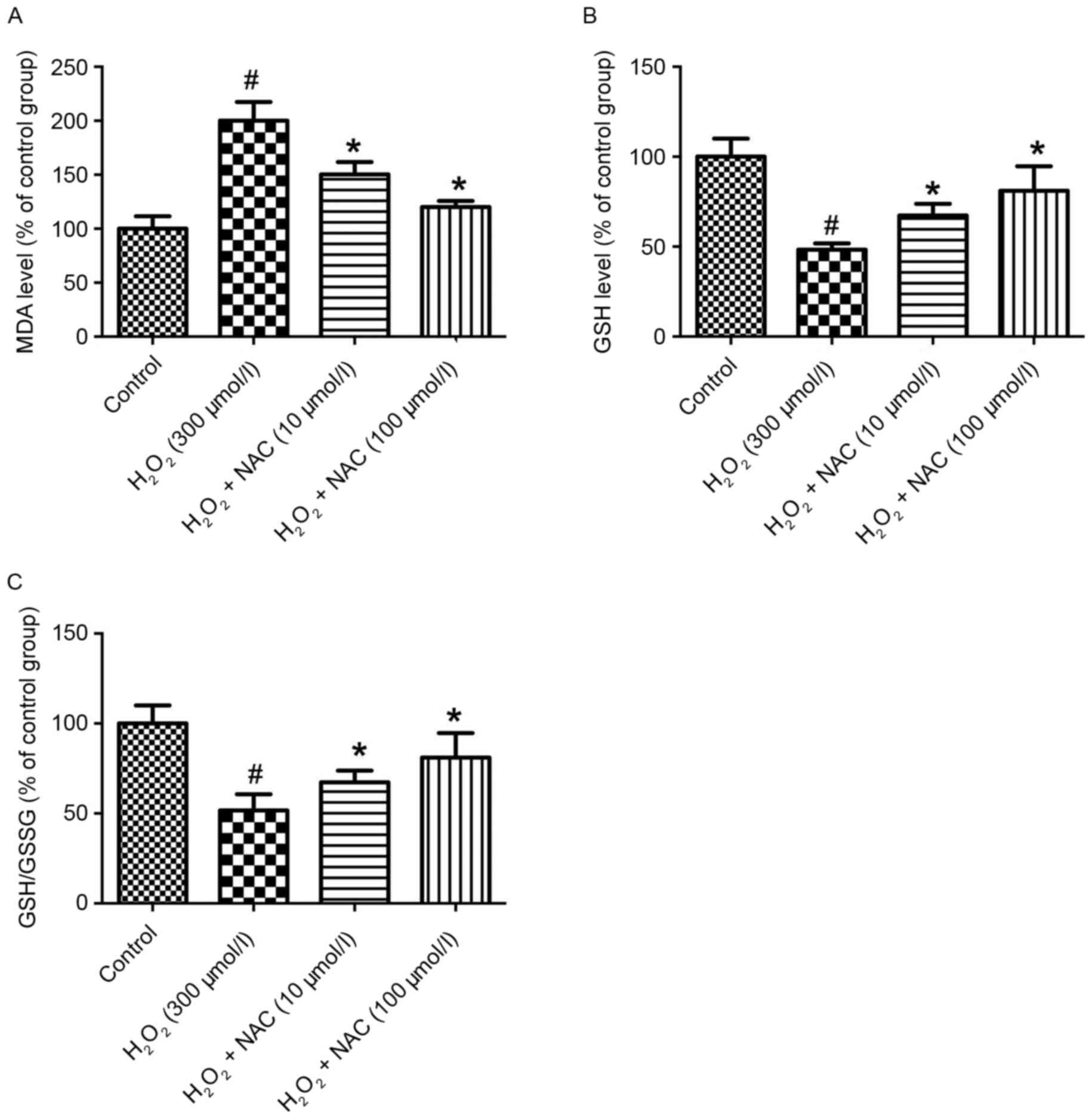
In the present study, the MDA level as a measure of
lipid peroxidation were significantly increased in the
H2O2 group (300 µmol/l) compared with the
control group (P<0.05; Fig.
3A). The MDA levels were significantly reduced in the NAC-low
(L; 10 µmol/l, low-concentration of NAC) and NAC-H (100 µmol/l,
high concentration of NAC) treatment groups vs. the
H2O2 group (P<0.05; Fig. 3A). Similarly, no significant
difference between the NAC-L and NAC-H groups was identified with
regard to reducing the MDA level (P>0.05; Fig. 3A). To investigate the effect of NAC
on anti-oxidative stress in the AD cell model, the biomarkers of
oxidative stress, including GSH and GSH disulfide (GSSG) were
assessed. The level of GSH activity by the aid of GSH/GSSG in the
H2O2 group (300 µmol/l) was significantly
decreased compared with the control group (P<0.05; Fig. 3B and C). Additionally, treatment
with NAC significantly alleviated GSH activity compared with the
H2O2 group (P<0.05; Fig. 3B and C). Furthermore, the NAC-H
group demonstrated significantly increased GSH activity compared
with the rats receiving NAC-L (P<0.05; Fig. 3B and C).
NAC ameliorates
H2O2-induced cell impairment by decreasing
ROS production
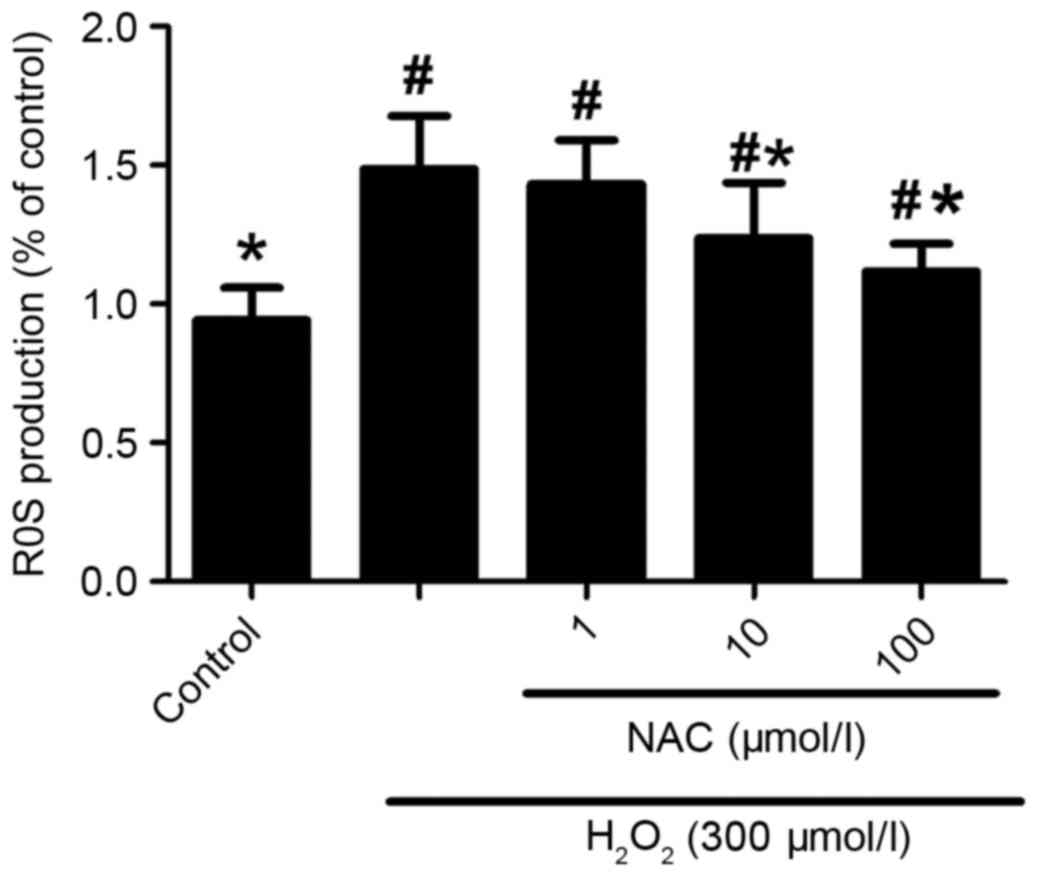
Oxidative stress is crucial in the pathogenesis of
AD. In the current study, by using ROS fluorescent dye, DCFH-DA,
the results demonstrated that intracellular DCF fluorescence was
significantly increased in the presence of 300 µmol/l
H2O2 compared with the control sample, which
was abolished by treatment with 10 and 100 µmol/l NAC (P<0.05;
Fig. 4), but not 1 µmol/l NAC
(P>0.05; Fig. 4). Taken
together, these data indicate that the neuroprotective effects of
NAC against H2O2-induced neurotoxicity
involve limiting oxidative stress injury.
NAC ameliorates
H2O2-induced injury by inhibition of MAPK
signal transduction
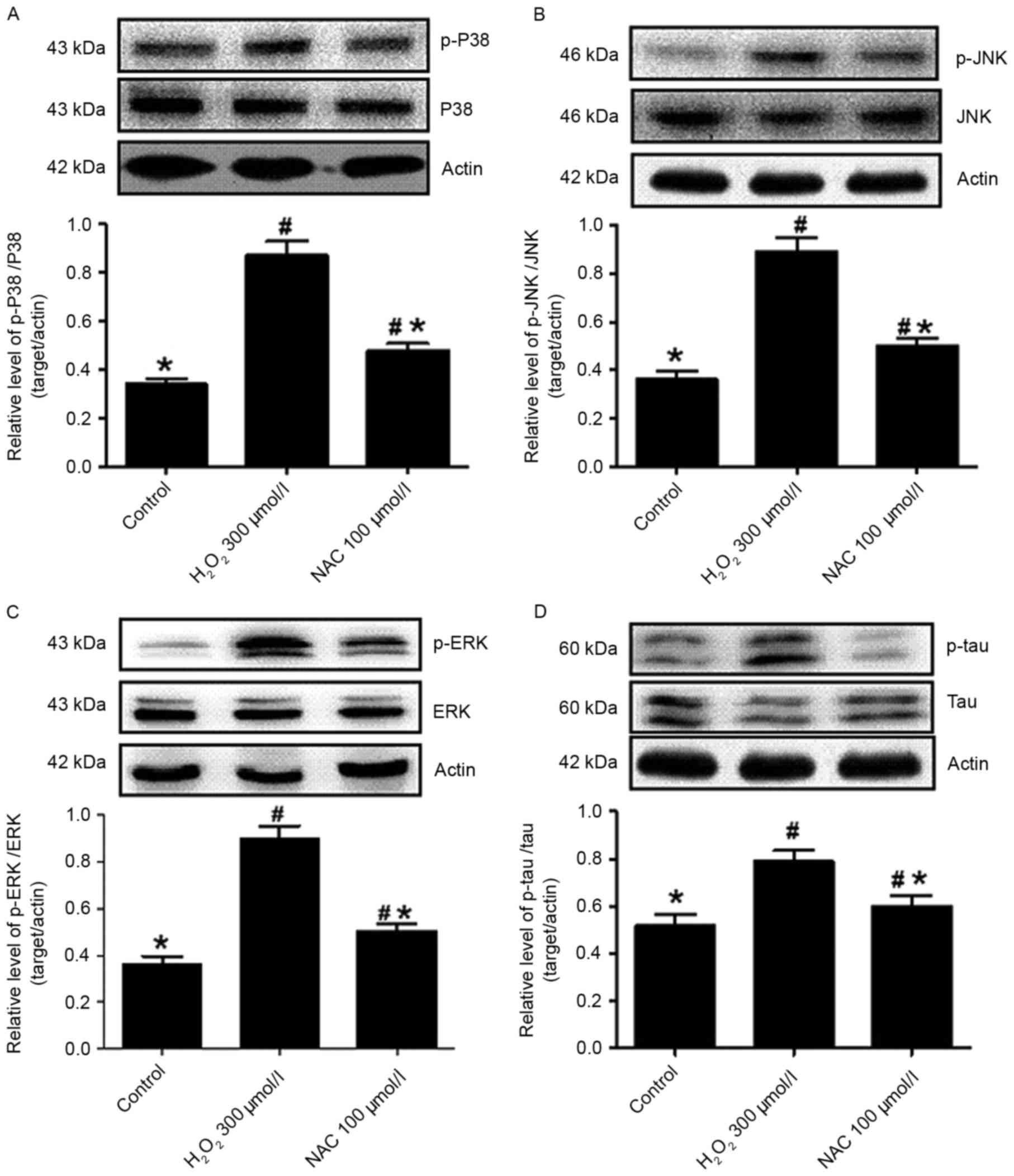
Enhanced levels of phosphorylated (p)-p38 were
detected in the presence of 300 µmol/l H2O2
injury (P<0.05 vs. control; Fig.
5A), while treatment with 100 µmol/l NAC caused the decline of
p-p38 levels (P<0.05 vs. H2O2 incubation
alone; Fig. 5A). Thus, the
protective effects of NAC involved the attenuation of p38 protein
phosphorylation. Furthermore, the levels of JNK protein
phosphorylation were analyzed in primary hippocampus neurons in the
absence or presence of NAC. p-JNK levels were significantly
increased in cells exposed to H2O2 (P<0.05
vs. control; Fig. 5B) and
significantly decreased following the addition of 100 µmol/l NAC to
the culture (P<0.05 vs. H2O2 alone;
Fig. 5B). Similarly, p-ERK
expression levels were increased in
H2O2-induced hippocampus neurons (P<0.05
vs. control; Fig. 5C), indicating
that ERK activity had increased. In addition, NAC (100 µmol/l)
significantly reduced the induction of p-ERK following
H2O2 incubation when compared with the
control group (P<0.05; Fig.
5C).
NAC decreased tau phosphorylation
induced by H2O2 injury
In the present study, western blot analyses
demonstrated that an increased level of p-tau was observed in the
presence of 300 µmol/l H2O2 compared with
control hippocampus neurons (P<0.05; Fig. 5D), while the expression levels of
p-tau were decreased in the NAC (100 µmol/l) treatment group
compared with the H2O2 alone group
(P<0.05; Fig. 5D). This clearly
indicated that H2O2 resulted in tau
phosphorylation in the hippocampus neurons and that NAC ameliorates
the levels of p-tau.
Discussion
In the present study, NAC was demonstrated to
protect differentiated primary rat hippocampus neurons against
H2O2-mediated toxicity as evidenced by
enhanced cell viability. While H2O2 (300
µmol/l) markedly decreased cell viability, exposure of cells to NAC
(100 µmol/l) overcame the negative effect of oxidative stress on
cell survival, and increased it by ~3-fold when compared with the
intact control cells. The results demonstrated that treatment with
NAC reduced the percentage of cell death that was observed
following incubation with H2O2. Using MTT
assay and light microscopy for the observation of cell viability,
NAC appeared to ameliorate cell death events induced by
H2O2 injury.
H2O2-induced cells displayed decreases in
neurite number and their length; NAC treatment was demonstrated to
restore the number of neurites and significantly augment their
length (Fig. 2). In addition, NAC
was observed to mitigate the excessive production of ROS,
indicating that the neuroprotective effects of the compounds in
this AD-like cellular model were probably associated with
inhibition of H2O2-induced oxidative stress
injury. Furthermore, NAC reduced H2O2-induced
MDA over-expression and upregulated the level of GSH. In the
current study, another mechanism underlying the neuroprotective
action of NAC likely includes its ability to inhibit MAPK signal
transduction following H2O2 exposure. In
addition, the present study demonstrated for the first time, to the
best of our knowledge, that NAC protects cells against
H2O2-mediated toxicity by attenuating the
increase in tau phosphorylation. These results indicate that NAC
may serve as a neuroprotective agent for
H2O2-associated injury.
Emerging evidence has suggested that oxidative
stress damage is closely associated with neurodegeneration,
including AD (6). Although whether
oxidative stress is involved in the onset of AD remains unclear,
oxidative stress is pivotal in disease progression, particularly in
cellular and tissue damage (17).
During the process of the oxidative stress reaction, Aβ passes
through the neuronal membrane, resulting in the overproduction of
ROS, which may destroy various classes of biological molecules,
such as lipids, proteins and DNA (18). Therefore, the ROS levels were used
to evaluate the extent of oxidative stress damage. In the present
study, NAC markedly reduced the excessive production of ROS levels
in a dose-dependent manner (10 and 100 µmol/l). These results
indicate that the neuroprotective effects of NAC in this cellular
model may be associated with antioxidant properties. In addition,
the data were in accordance with previous findings, which reported
the neuroprotective action of NAC in various in vivo and
in vitro studies (11).
The mammalian family of MAPKs include ERK, p38, and
JNK, with each MAPK signaling pathway consisting of at least three
components (19). These signaling
pathways regulate a variety of cellular activities, including cell
proliferation, differentiation, survival and death. The activated
MAPK signaling pathways are proposed to contribute to AD
pathogenesis via various mechanisms, including induction of
neuronal death (20), and
transcriptional and enzymatic activation of β- and γ-secretases
(21). In addition, under
conditions of oxidative stress, JNK and p38 are activated and
induce the expression of the β-secretase gene, suggesting a pivotal
role in cell viability in AD (22). Meanwhile, γ-secretase activity was
found to be blocked by a JNK inhibitor, thus implicating the JNK
signaling pathway in the regulation of γ-secretase activity.
Furthermore, a previous study demonstrated that the detrimental
effects of ERK resulted from promoting oxidative stress (23). In the current study, NAC was shown
to protect cells against H2O2-induced
toxicity by attenuating the increased levels of p38, JNK and ERK
phosphorylation. These results indicated that inhibition of MAPK
signal transduction by NAC was crucial in the survival against
oxidative stress in primary rat hippocampus neurons. It is well
known that extensively phosphorylated tau protein forms pathologic
inclusions, containing fibrillar aggregates, and are present in AD
(24). Tau is proposed as one of
the microtubule stabilizing proteins exerting a crucial role in the
facilitation of tubulin assembly into microtubules, thus
contributing to maintenance of normal cellular morphology (1). Abnormally hyperphosphorylated-tau
possesses lower affinity for microtubules, which promotes
cytoskeleton rearrangements with consequent impairments of axonal
transport and intracellular trafficking (25). Results from the present study
indicated that abrogation of tau hyperphosphorylation by 100 µmol/l
NAC may eventually contribute to restoration and even improvement
of cell morphology.
From previous studies, NAC maintained intracellular
GSH levels and may be beneficial for a range of neuronal cell types
against various oxidative stress stimuli in vitro (26). In addition, Hart et al
(27) indicated that NAC may
reduce neuronal death by blocking attempted entry into the cell
cycle, by improving free radical surveillance and scavenging ROS
levels, or by preserving mitochondrial function, regenerating
endogenous antioxidants and repairing oxidative damage (27). Recently, Adair et al
(28) performed a controlled
clinical trial where NAC or placebo was administered in a
double-blind fashion to patients with probable AD. The authors
observed that NAC exerted a positive effect on nearly every outcome
measure, although significant differences were obtained only for a
subset of cognitive tasks (28).
In the current study, the findings indicated that NAC attenuated
H2O2-induced injury by inhibition of MAPK
signal transduction and antioxidative action.
In conclusion, NAC exerted a neuroprotective effect
against H2O2-induced toxicity in primary
hippocampus neurons. The protective ability of NAC most likely
results from inhibition of oxidative stress and from reducing cell
death. Another potential mechanism by which this compound protects
cells from oxidative stress toxicity may be associated with the
downregulation of MAPK signal transduction and tau
phosphorylation.
Acknowledgements
Not applicable.
Funding
The current study was supported by the Projects of
Wenzhou City Committee of Science and Technology (Wenzhou, China;
Y20100146).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WW and YMZ conceived and designed the experiment.
CLX and XDX performed the experiments and acquisition of data. BHL
and YMZ analyzed and interpreted the data. YMZ wrote the article.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were reviewed and
approved by the Ethical Committee of Wenzhou Medical University
(Wenzhou, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Giacobini E and Gold G: Alzheimer disease
therapy-moving from amyloid-β to tau. Nat Rev Neurol. 9:677–686.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Steel K: Alzheimer's disease. N Engl J
Med. 362:1844–1845. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schneider LS, Dagerman KS, Higgins JP and
McShane R: Lack of evidence for the efficacy of memantine in mild
Alzheimer disease. Arch Neurol. 68:991–998. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lang AE: Clinical trials of
disease-modifying therapies for neurodegenerative diseases: The
challenges and the future. Nat Med. 16:1223–1226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pimplikar SW: Reassessing the amyloid
cascade hypothesis of Alzheimer's disease. Int J Biochem Cell Biol.
41:1261–1268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Butterfield DA, Swomley AM and Sultana R:
Amyloid β-peptide (1–42)-induced oxidative stress in Alzheimer
disease: Importance in disease pathogenesis and progression.
Antioxid Redox Signal. 19:823–835. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tavakkoli M, Miri R, Jassbi AR, Erfani N,
Asadollahi M, Ghasemi M, Saso L and Firuzi O: Carthamus, Salvia and
Stachys species protect neuronal cells against oxidative
stress-induced apoptosis. Pharm Biol. 52:1550–1557. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ando K, Uemura K, Kuzuya A, Maesako M,
Asada-Utsugi M, Kubota M, Aoyagi N, Yoshioka K, Okawa K, Inoue H,
et al: N-cadherin regulates p38 MAPK signaling via association with
JNK-associated leucine zipper protein: Implications for
neurodegeneration in Alzheimer disease. J Biol Chem. 286:7619–7628.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Savage MJ, Lin YG, Ciallella JR, Flood DG
and Scott RW: Activation of c-Jun N-terminal kinase and p38 in an
Alzheimer's disease model is associated with amyloid deposition. J
Neurosci. 22:3376–3385. 2002.PubMed/NCBI
|
|
10
|
Ferrer I, Gomez-Isla T, Puig B, Freixes M,
Ribé E, Dalfó E and Avila J: Current advances on different kinases
involved in tau phosphorylation, and implications in Alzheimer's
disease and tauopathies. Curr Alzheimer Res. 2:3–18. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Unnithan AS, Jiang Y, Rumble JL, Pulugulla
SH, Posimo JM, Gleixner AM and Leak RK: N-acetyl cysteine prevents
synergistic, severe toxicity from two hits of oxidative stress.
Neurosci Lett. 560:71–76. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vedunova MV, Mitroshina EV, Sakharnova TA,
Bobrov MY, Bezuglov VV, Khaspekov LG and Mukhina IV: Effect of
N-arachidonoyl dopamine on activity of neuronal network in primary
hippocampus culture upon hypoxia modelling. Bull Exp Biol Med.
156:461–464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garrido J, Gaspar A, Garrido EM, Miri R,
Tavakkoli M, Pourali S, Saso L, Borges F and Firuzi O: Alkyl esters
of hydroxycinnamic acids with improved antioxidant activity and
lipophilicity protect PC12 cells against oxidative stress.
Biochimie. 94:961–967. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu Y, Wang JR, Sun PH, Guo Y, Zhang ZJ,
Jin GZ and Zhen X: Neuroprotective effects of atypical D1 receptor
agonist SKF83959 are mediated via D1 receptor-dependent inhibition
of glycogen synthase kinase-3 beta and a receptor-independent
anti-oxidative action. J Neurochem. 104:946–956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mihara M and Uchiyama M: Determination of
malonaldehyde precursor in tissues by thiobarbituric acid test.
Anal Biochem. 86:271–278. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bass DA, Parce JW, Dechatelet LR, Szejda
P, Seeds MC and Thomas M: Flow cytometric studies of oxidative
product formation by neutrophils: A graded response to membrane
stimulation. J Immunol. 130:1910–1917. 1983.PubMed/NCBI
|
|
17
|
Melo A, Monteiro L, Lima RM, Oliveira DM,
Cerqueira MD and El-Bachá RS: Oxidative stress in neurodegenerative
diseases: Mechanisms and therapeutic perspectives. Oxid Med Cell
Longev. 2011:4671802011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dumont M and Beal MF: Neuroprotective
strategies involving ROS in Alzheimer disease. Free Radic Biol Med.
51:1014–1026. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raman M, Chen W and Cobb MH: Differential
regulation and properties of MAPKs. Oncogene. 26:3100–3112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marques CA, Keil U, Bonert A, Steiner B,
Haass C, Muller WE and Eckert A: Neurotoxic mechanisms caused by
the Alzheimer's disease-linked Swedish amyloid precursor protein
mutation: Oxidative stress, caspases, and the JNK pathway. J Biol
Chem. 278:28294–28302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen C, Chen Y, Liu H, Zhang K, Zhang T,
Lin A and Jing N: Hydrogen peroxide promotes Abeta production
through JNK-dependent activation of gamma-secretase. J Biol Chem.
283:17721–17730. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tamagno E, Guglielmotto M, Giliberto L,
Vitali A, Borghi R, Autelli R, Danni O and Tabaton M: JNK and
ERK1/2 pathways have a dual opposite effect on the expression of
BACE1. Neurobiol Aging. 30:1563–1573. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sawe N, Steinberg G and Zhao H: Dual roles
of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci
Res. 86:1659–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takei Y, Teng J, Harada A and Hirokawa N:
Defects in axonal elongation and neuronal migration in mice with
disrupted tau and map1b genes. J Cell Biol. 150:989–1000. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alonso AC, Zaidi T, Grundke-Iqbal I and
Iqbal K: Role of abnormally phosphorylated tau in the breakdown of
microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 91:pp.
5562–5566. 1994; View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Munoz AM, Rey P, Soto-Otero R, Guerra MJ
and Labandeira-Garcia JL: Systemic administration of
N-acetylcysteine protects dopaminergic neurons against
6-hydroxydopamine-induced degeneration. J Neurosci Res. 76:551–562.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hart AM, Terenghi G, Kellerth JO and
Wiberg M: Sensory neuroprotection, mitochondrial preservation, and
therapeutic potential of N-acetyl-cysteine after nerve injury.
Neuroscience. 125:91–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adair JC, Knoefel JE and Morgan N:
Controlled trial of N-acetylcysteine for patients with probable
Alzheimer's disease. Neurology. 57:1515–1517. 2001. View Article : Google Scholar : PubMed/NCBI
|