Introduction
Breast cancer is a heterogeneous disease and is the
most frequently diagnosed type of cancer in women, with an
estimated 1.38 million new cases per year worldwide (1). Breast cancer patients with the same
stage of disease may have different treatment responses and overall
outcome (2). In breast cancer,
epigenetic modifications are often noticed, including aberrant DNA
hypermethylation (3). As
previously reported, DNA methylation often occurs at carbon-5 of
cytosine residues in CpG dinucleotides (4). Methylation changes in CpG islands
(CGI) and CpG shores (low CpG density areas ~2 kb close to CGI)
affect gene expression and reprogramming (5,6). In
general, hypermethylation of CpG sites at tumor suppressor gene
promoters and hypomethylation at oncogene promoters is thought to
be involved in cancer (7). CpG
islands are often observed in the promoter region and serve a
crucial role in regulating key cellular functions (8). Hypermethylation in gene promoters
seems to be an early event in carcinogenesis and the number of
genes affected increases with breast cancer progression (9). DNA methylation has long been
considered a key regulator of gene expression. The genetic basis of
gene expression has been investigated across tissues and
populations (10). It plays an
important regulatory role in eukaryotic genomes. Alterations in
methylation can affect transcription and phenotypic variation
(11). A previous study indicated
that genetic variation may have a substantial impact on local
methylation patterns (12).
The culture of mammalian cells in vitro
provides a defined platform for investigating cell and tissue
physiology and pathophysiology outside of the organism (13). Traditionally, this has been done by
culturing single cell populations on two-dimensional (2D)
substrates such as tissue culture polystyrene (TCPS) or the surface
of tissue analogs (14).
Experiments with these 2D cell constructs have provided the base
for preliminary interpretation of complex biological phenomena,
including molecular biology, stem cell differentiation, and tissue
morphogenesis. Furthermore, 2D experiments have given rise to
seminal findings in the dynamic association between cell function
and interactions with the cellular microenvironment (15). However, previous work demonstrated
that cells often exhibit unnatural behavior when excised from
native three-dimensional (3D) tissues and confined as a monolayer
(16). 2D culture is technically
easier and simple, but there is a lack of a natural
microenvironment. Tumor cell growth is easily affected by internal
and external environments and the cost of 2D culture is high. 3D
culture can simulate the complex growth environment in the body,
tumor tissue complex signal transduction pathways and the formation
of new blood vessels (17). It is
similar to Ti culture, it can simulate cell complex growth
environment and is easy to establish, but it is still different
from the internal growth environment for tumor cells. However,
whether changes in DNA methylation state are influenced by the cell
culture method in breast cancer is still unknown.
In the present study, it was aimed to investigate
the influence of breast cancer cell culture method on DNA
methylation state. Results indicated that methylation status did
not change in breast cancer cells cultured under either 2D, 3D or
Ti conditions.
Materials and methods
Cell culture
The breast cancer cell line MCF-7 was obtained from
Ruijin Hospital, Affiliated to Shanghai Jiaotong University
(Shanghai, China). Cells were cultured in Dulbecco's modified
Eagle's medium (Invitrogen; Thermo Fisher Scientific, Inc. Waltham,
MA, USA) supplemented with 10% fetal bovine serum (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany). Subsequently, cells were cultured
in 2D (density, 60,000/cm2), 3D (density,
70,000/cm2) and Ti adhesion substrates (density,
60,000/cm2) in 5% CO2 at 37°C. For 2D
substrates, 2% alginate solution was mixed with calcium sulfate
(Sigma-Aldrich; Merck KGaA) and cast between glass plates, from
which topographically flat hydrogel disks were punched out. Excess
calcium was leached out by changing the medium every day for 4 days
prior to seeding. The 3D adhesion substrate was prepared by
suspending cells in 2% alginate solution. A custom-designed
encapsulation unit was used, and the alginate/cell suspension was
extruded into an isotonic 5.0% (w/v) CaCl2 cross-linking
solution (Sigma-Aldrich; Merck KGaA); the generated beads were
washed with PBS to remove excess Ca2+ and were
dynamically cultured in spinner flasks (Bellco Glass, Inc.,
Vineland, NJ, USA). For Ti culture, cells were added to in
situ culture flasks with 3 ml Active Messages 2.0 (AM-II;
Gibco; Thermo Fisher Scientific, Inc.). Finally, flasks were placed
into a humidified cell incubator containing 5% CO2 at
37°C for 5–7 days.
Total RNA isolation and chip
genome-wide methylation detection
The RNeasy mini kit (Qiagen, Inc., Valencia, CA,
USA) was used to isolate total RNA from 6×103/ml cells
according to the manufacturer's protocol. Subsequently, the DNA was
performed whole-genome detection by DNA methylation 450k BeadChips
(Illumina Inc., San Diego, CA, USA). The extracted genomic DNA was
processed by hydrosulfite transformation using EZ DNA Methylation
kit (Zymo Research Corp., Irvine, CA, USA). Bisulfite-converted
genomic DNA is amplified using locus-specific PCR primers flanking
an oligonucleotide probe with a 5′ fluorescent reporter dye (6FAM)
and a 3′ quencher dye (TAMRA) (18). Amplified DNA was cut into segments
by DNA restriction endonucleases at 37°C, and the DNA fragments
were precipitated by isopropanol. DNA pellet after centrifugation
at 12,000 g, at room temperature for 5 min was resuspended in
buffer RA1, and the resuspended DNA samples with concentration of
2×103 µg/µl were dispersed on BeadChip chips and
Illumina Human HT-12 V4.0 expression BeadChip (Illumina, Inc.) was
used for hybridization at 37°C (19). Arrays were scanned on the Illumina
iScan system, and raw data was imported and analyzed with the
BeadStudio software (version 3.1.3.0 Illumina, Inc). Prior to use,
Illumina data we reserved on the basis of the MIAME guidelines in
Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo/).
DNA bisulfite modification
A total of 1.5 µg DNA was denatured in 50 µl of 0.2
M NaOH at 37°C for 10 min. Then, 30 µl of freshly prepared 10 mM
hydroquinone (Sigma-Aldrich; Merck KGaA) and 520 µl of 3 M sodium
bisulfite (Sigma; Merck KGaA) at pH 5.0 were added and mixed. The
samples were overlaid with mineral oil to prevent evaporation and
incubated at 50°C for 16 h. The bisulfite-treated DNA was isolated
using Wizard DNA Clean-Up System (Promega; Thermo Fisher
Scientific., Inc.). The DNA was eluted by 50 µl of warm water and
5.5 µl of 3 M NaOH were added at 37°C for 5 min. The DNA was
ethanol precipitated with glycogen as a carrier and resuspended in
20 µl of water. Bisulfite-treated DNA was stored at −20°C until
further use.
Quantitative methylation-specific
polymerase chain reaction (QMSP) for detecting differential
methylation
The Multisource Genomic DNA Miniprep kit (Axygen
Scientific, Inc. Union City, CA, USA) was used to extract total DNA
from 60,000 cells/well, following the manufacturer's protocol. The
CpGenome Universal DNA Modification kit (Chemicon International,
Inc., Temecula, CA, USA) was used for DNA bisulfite modification.
To determine the methylation status of mutL homolog (MLH),
phosphatase and tensin homolog (PTEN), runt-related transcription
factor (RUNX), Ras association domain family (RASSF), cadherin 1
(CDH1), O-6-methylguanine-DNA methyltransferase (MGMT) and P16.
QMSP was performed in a TaqMan probe system using an Applied
Biosystems 7900HT Fast Real-Time PCR System in a total volume of 20
µl reaction mixture containing 2 µl of bisulfite template DNA, 250
nM of each primer (primer sequences of MLH, PTEN, RUNX, CDH1, MGMT
and P16 are in Table I), 225 nM
TaqMan probe, and 10 µl of FastStart Universal Probe Master (ROX;
Roche Diagnostics, Roche Applied Science, Mannheim, Germany).
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Primer Sequences
(5′-3′) |
|---|
| MLH |
|
| R |
ATGGCCTGAATGGAGCCCCAGGAGAGG |
| F |
TCCATTCAGGCCATCGCCTGTGCTGAG |
| PTEN |
|
| R |
TTTCATGGTGTTTTATCCCTC |
| F |
TTTCCTGCAGAAAGACTTGA |
| RUNX |
|
| R |
CCTGACGAAGTGCCATAGTAGA |
| F |
CCACCACTCACTACCACACCTA |
| RASSF |
|
| R |
TTTGTGAGAGTGTGTTTAGTTTTG |
| F |
CCCAATTAAACCCATACTTCA |
| CDH1 |
|
| R |
TCCCCAAAACGAAACTAACGAC |
| F |
AATTTTAGGTTAGAGGGTTATCGCGT |
| MGMT |
|
| R |
CAACATCACTAACACCTAACC |
| F |
CCTAATGTTGGGATAGTT |
| P16 |
|
| R |
ACCCGACCCCGAACCGCGACCGTAA |
| F |
TTATTAGAGGGTGGGGCGGATCGCGTCG |
Analysis of differential gene
transcription
Principal component analysis (PCA) was used to
analyze data obtained from QMSP in which the content contains some
inter-correlated quantitative dependent variables. Partek Genomics
Suite 6.5 (Partek, Inc., St Louis, MO, USA) was used to analyze the
gene expression data. The analyzed data were corrected and
normalized by quantile normalization and summarization. PCA was
used for global visualization of all data sets.
Functional enrichment and pathway
enrichment analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) online tool was used to conduct Gene
Ontology (GO; www.geneontology.org/) and KEGG (Kyoto Encyclopedia of
Genes and Genomes) pathway enrichment analysis (www.genome.jp/kegg/). DAVID is used to convert
collected data into biological meaning and contributes to the
explanation of data sets on a genome-wide scale. GO terms and KEGG
pathways of which P<0.1 were chosen as previously described
(20).
Statistical analysis
SPSS 18.0 (SPSS, Inc., Chicago, IL, USA) and
GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) were
used to analyze the data. A two-tailed t-test was used to
differentiate the mean methylation scores between two samples. A
paired t-test and one-way analysis of variance were applied to
determine the differences of average sib pair in methylation
scores. The network representation was generated using GeneGO
MetaCore software (version 4.3; www.genego.com/.metacore.php, GeneGo, Inc., Encinitas,
CA, USA). The Venn diagram is a graphic organizer constructed by
overlapping circles to indicate features common or unique to two or
more concepts (21). Methylation
diversity was obtained by Genomestudio software version 2010.1.
Results
Analysis of differential gene
transcription
Partek Genomics Suite 6.5 (Partek, Inc.) was used to
analyze the gene expression data. The data were subsequently
corrected and normalized by quantile normalization and
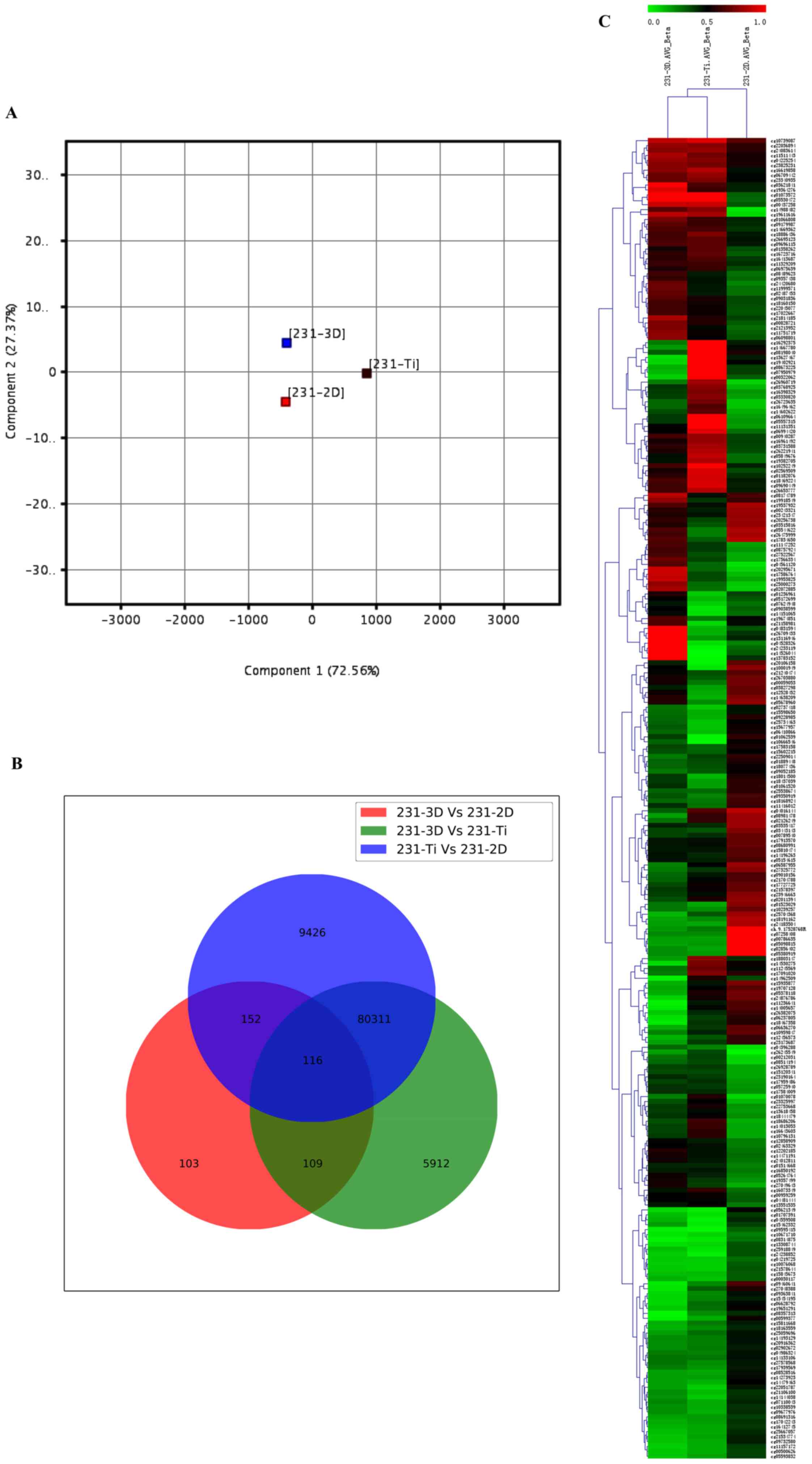
summarization. Cells were cultured at 2D, 3D and Ti substrate. PCA
was used for global visualization of all data, which revealed the
close connections between the 3D, 2D and Ti groups (Fig. 1A). Gene lists were established with
a P≤0.05. A Venn diagram of bisulfite modification DNA in 3D group,
2D group and Ti group was created (Fig. 1B). The results revealed that 116
genes were common among the 231-3D vs. 231-2D group, 231-3D vs.
231-Ti group and 231-Ti vs. 231-2D group. Other than the 116 common
genes, 152 genes were shared in the 231-3D vs. 231-2D group and the
231-Ti vs. 231-2D group, 109 genes were common in the 231-3D vs.
231-2D group and the 231-3D vs. 231-Ti group, and 80,311 genes were
common in the 231-3D vs. 231-Ti group and the 231-Ti vs. 231-2D
group. Unsupervised clustering analysis of the CpG location
indicated that 268 CpGs presented different levels of methylation
in the three groups of samples, and 116 CpG were highly methylated
in the three groups of samples (Fig.
1C).
Analysis on different CpG
location
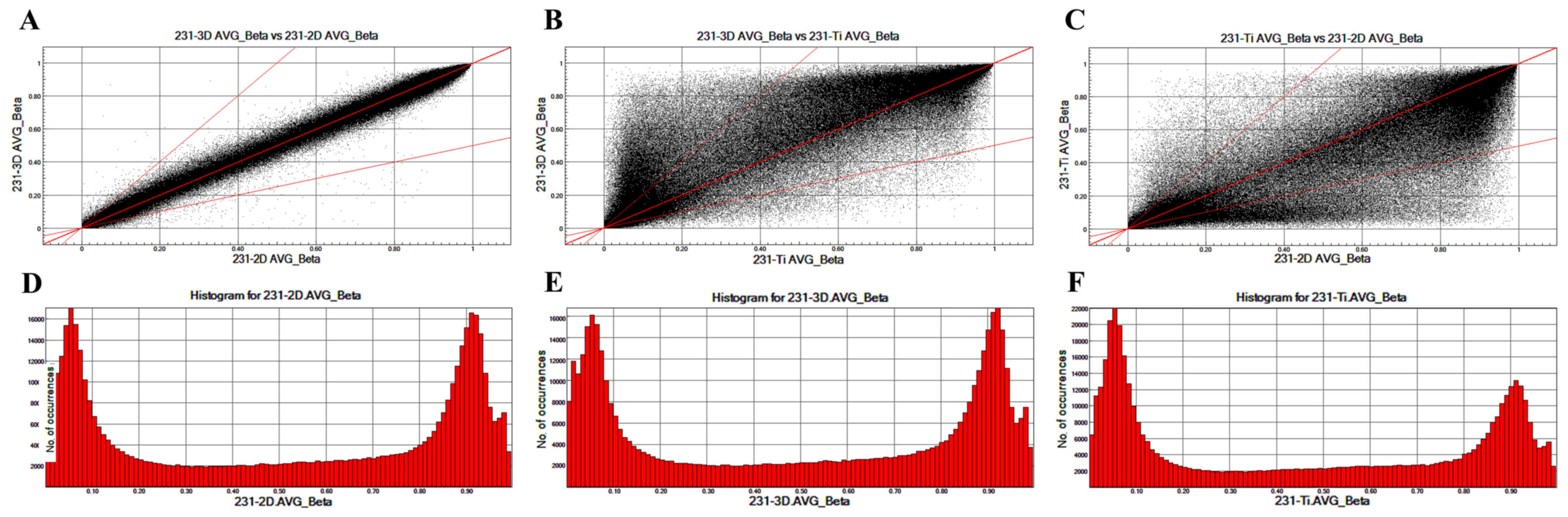
To analyze the different CpG locations in breast
cancer cells cultured in 2D, 3D and Ti substrates, scatter plots
were prepared that compared the CpG sites in 231-3D, 231-2D and
231-Ti group (Fig. 2). The
methylation patterns between 231-3D vs. 231-2D (Fig. 2A) are more similar compared with
those between 231-3D vs. 231-Ti (Fig.
2B) or 231-Ti vs. 231-2D (Fig.
2C). Fig. 2D-F displays the
column distribution of β-values in the three groups of samples.
Functional analysis of methylated
DNA
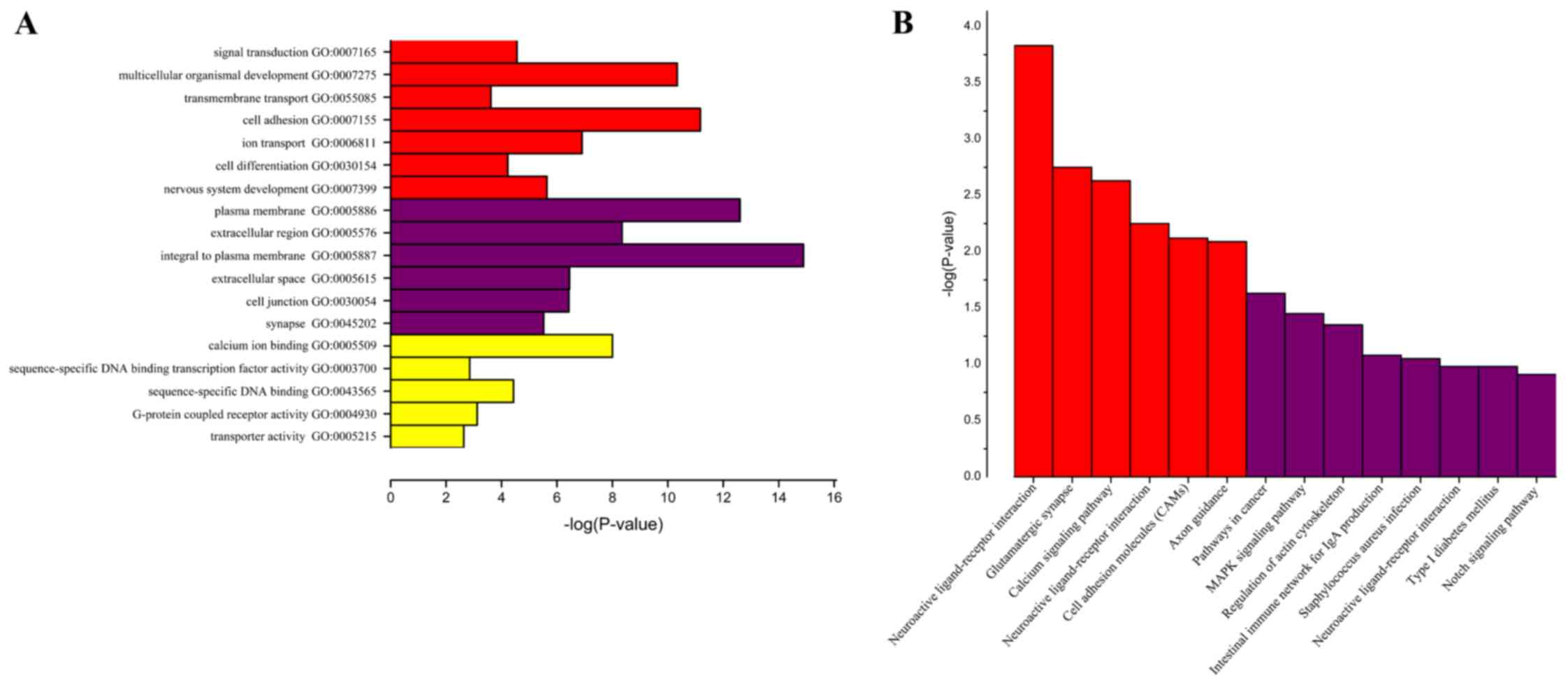
To identify the biological functions, cellular
components and molecular functions in which the identified genes
may serve a role, GO term analysis was performed. GO term analysis
identified numerous genes that were expressed differentially in
different cellular processes, including signal transduction,
transmembrane transport, cell differentiation, sequence-specific
DNA binding transcription factor activity, sequence-specific-DNA
binding, G-protein coupled receptor activity and transporter
activity (Fig. 3A). To further
refine the biological functions of genes corresponding to
differential methylation, KEGG enriched pathway analysis was used
to systematically analyze gene functions based on networks of genes
and molecules. Pathway analyses of the corresponding genes
identified 14 significantly over-represented cellular pathways
(Fig. 3B), of which 8 were more
significant, including pathways in cancer, mitogen-activated
protein kinase (MAPK) signaling pathway, regulation of actin
cytoskeleton, intestinal immune network for immunoglobulin (Ig)A
production, Staphylococcus aureus infection, neuroactive
ligand-receptor interaction, type 1 diabetes mellitus and Notch
signaling pathway. To determine whether the different cell culture
methods effected methylation of important genes, QMSP analysis was
used. To determine whether the different cell culture methods have
an effect on genomic DNA methylation, the methylation levels of
MLH, PTEN, RUNX, RASSF, CDH1, MGMT and P16 genes was investigated
in breast cancer cell lines MCF-7 cultured in 2D, 3D or Ti culture
pattern, respectively. By methylation analysis, methylation levels
of MLH, PTEN, RUNX, RASSF, CDH1, MGMT and P16 genes had no
significant difference between 2D, 3D and Ti culture pattern (data
not shown).
Discussion
DNA hypermethylation is reported to serve a role in
many cancers such as breast cancer. During the progression of
cancer, hypermethylation of CpG islands serves a key role in
silencing tumor regulatory genes (22). The observation of epigenetic
changes indicated that DNA hypermethylation is a key factor
influencing the progression of breast cancer (23). DNA hypermethylation often occurs in
cancer cells and specific sets of genes (24).
PCA is a very useful method to analyze data tables,
and data are expressed with some inter-correlated quantitative
dependent variables (25).
Previous studies have used PCA to extract information and to
display the pattern of similarity between the number of variables
and the number of observations in PCA maps (26). In the present study, PCA was used
to analyze the methylation differences of DNA in MCF-7 breast
cancer cells under 2D, 3D or Ti adhesive substrate culturing
conditions. The results indicated that the differentially
methylated DNA in the three groups was closely related with each
other. In addition, a total of 116 differentially methylated sites
were identified as commonly occurring in the 3 groups of samples,
and the common sections presented high rates of methylation.
Unsupervised clustering analysis was used to explore the
methylation status of CpGs. The results demonstrated that 268 CpGs
presented different levels of methylation in the three groups of
samples and 116 CpGs of which all appeared high methylation status.
Abnormal DNA methylation often occurs in CpG islands, and CpG
island shores serve a key role in harboring the changes of DNA
methylation (27). In breast
cancer, methylation of CpG island shores is associated with
clinical features (28). Scatter
plots comparing all CpG sites among the 231-3D, 231-2D and 231-Ti
groups were constructed to analyze the different CpG locations in
breast cancer cells cultured in 2D, 3D and Ti substrate. The
results revealed that the methylation pattern between 231-3D vs.
231-2D, 231-3D vs. 231-Ti and 231-Ti vs. 231-2D was no significant
different.
GO analysis results indicated that genes with
differential expression are involved in different cellular
processes, such as signal transduction, transmembrane transport,
cell differentiation, sequence-specific DNA binding transcription
factor activity, sequence-specific-DNA binding, G-protein coupled
receptor activity and transporter activity. Abnormalities in PTEN,
k-RAS, or β-catenin genes can alter several different signal
transduction pathways (29). RASSF
play an important role in transmembrane transport. They are Ras
effectors and are transported into the nucleus by classical nuclear
transport pathways (30). CDH1 is
involved in cell differentiation and has a capacity to control cell
fate by altering directional cell proliferation and apoptosis
(31). KEGG results demonstrated
that genes were enriched in 14 pathways including pathways in
cancer, MAPK signaling pathway, Regulation of actin cytoskeleton,
Intestinal immune network for IgA production, Staphylococcus
aureus infection, Neuroactive ligand-receptor interaction, Type
1 diabetes mellitus and Notch signaling pathway. Therefore, the
role of different culture methods on the methylated level of MLH,
PTEN, RUNX, RASSF, CDH1, MGMT and P16 gene was investigated, via
QMSP and demonstrated that the was no significant difference
between 2D, 3D and Ti culture pattern. PTEN suppresses tumor
development and metastasis, and is mutated in many cancers
(32). Abnormal expression of PTEN
was observed in many tumors (33).
In breast cancer, PTEN inhibits cell growth and induces apoptosis
(34). RUNX genes have attracted
increasing attention, owing to their roles in suppressing or
promoting tumors (35). In many
key pathways, RUNX may regulate lineage-specific gene expression
(27,36). RUNX family members may serve a
broader role in multistep breast tumorigenesis (37). In the progression of many cancers,
RASSF proteins are often downregulated and may suppress tumor
development and metastasis (38).
RASSF gene is often silenced by promoter methylation (39). The present study demonstrated that
methylation of MLH, PTEN, RUNX, RASSF, CDH1, MGMT and P16 genes had
no difference under 2D, 3D or Ti culture conditions.
In conclusion, changes in methylation status may be
associated with the occurrence and metastasis of breast cancer.
Growth environment of breast cancer cells have no influence on
methylation status of MLH, PTEN, RUNX, RASSF, CDH1, MGMT and P16
genes. The present findings may shed light to treating breast
cancer by identifying known and novel gene targets.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van't Veer LJ, Dai H, van de Vijver MJ, He
YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ,
Witteveen AT, et al: Gene expression profiling predicts clinical
outcome of breast cancer. Nature. 415:530–536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li S, Rong M and Iacopetta B: DNA
hypermethylation in breast cancer and its association with
clinicopathological features. Cancer Lett. 237:272–280. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yegnasubramanian S, Haffner MC, Zhang Y,
Gurel B, Cornish TC, Wu Z, Irizarry RA, Morgan J, Hicks J, DeWeese
TL, et al: DNA hypomethylation arises later in prostate cancer
progression than CpG island hypermethylation and contributes to
metastatic tumor heterogeneity. Cancer Res. 68:8954–8967. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ji H, Ehrlich LI, Seita J, Murakami P, Doi
A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al:
Comprehensive methylome map of lineage commitment from
haematopoietic progenitors. Nature. 467:338–342. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Doi A, Park IH, Wen B, Murakami P, Aryee
MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, et al:
Differential methylation of tissue- and cancer-specific CpG island
shores distinguishes human induced pluripotent stem cells,
embryonic stem cells and fibroblasts. Nat Genet. 41:1350–1353.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Portela A and Esteller M: Epigenetic
modifications and human disease. Nat Biotechnol. 28:1057–1068.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsai HC and Baylin SB: Cancer epigenetics:
Linking basic biology to clinical medicine. Cell Res. 21:502–517.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fackler MJ, McVeigh M, Evron E, Garrett E,
Mehrotra J, Polyak K, Sukumar S and Argani P: DNA methylation of
RASSF1A, HIN-1, RAR-beta, Cyclin D2 and Twist in in situ and
invasive lobular breast carcinoma. Int J Cancer. 107:970–975. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dimas AS, Deutsch S, Stranger BE,
Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C,
Gutierrez Arcelus M, Sekowska M, et al: Common regulatory variation
impacts gene expression in a cell type-dependent manner. Science.
325:1246–1250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murrell A, Heeson S, Cooper WN, Douglas E,
Apostolidou S, Moore GE, Maher ER and Reik W: An association
between variants in the IGF2 gene and Beckwith-Wiedemann syndrome:
Interaction between genotype and epigenotype. Hum Mol Genet.
13:247–255. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kerkel K, Spadola A, Yuan E, Kosek J,
Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, et al: Genomic
surveys by methylation-sensitive SNP analysis identify
sequence-dependent allele-specific DNA methylation. Nat Genet.
40:904–908. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li XJ, Valadez AV, Zuo P and Nie Z:
Microfluidic 3D cell culture: Potential application for
tissue-based bioassays. Bioanalysis. 4:1509–1525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baharvand H, Hashemi SM, Kazemi Ashtiani S
and Farrokhi A: Differentiation of human embryonic stem cells into
hepatocytes in 2D and 3D culture systems in vitro. Int J Dev Biol.
50:645–652. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tibbitt MW and Anseth KS: Hydrogels as
extracellular matrix mimics for 3D cell culture. Biotechnol Bioeng.
103:655–663. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu M, Huang S, Yu KJ and Clyne AM: Dextran
and polymer polyethylene glycol (PEG) coating reduce both 5 and 30
nm iron oxide nanoparticle cytotoxicity in 2D and 3D cell culture.
Int J Mol Sci. 13:5554–5570. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weigelt B, Ghajar CM and Bissell MJ: The
need for complex 3D culture models to unravel novel pathways and
identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev.
69–70:42–51. 2014. View Article : Google Scholar
|
|
18
|
Hájková H, Fritz MH, Haškovec C, Schwarz
J, Šálek C, Marková J, Krejčík Z, Dostálová Merkerová M, Kostečka
A, Vostrý M, et al: CBFB-MYH11 hypomethylation signature and PBX3
differential methylation revealed by targeted bisulfite sequencing
in patients with acute myeloid leukemia. J Hematol Oncol. 7:662014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu X, Jia X, Yuan H, Ma K, Chen Y, Jin Y,
Deng M, Pan W, Chen S, Chen Z, et al: DNA methyltransferase 1
functions through C/ebpa to maintain hematopoietic stem and
progenitor cells in zebrafish. J Hematol Oncol. 8:152015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma CH, Lv Q, Cao Y, Wang Q, Zhou XK, Ye BW
and Yi CQ: Genes relevant with osteoarthritis by comparison gene
expression profiles of synovial membrane of osteoarthritis patients
at different stages. Eur Rev Med Pharmacol Sci. 18:431–439.
2014.PubMed/NCBI
|
|
21
|
Chen H and Boutros PC: VennDiagram: A
package for the generation of highly-customizable Venn and Euler
diagrams in R. BMC Bioinformatics. 12:352011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lewis CM, Cler LR, Bu DW, Zöchbauer-Müller
S, Milchgrub S, Naftalis EZ, Leitch AM, Minna JD and Euhus DM:
Promoter hypermethylation in benign breast epithelium in relation
to predicted breast cancer risk. Clin Cancer Res. 11:166–172.
2005.PubMed/NCBI
|
|
24
|
Stirzaker C, Song JZ, Davidson B and Clark
SJ: Transcriptional gene silencing promotes DNA hypermethylation
through a sequential change in chromatin modifications in cancer
cells. Cancer Res. 64:3871–3877. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abdi H and Williams LJ: Principal
component analysis. Wiley Interdiscip Rev Comput Stat. 2:433–459.
2010. View
Article : Google Scholar
|
|
26
|
Bro R and Smilde AK: Principal component
analysis. Anal Methods. 6:2812–2831. 2014. View Article : Google Scholar
|
|
27
|
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z,
Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al:
The human colon cancer methylome shows similar hypo- and
hypermethylation at conserved tissue-specific CpG island shores.
Nat Genet. 41:178–186. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Farkas SA, Milutin-Gašperov N, Grce M and
Nilsson TK: Genome-wide DNA methylation assay reveals novel
candidate biomarker genes in cervical cancer. Epigenetics.
8:1213–1225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matias-Guiu X, Catasus L, Bussaglia E,
Lagarda H, Garcia A, Pons C, Muñoz J, Argüelles R, Machin P and
Prat J: Molecular pathology of endometrial hyperplasia and
carcinoma. Hum Pathol. 32:569–577. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumari G, Singhal PK, Rao MR and
Mahalingam S: Nuclear transport of Ras-associated tumor suppressor
proteins: Different transport receptor binding specificities for
arginine-rich nuclear targeting signals. J Mol Biol. 367:1294–1311.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reardon SN, King ML, MacLean JA II, Mann
JL, DeMayo FJ, Lydon JP and Hayashi K: CDH1 is essential for
endometrial differentiation, gland development, and adult function
in the mouse uterus. Biol Reprod. 86(141): 1–10. 2012. View Article : Google Scholar
|
|
32
|
Simpson L and Parsons R: PTEN: Life as a
tumor suppressor. Exp Cell Res. 264:29–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maehama T and Dixon JE: PTEN: A tumour
suppressor that functions as a phospholipid phosphatase. Trends
Cell Biol. 9:125–128. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stemke-Hale K, Gonzalez-Angulo AM, Lluch
A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, et
al: An integrative genomic and proteomic analysis of PIK3CA, PTEN
and AKT mutations in breast cancer. Cancer Res. 68:6084–6091. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Blyth K, Cameron ER and Neil JC: The RUNX
genes: Gain or loss of function in cancer. Nat Rev Cancer.
5:376–387. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Levanon D and Groner Y: Structure and
regulated expression of mammalian RUNX genes. Oncogene.
23:4211–4219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lau QC, Raja E, Salto-Tellez M, Liu Q, Ito
K, Inoue M, Putti TC, Loh M, Ko TK, Huang C, et al: RUNX3 is
frequently inactivated by dual mechanisms of protein
mislocalization and promoter hypermethylation in breast cancer.
Cancer Res. 66:6512–6520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Allen NP, Donninger H, Vos MD, Eckfeld K,
Hesson L, Gordon L, Birrer MJ, Latif F and Clark GJ: RASSF6 is a
novel member of the RASSF family of tumor suppressors. Oncogene.
26:6203–6211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Djos A, Martinsson T, Kogner P and Carén
H: The RASSF gene family members RASSF5, RASSF6 and RASSF7 show
frequent DNA methylation in neuroblastoma. Mol Cancer. 11:402012.
View Article : Google Scholar : PubMed/NCBI
|