Introduction
Traumatic brain injury (TBI) has been reported to
induce the release of arachidonic acid (AA) from cell membranes
(1). AA levels have been indicated
to be increased by 1,093% within 48 h following TBI and to remain
elevated for several days (2).
Cytochrome P450 (CYP) enzymes, particularly CYP4A isozymes,
catalyze the ω-hydroxylation of AA to 20-hydroxyeicosatetraenoic
acid (20-HETE) (3). Previous
studies have suggested that 20-HETE may participate in various
physiological and pathological processes, including the regulation
of vascular tone, cerebral blood flow, cellular proliferation and
inflammation; 20-HETE has been identified as a potent
vasoconstrictor in cerebral vasculature and it has been reported to
suppress inflammatory reactions (4–6).
However, contradictory results have indicated that 20-HETE
increased the vascular production of reactive oxygen species (ROS)
and promoted the activation of nuclear factor-κB in cerebrovascular
endothelial cells, and consequently deteriorated cerebral
inflammation (7).
TBI consists of two stages of pathophysiological
injury: The primary stage, characterized by brain contusion,
parenchymal hemorrhage, subarachnoid hemorrhage and diffuse axonal
injury; and the secondary stage, characterized by edema,
herniation, ischemia and infarction (8). Secondary injury mechanisms have been
reported to contribute to ongoing injury, during which ROS
production is elevated. ROS have been implicated in the pathology
of acute and chronic brain injuries, as they have been demonstrated
to disrupt the blood brain barrier (BBB) and increase cerebral
vascular permeability, thus leading to the development of brain
edema (8).
Matrix metalloproteinase (MMP)-9 has been reported
to serve critical roles during tissue morphogenesis and repair, in
cell death, and it has been associated with the prognosis of
neurological diseases (9,10). In animal and human TBI studies,
MMP-9 has been demonstrated to contribute to the pathophysiology of
brain edema, and its overexpression was associated with an increase
in BBB permeability through the degradation of tight junction
proteins, such as occludin and zonula occludens (ZO)-1 (11–13).
MMP-9 has been reported to be activated by ROS, in addition to by
the c-Jun N-terminal kinase (JNK) intracellular signaling pathway
during BBB breakdown (13,14), whereas JNK inhibition was
identified to protect BBB integrity, via suppression of MMP-9
activation (15). Therefore, it
may be hypothesized that 20-HETE can increase the production of ROS
and, through the activation of the JNK signaling pathway, increase
the expression of MMP-9 and promote the degradation of occludin and
ZO-1, eventually resulting in BBB dysfunction. The present study
investigated the roles of 20-HETE in BBB dysfunction and brain
edema development, following the experimental induction of TBI, and
explored the molecular mechanisms underlying these events.
Materials and methods
Animals and materials
Male Sprague-Dawley (SD) rats (n=89; age, 4–6 weeks;
weight, 180–260 g) were obtained from Shanghai SLAC Laboratory
Animal Co., Ltd. (Shanghai, China). Rats were housed under 12/12 h
light/dark cycles at 22±1°C, a relative humidity of 40–70% and
given food and water ad libitum. The present study was
approved by the Institutional Animal Care and Use Committee of
Shanghai Jiao Tong University School of Biomedical Engineering
(Shanghai, China).
Antibodies against MMP-9 (cat. no. ab119906),
occludin (cat. no. ab31721) and ZO-1 (cat. no. ab190085) were
purchased from Abcam (Cambridge, UK), and antibodies against rat
polyclonal anti-phosphorylated (p)-JNK (cat. no. 9252S) and rat
polyclonal anti-p-c-Jun antibodies (cat. no. 2315S) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Animal model
SD rats were randomly divided into 2 groups: TBI and
sham groups (n=5 rats/group). Rats in the TBI group were
anesthetized with ketamine (75 mg/kg) and xylazine (10 mg/kg;
Shanghai Rantai Biological Company, Shanghai, China), and mounted
on a stereotaxic frame (Stoelting Co., Wood Dale, IL, USA). A 15
mm-long midline scalp incision was made and a craniotomy (6 mm in
diameter) was then performed over the central aspect of the right
parietal cortex, 2 mm lateral to the sagittal suture. Care was
taken to keep the dura intact following exposure. A controlled
cortical injury model of TBI was then established, using an impact
device (PinPoint Precision Cortical Impactor PCI 3000; Hatteras
Instruments, Inc., Cary, NC, USA) with a 2.5-mm rounded steel
impactor tip. A moderate injury was inflicted in the right parietal
cortex, using a deformation depth of 2.5 mm, a velocity of 1.5
m/sec and a duration time of 85 msec, and the incision was closed.
Following recovery of ~1 h from the injury, the rats were returned
to their cages and allowed free access to food and water. Rats in
the sham group underwent surgery, similar to the TBI rats, however
received no injury. Rats were maintained for 3, 24 and 72 h,
following the induction of TBI; these rats formed the TBI groups
(n=5 rats/group). In addition, rats were excluded from the study if
dural integrity was compromised. To obtain rat brains, the foramen
magnum was first transected with scissors. The parietal bone was
carefully opened through the foramen magnum and parietal hemostatic
forceps were inserted in order to break both sides of the parietal
bone with oblique insertion scissors. The olfactory bulb on the
parietal was also carefully removed, and one side of the optic
nerve was cut in order to probe into the base of the skull; the
whole brain could then be tilted and removed. Rat brains were fixed
in the 4% paraformaldehyde for 24 h at 4°C, then embedded in
paraffin and sliced into 10 mm sections.
BBB permeability
BBB permeability was investigated at 3, 24 and 72 h
following TBI by dynamic contrast-enhanced magnetic resonance
imaging (DCE-MRI) (8). Rats
underwent MRI using a clinical 3.0 T MRI scanner with an
eight-channel head coil (Intera-achieva SMI-2.1; Philips
Healthcare, DA Best, The Netherlands). MRI pulse sequences were as
follows: T2 weighted (T2W) image, T2 map, post-T1 weighted image
and DCE-MRI. The image processing software (Cine Tool; GE
Healthcare Life Sciences, Little Chalfont, UK) was used to obtain
the quantitative parameter volume transfer constant
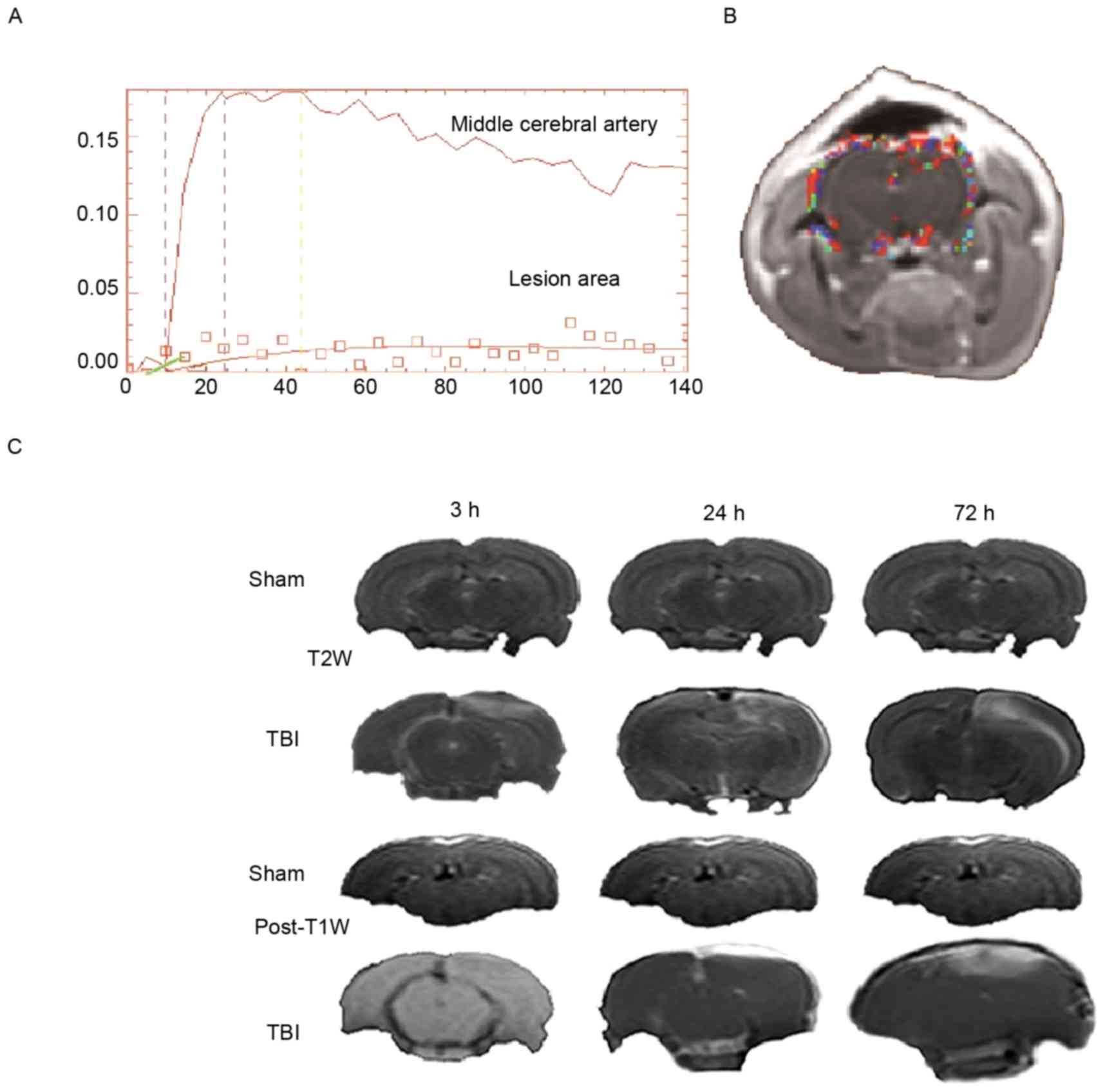
(Ktrans). Ktrans was calculated based on a
modified two compartment model (Fig.
1A), where all of the contrast media in the tissue were
primarily located in the two chambers, namely, the intravascular
space and the extravascular space. In this model, Ktrans
was calculated by measuring the accumulation of
gadolinium-diethylenetriaminepentacetate in the
extravascular-extracellular space. Ktrans was calculated
by manually outlining the regions of interest (ROIs). A total of 3
ROIs at all time points were manually drawn in a 2-mm single slice
T2W scan, as presented in Fig. 1B
(obtained at 72 h). ROIs for the sham and TBI groups included the
focal lesion and the contralateral brain areas. The hyperintensity
areas of T2W images were manually outlined (Fig. 1C) following TBI, using the signal
intensity difference of T2W (the difference threshold was 300) to
define the border between the hyperintensity area and
healthy-looking tissue. Additional T2 maps (scaling of 0–150 ms)
were used to define the border between the hyperintensity area and
healthy-looking tissue when the border was ill-defined in T2W
images. The focal lesion area refers to the outlined hyperintensity
area which may include the cortex, hippocampal tissues, etc. This
procedure was performed by two radiologists.
Brain edema
Brain edema was assessed via measuring the tissue
water content, as previously described (8). Briefly, The SD rats were subjected to
deep anesthesia. After the diaphragm was cut off, the chest wall
was cut along the anterior axillary line. The heart was exposed
after the chest wall was turned up, and a syringe was inserted via
the left ventricle in the ascending aorta, the insertion depth is
~1 mm, and then the brain was decapitated. The wet weight of a 3-mm
coronal tissue section of the ipsilateral cortex, centered on the
impact site, was measured immediately following sacrifice at 3, 24,
72 h and 7 days post-TBI. Tissue samples were then dried in an oven
at 100°C for 48 h to obtain the dry weight. Tissue water content
was calculated using the following formula: Water content = [(wet
weight - dry weight) / wet weight] × 100%.
Oxidative stress indices
The levels of malondialdehyde (MDA; cat. no. A003-1)
and superoxide dismutase (SOD; cat. no. A001-3), and the total
antioxidative capacity (T-AOC; cat. no. A015) were measured using
commercially available kits (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). Briefly, following TBI (3, 24 and 72 h,
and 7 days post-TBI), the left-brain hemispheres were removed and
homogenized in ice-cold PBS. The brain tissue was poured into the
glass homogenate tube, and the remaining 1/3 homogenate medium or
saline was also added to the glass homogenate tube. The tube in the
left hand was inserted into the homogenate containing the mixture
of water containers. The rod in the right hand was vertically
inserted into the casing for dozens of times (6–8 min) upper and
lower to make the tissue homogenate. The samples were centrifuged
at 2,000 × g for 10 min at 4°C and the supernatants were used to
measure MDA, SOD and T-AOC, according to the manufacturer's
protocol.
Liquid chromatography-mass
spectrometry (LC-MS)
SD rats were anesthetized with ketamine (75 mg/kg)
and xylazine (10 mg/kg). Brain tissue samples from the area of
injury were obtained and frozen immediately at −80°C. The brain
samples were homogenized as mentioned above on ice and 100 g
homogenate were used for proteomic screening. Deuterated
20-HETE-d6 (2 µl) (SCIEX, Framington, MA, USA) was added
to the homogenates as an internal standard. Subsequently, the
samples (each sample was the homogenate from the brain of one rat
with PBS) were injected into the LC-MS (TripleTOF®
5600+; SCIEX) following evaporation and reconstitution in the
mobile phase with buffer A (0.1% formic acid in water) and buffer B
(98% acetonitrile and 0.1% formic acid in water). The samples were
quantified by LC-MS (curtain gas: 44 Psi; collision gas: 12 Psi;
ionspray voltage: 5,000 V; nebulizer current: 6 A; temperature:
55°C; ion source gas1: 90 Psi; ion source gas2: 90 Psi) and the
levels of 20-HETE were measured (Mass Frontier Software v5.0;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) in each
sample.
Western blot analysis
Brains were removed and the 2 hemispheres were
separated. The lesion areas were homogenized in a buffer containing
Tris 50 mM (pH=7.4), NaCl 150 mM, 1% Triton X-100, EDTA 1 mM, and
phenylmethylsulfonyl fluoride 2 mM. The homogenates were
centrifuged at 500 × g for 15 min at 4°C, and the supernatants were
collected. Protein concentration in the supernatants was determined
using a bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.), with bovine serum albumin as the standard. Equal
amounts of extracted protein samples (50 µg) were separated by 12%
SDS-PAGE and transferred onto nitrocellulose membranes, which were
then blocked with 5% nonfat milk for 2 h at room temperature.
Membranes were probed (4°C overnight) with primary antibodies
against occludin, ZO-1, MMP-9, p-JNK and p-c-Jun (1:400). They were
then incubated at 37°C for 1 h with horseradish
peroxidase-conjugated secondary antibodies (cat. nos. 140253,
33725, 3007, 6254 and 6093; Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA). Protein bands were visualized using an
enhanced chemiluminescence detection system (Amersham; GE
Healthcare Life Sciences, Little Chalfont, UK). Blots were
semi-quantified using Multi Gauge software (ias3000, Fuji, Tokyo,
Japan) and the results were normalized to those of β-actin (cat.
no. ab8226; 1:1,000; Abcam), which was used as the loading
control.
Statistical analysis
All analyses were performed with SPSS v17.0 software
(SPSS, Inc., Chicago, IL, USA). Normality testing was performed
using the Shapiro-Wilk test and equality of variance was assessed
using Levene's test. The statistical significance of the
differences between groups was assessed using two-way analysis of
variance with a Bonferroni post hoc test. Data are expressed as the
mean ± standard deviation of 3 repeated experiments. P<0.05 was
considered to indicate a statistically significant difference.
Results
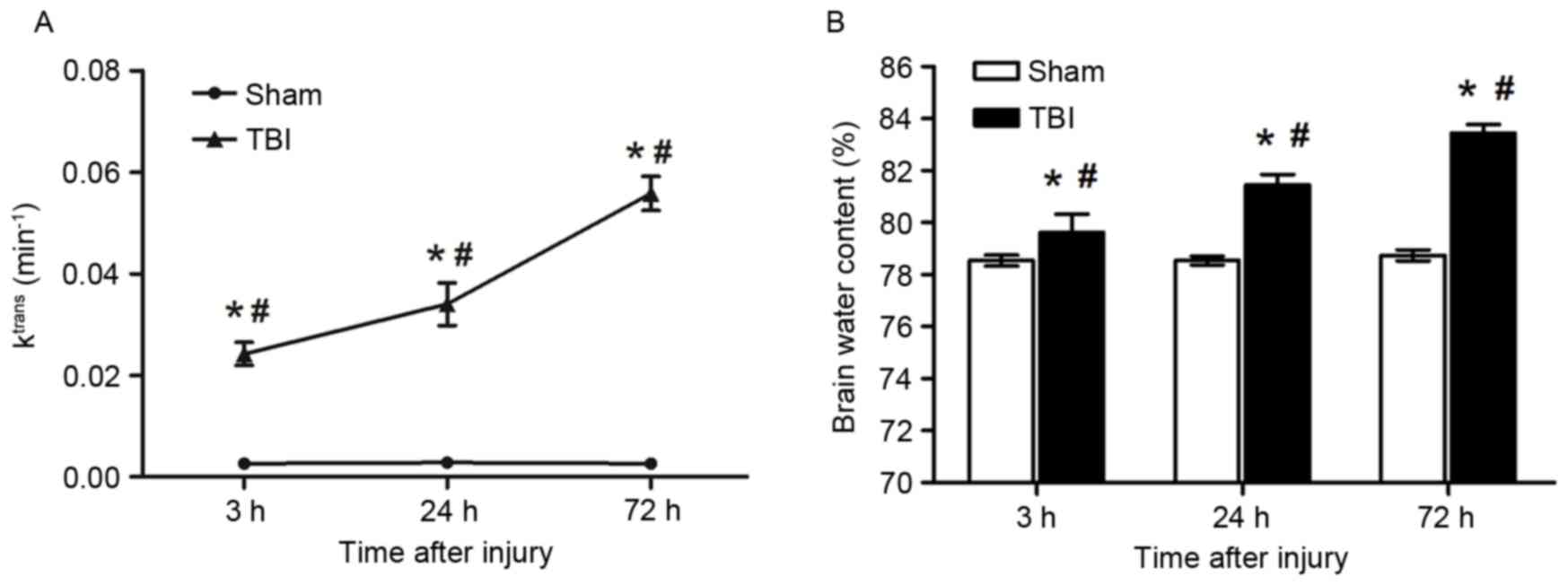
Alterations in Ktrans
An immediate and sustained increase in
Ktrans post-injury was observed among rats in the TBI
group compared with the sham group. As demonstrated in Fig. 2A, Ktrans
(min−1) was identified to be significantly increased at
3 (0.024±0.002 vs. 0.003±0.001 min−1, P<0.01), 24
(0.034±0.004 vs. 0.003±0.001 min−1, P<0.01) and 72 h
(0.056±0.003 vs. 0.003±0.003 min−1, P<0.05) following
the induction of TBI compared with the sham group. In addition, the
increase in Ktrans appeared to be time-dependent, as
Ktrans values were significantly different between the
various time points within the TBI group (P<0.05; Fig. 2A). The increase of
Ktrans suggests the extent of the disruption of the
BBB.
Brain edema
Brain water content was demonstrated to be increased
as early as 3 h following injury compared with the sham group
(79.62±0.70 vs. 78.54±0.21%, P<0.05), thus indicating the extent
of brain edema. The significant increase in brain water was
sustained at 24 (81.44±0.40 vs. 78.54±0.21%, P<0.05) and 72 h
(83.44±0.33 vs. 78.74±0.21%, P<0.05) following the induction of
TBI compared with the sham group (Fig.
2B). In addition, brain water contents among rats in the TBI
group were identified to be significantly increased 24 and 72 h
post-injury compared with at 3 h post-injury (P<0.05 vs. 3 h
post-TBI; Fig. 2B).
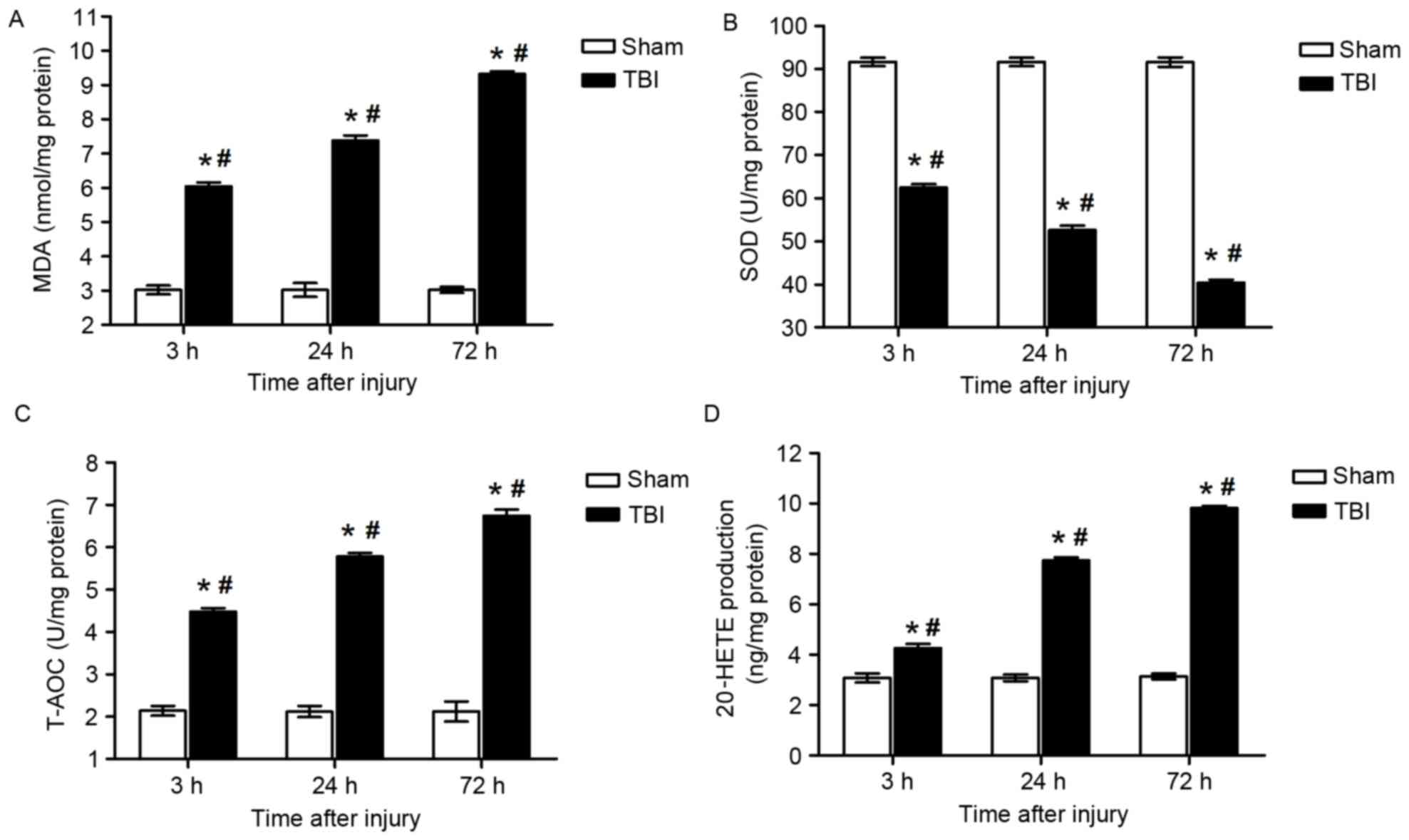
Oxidative stress indices
MDA levels in brain tissue samples from the lesion
area were demonstrated to be increased at 3 (6.04±0.11 vs.
3.02±0.13 nmol/mg, P<0.05), 24 (7.38±0.15 vs. 3.02±0.20 nmol/mg,
P<0.05) and 72 h (9.32±0.08 vs. 3.02±0.08 nmol/mg, P<0.05)
post-injury compared with the sham group (Fig. 3A). Notably, MDA levels were
identified to be significantly different between the various time
points within the TBI group (P<0.05; Fig. 3A), thus suggesting an increase in
ROS production following TBI.
The present results demonstrated that SOD activity
in the lesion area was significantly suppressed 3 (62.44±0.84 vs.
91.68±0.99 U/mg, P<0.01), 24 (52.62±1.01 vs. 91.67±0.98 U/mg,
P<0.05) and 72 h (40.43±0.72 vs. 91.64±1.09 U/mg, P<0.05)
following the induction of TBI compared with the sham group
(Fig. 3B). Similarly, the decrease
in SOD activity was also identified to be time-dependent within
rats in the TBI group (P<0.05; Fig.
3B).
Brain tissue from the lesion area also exhibited
reduced T-AOC 3 (6.74±0.15 vs. 2.14±0.11 U/mg, P<0.05), 24
(5.78±0.08 vs. 2.12±0.13 U/mg, P<0.05) and 72 h (4.48±0.08 vs.
2.12±0.24 U/mg, P<0.05) following TBI compared with the sham
group (Fig. 3C). Similarly, the
decrease in T-AOC was also identified to be time-dependent within
rats in the TBI group (P<0.05; Fig.
3C).
20-HETE production
LC-MS analysis demonstrated that the levels of
20-HETE were significantly upregulated in brain samples isolated
from the area of injury at 3 (4.26±0.17 vs. 3.08±0.18 ng/mg,
P<0.05), 24 (7.74±0.11 vs. 3.08±0.13 ng/mg, P<0.05) and 72 h
(9.82±0.08 vs. 3.14±0.12 ng/mg, P<0.05) post-TBI compared with
the sham group (Fig. 3D).
Furthermore, the increase in 20-HETE production was also identified
to be time-dependent within rats in the TBI group (P<0.05;
Fig. 3D). The increase of 20-HETE
suggests that the increase of the metabolite of AA following
TBI.
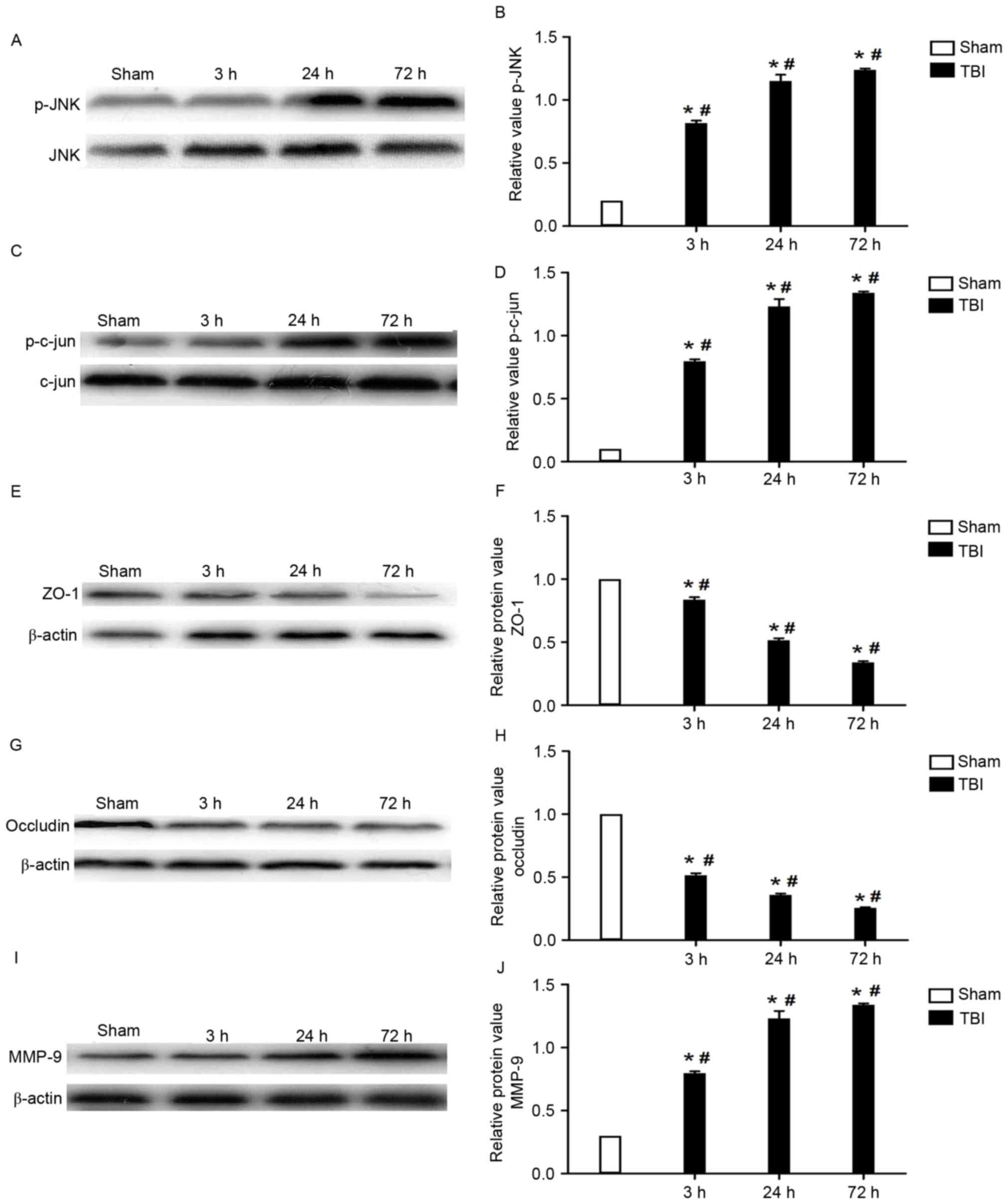
Expression of JNK, c-Jun, ZO-1,
occludin and MMP-9
The protein expression levels of JNK, c-Jun, ZO-1,
occludin and MMP-9 in brain tissue samples isolated from the area
of injury were assessed using western blot analysis. The present
results demonstrated that the protein expression levels of p-JNK
and p-c-Jun were significantly increased following the induction of
TBI compared with the sham group (P<0.05), and the increase
appeared to be time-dependent (Fig.
4A-D). Conversely, the expression of the tight junction
proteins ZO-1 and occludin was observed to be significantly
downregulated in TBI rats compared with that of rats from the sham
group (P<0.05). Similarly, the decrease in ZO-1 and occludin
protein expression levels appeared to be time-dependent (Fig. 4E-H). As presented in Fig. 4I and J, the protein expression
levels of MMP-9 were significantly upregulated, in a time-dependent
manner, following the induction of TBI compared with the sham group
(P<0.05).
Discussion
In the present study, brain water content was
observed to be increased as early as 3 h following the induction of
TBI, in accordance with previous reports demonstrating the
development of brain edema 3 h following the onset of injury
(8,16). Notably, the present study
demonstrated that 20-HETE levels were significantly increased 3, 24
and 72 h following the induction of TBI compared with the sham
group, thus suggesting that 20-HETE may contribute to the
post-traumatic development of brain edema. These results suggested
that 20-HETE may compromise the integrity and enhance the
permeability of the BBB, thus participating in the development of
brain edema following TBI.
Traumatic brain edema has been reported to consist
of cytotoxic and vasogenic edema (8). Cytotoxic edema has been suggested to
occur within 30 min following the brain insult, and has been
attributed to the activation of inflammatory processes, the
increase in oxidative stress and the enzymatic degradation of the
extracellular matrix in the BBB (16,17).
Conversely, vasogenic edema has been reported to appear several h
following the onset of TBI (16,17).
The results of the present study suggested that 20-HETE may
influence the brain water contents during the initial 3 h following
injury, probably through the potentiation of pathways contributing
to the development of cytotoxic edema at the early post-injury
phase.
To further investigate the molecular mechanisms
underlying the implication of 20-HETE in the development of brain
edema, ROS generation was assessed post-TBI. The present results
demonstrated that ROS production was significantly increased
following the induction of TBI compared with the sham group, and
suggested that the aberrant ROS generation may contribute to the
dysregulation of the BBB. Furthermore, these observations suggested
that the BBB-compromising effects of 20-HETE during the development
of brain edema may be mediated by an increase in oxidative
stress.
Previous studies have suggested a strong association
between oxidative stress and MMP-9 in the pathophysiology of BBB
damage following TBI (18,19), as oxidative stress has been
reported to trigger molecular cascades that mediate MMP-9
activation (20). MMP-9 activation
can lead to the degradation of tight junction proteins, such as
occludin and ZO-1, and thus increase BBB permeability following
injury (21). The present study
identified that the expression of MMP-9 was enhanced following the
induction of TBI, in accordance with previous reports (20,21).
In addition, the expression of the tight junction proteins occludin
and ZO-1 was significantly downregulated following TBI. These
results suggested that 20-HETE may modulate the expression of MMP-9
and subsequently of BBB tight junction proteins, and thus
contribute to the development of brain edema post-TBI.
The JNK signaling pathway has been implicated in
neuronal injury triggered by TBI-induced increases in oxidative
stress (22,23), whereas the inhibition of JNK has
been identified to prevent BBB disruptions, via inhibiting the
activation of MMP-9 (15). These
observations suggested that the JNK pathway may serve an important
role in the regulation of tight junction proteins and the
maintenance of BBB integrity. The present study demonstrated that
the activation of JNK and its downstream transcription factor c-Jun
was enhanced following TBI, thus suggesting that 20-HETE may
regulate MMP-9 expression through the modulation of the JNK
intracellular signaling pathway. Yu et al (24) reported that 20-HETE upregulated the
expression of MMP-9 through phosphatidylinositol-4,5-bisphosphate
3-kinase and extracellular signal-regulated kinase-mediated
signaling pathways in human non-small cell lung cancer cells, thus
suggesting an association between 20-HETE and MMP-9. However, the
molecular mechanisms underlying the regulatory effects of 20-HETE
on MMP-9 expression may differ across species and cell types. The
results of the current study suggested that increased ROS
generation may be implicated in the mechanisms underlying the
effects of 20-HETE on MMP-9; however, Yu et al (24) reported that the effects of 20-HETE
on MMP-9 appeared to be independent of ROS. Fordsmann et al
(25) demonstrated that the
20-HETE inhibitor HET0016 ameliorated the reduction in cerebral
blood flow in a rat brain injury model. Therefore, the mechanisms
underlying the 20-HETE-mediated BBB impairment and brain edema
development may also involve the disruption of cerebral blood
flow.
In conclusion, the present results suggested that
20-HETE may be involved in the compromise of BBB integrity and the
development of brain edema following TBI. In addition, the present
study suggested that the molecular mechanisms underlying the
implication of 20-HETE on BBB dysfunction may involve an increase
in oxidative stress and in the expression of MMP-9, thus
contributing to the degradation of tight junction proteins,
possibly through the activation of the JNK signaling pathway. Thus,
based on this study, the inhibition of 20-HETE may be used in
future experiments and another mechanism and treatment choice may
be explored in clinical experiments.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81271540 and
81301213).
References
|
1
|
Birnie M, Morrison R, Camara R and Strauss
KI: Temporal changes of cytochrome P450 (Cyp) and
eicosanoid-related gene expression in the rat brain after traumatic
brain injury. BMC Genomics. 14:3032013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang S, Ma Y, Liu Y, Que H, Zhu C and Liu
S: Arachidonic acid: A bridge between traumatic brain injury and
fracture healing. J Neurotrauma. 29:2696–2705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnson AL, Edson KZ, Totah RA and Rettie
AE: Cytochrome P450 ω-hydroxylases in inflammation and cancer. Adv
Pharmacol. 74:223–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Harder DR, Gebremedhin D, Narayanan J,
Jefcoat C, Falck JR, Campbell WB and Roman R: Formation and action
of a P-450 4A metabolite of arachidonic acid in cat cerebral
microvessels. Am J Physiol. 266:H2098–H2107. 1994.PubMed/NCBI
|
|
5
|
Zhu J, Wang B, Lee JH, Armstrong JS,
Kulikowicz E, Bhalala US, Martin LJ, Koehler RC and Yang ZJ:
Additive neuroprotection of a 20-HETE inhibitor with delayed
therapeutic hypothermia after hypoxia-ischemia in neonatal piglets.
Dev Neurosci. 37:376–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Garcia V, Cheng J, Weidenhammer A, Ding Y,
Wu CC, Zhang F, Gotlinger K, Falck JR and Schwartzman ML:
Androgen-induced hypertension in angiotensinogen deficient mice:
Role of 20-HETE and EETS. Prostaglandins Other Lipid Mediat.
116-117:124–130. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Toth P, Csiszar A, Sosnowska D, Tucsek Z,
Cseplo P, Springo Z, Tarantini S, Sonntag WE, Ungvari Z and Koller
A: Treatment with the cytochrome P450 ω-hydroxylase inhibitor
HET0016 attenuates cerebrovascular inflammation, oxidative stress
and improves vasomotor function in spontaneously hypertensive rats.
Br J Pharmacol. 168:1878–1888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei XE, Zhang YZ, Li YH, Li MH and Li WB:
Dynamics of rabbit brain edema in focal lesion and perilesion area
after traumatic brain injury: A MRI study. J Neurotrauma.
29:2413–2420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cai H, Mu Z, Jiang Z, Wang Y, Yang GY and
Zhang Z: Hypoxia-controlled matrix metalloproteinase-9
hyperexpression promotes behavioral recovery after ischemia.
Neurosci Bull. 31:550–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu FF, Sun S, Ho AS, Lee D, Kiang KM,
Zhang XQ, Wang AM, Wu EX, Lui WM, Liu BY and Leung GK: Effects of
progesterone vs. dexamethasone on brain oedema and inflammatory
responses following experimental brain resection. Brain Inj.
28:1594–1601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim JY, Ko AR, Hyun HW and Kang TC: ETB
receptor-mediated MMP-9 activation induces vasogenic edema via ZO-1
protein degradation following status epilepticus. Neuroscience.
304:355–367. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu G, Wu J, Jiao Y, Wang L, Wang F and
Zhang Y: Rosiglitazone infusion therapy following minimally
invasive surgery for intracerebral hemorrhage evacuation decreases
matrix metalloproteinase-9 and blood-brain barrier disruption in
rabbits. BMC Neurol. 15:372015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang GY, Wang N and Liao HN: Effects of
muscone on the expression of P-gp, MMP-9 on blood-brain barrier
model in vitro. Cell Mol Neurobiol. 35:1105–1115. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Y, Wang D, Wang H, Qu Y, Xiao X and
Zhu Y: The protective effect of HET0016 on brain edema and
blood-brain barrier dysfunction after cerebral
ischemia/reperfusion. Brain Res. 1544:45–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Urrutia A, Rubio-Araiz A, Gutierrez-Lopez
MD, ElAli A, Hermann DM, O'Shea E and Colado MI: A study on the
effect of JNK inhibitor, SP600125, on the disruption of blood-brain
barrier induced by methamphetamine. Neurobiol Dis. 50:49–58. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jungner M, Siemund R, Venturoli D,
Reinstrup P, Schalén W and Bentzer P: Blood-brain barrier
permeability following traumatic brain injury. Minerva Anestesiol.
82:525–533. 2016.PubMed/NCBI
|
|
17
|
Hanrahan F and Campbell M: Frontiers in
neuroscience neuroinflammationTranslational research in traumatic
brain injury. Laskowitz D and Grant G: CRC Press/Taylor and Francis
Group (c) 2016 by Taylor & Francis Group LLC; Boca Raton (FL):
2016
|
|
18
|
Gasche Y, Copin JC, Sugawara T, Fujimura M
and Chan PH: Matrix metalloproteinase inhibition prevents oxidative
stress-associated blood-brain barrier disruption after transient
focal cerebral ischemia. J Cereb Blood Flow Metab. 21:1393–1400.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lapchak PA, Chapman DF and Zivin JA:
Metalloproteinase inhibition reduces thrombolytic (tissue
plasminogen activator)-induced hemorrhage after thromboembolic
stroke. Stroke. 31:3034–3040. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gu JH, Ge JB, Li M, Xu HD, Wu F and Qin
ZH: Poloxamer 188 protects neurons against ischemia/reperfusion
injury through preserving integrity of cell membranes and blood
brain barrier. PLoS One. 8:e616412013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang ZG, Cheng Y, Yu XC, Ye LB, Xia QH,
Johnson NR, Wei X, Chen DQ, Cao G, Fu XB, et al: bFGF protects
against blood-brain barrier damage through junction protein
regulation via PI3K-Akt-Rac1 pathway following traumatic brain
injury. Mol Neurobiol. 53:7298–7311. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Y, Wang H and Zhu Y, Chen L, Qu Y and
Zhu Y: The protective effect of nordihydroguaiaretic acid on
cerebral ischemia/reperfusion injury is mediated by the JNK
pathway. Brain Res. 1445:73–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mehta SL, Manhas N and Raghubir R:
Molecular targets in cerebral ischemia for developing novel
therapeutics. Brain Res Rev. 54:34–66. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu W, Chen L, Yang YQ, Falck JR, Guo AM,
Li Y and Yang J: Cytochrome P450 ω-hydroxylase promotes
angiogenesis and metastasis by upregulation of VEGF and MMP-9 in
non-small cell lung cancer. Cancer Chemother Pharmacol. 68:619–629.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fordsmann JC, Ko RW, Choi HB, Thomsen K,
Witgen BM, Mathiesen C, Lønstrup M, Piilgaard H, MacVicar BA and
Lauritzen M: Increased 20-HETE synthesis explains reduced cerebral
blood flow but not impaired neurovascular coupling after cortical
spreading depression in rat cerebral cortex. J Neurosci.
33:2562–2570. 2013. View Article : Google Scholar : PubMed/NCBI
|