Introduction
Cervical cancer (CC) is one of the most lethal
cancers with increasing incidence and mortality over the past
decades, and is the second most common female malignant disease
worilwide (1). According to the
latest world cancer statistics, approximately 529,800 female are
diagnosed with CC and approximately 275,100 die worldwide each
year, making CC the second fastest growing cancer and a serious
threat to women's health (2).
Meanwhile, the age of CC incidence has progressively decreased,
which has attracted wide attention. Recent studies have shown that
lifestyle, environmental pollution, population aging genetic
predisposition, HPV infection and the impact of hormones are the
important causes of CC (3).
Although the morbidity and mortality of CC has declined in the past
30 years, the 5-year survival rate of advanced-stage patients still
below 40% (4). Therefore, in order
to improve the cure percentage of CC, it is important to understand
its molecular mechanism and identify effective diagnostic and
prognostic biomarkers.
Long non-coding RNA (lncRNA) is a non-coding RNA
more than 200 nucleotides in length (5). More and more evidence has showed that
lncRNAs is an important part of a complex gene regulatory network
which regulates gene expression at the epigenetics and
transcriptome levels (6). The
lncRNAs are differently expressed in many kinds of cancers
(7,8), including gastric, lung and ovarian
cancer (9–11). In addition, abnormal expression of
lncRNAs has been related to metastasis, recurrence, and prognosis
of various human tumors (12).
More importantly, Compared with protein coding mRNAs and miRNA,
lncRNAs have greater tissue specificity (13). Thus, discovery of differentially
expressed lncRNAs in CC may be important for the diagnosis and the
identifications for this disease.
Recently, the hypothesis of competing endogenous
RNAs (ceRNAs) has suggested that RNA transcripts interact via miRNA
response elements. Increasing evidences indicates that lncRNAs,
mRNAs and pseudogene acting as ceRNAs can be regulated by MREs and
play key funtions in metastasis, tumorigenesis and progression of
tumors (14). Meanwhile, ceRNA
activity also plays an important roles in the transcriptome and
increasing evidence has shown that genetic information is closely
related to pathological change in most cancers (15).
HPV infection alone is not be the only factor CC
formation. Host genetic variations also play an important roles in
the development of CC (16). With
the development of high-throughput gene sequencing technologies and
molecular biology methods, we can use these new tools for the
discovery and identification of cancer biomarkers (17,18).
However, studies to date have lacked the integrated analysis of
large samples and the sensitivity of CC-specific lncRNAs
biomarkers. In addition, small sample studies do not have the
statistical power to explain the relationships between abnormal
lncRNAs and CC patients' clinical features Recently, The Cancer
Genome Atlas (TCGA) (http://cancergenome.nih.gov) database has collected
and provided a large sample size of CC genome sequencing data. The
aim of our study was to solve the problem of small sample size and
improve the accuracy and reliability of results by using TCGA RNA
sequencing data from CC patients to find CC-related lncRNAs. In
this study, we collected whole transcriptome RNA sequencing data of
307 CC tissues specimens and six adjacent nontumor tissue specimens
through the TCGA database. To the best of our knowledge, our study
is the first time to investigate the CC-related lncRNA expression
profiles through the use of a large-scale samples RNA sequencing
database. Subsequently, the reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) was used to validate part of
the bioinformatics analysis results by 31 pairs of newly diagnosed
CC clinical samples. This new method of finding CC-related lncRNAs
through the ues of ceRNA network can help determine the potential
functions of lncRNAs in CC progression and development.
Materials and methods
Patients and samples
Following the TCGA guidelines, we downloaded RNA
sequencing data and clinical pathological information from 307
cases of cervical squamous cell carcinoma (CESC) in the TCGA
database (up to Decenber 1, 2016). Then, we excluded cases without
completed analysis data, with a histologic diagnosis that was not
CESC, with more than two malignant tumors, and those which had
received preoperative chemoradiation. Finally, 289 CC patients
remained for analysis based on the above exclusion criteria. From
these patients, RNA sequencing data from 289 tumor tissues and six
nontumor tissues were obtained. Using the international Federation
of Gynecology and Obstetrics (FIGO) staging system, we divided the
patients into three groups, FIGO stage I were 158 patients, FIGO
stage II, 68 patients; and FIGO stage III–IV, 63 patients.
In addition, 31 tissue specimens (tumor tissues and
adjacent normal tissue) were collected between 2016 and 2017 at the
Zhongda Hospital of Southeast University (Nanjing, China) form CC
patients, aged 23–64 years for RT-qPCR analysis. Tissues specimens
were rapidly frozen in RNAlater (Ambion; Thermo Fisher Scientific,
Inc., Austin, TX, USA) and were stored in liquid nitrogen for
subsequent RNA extraction and RT-qPCR analysis. These 31 patients
were diagnosed of CC based on the histopathology and clinical
history. All patients signed informed consent, and this study also
was approved by the ethics committee of Zhongda Hospital Southeast
University.
RNA sequence data collects and
analysis
The CESC-RNA sequencing data (level 3) and clinical
information were downloaded from TCGA database until December 1,
2016. The TCGA database provides normalized count data for RNA
sequencing through the RNASeqV2 system, which contained the lncRNA
and mRNA sequencing data. Meanwhile, CESC miRNA sequencing data
also were obtained through the TCGA database. Level 3 miRNA
sequencing base data were obtained through Illumina HiSeq 2000
miRNA sequencing platforms (Illumina, Inc., San Diego, CA, USA).
The RNA sequencing data from these CESC patients tissues specimens
had previously been normalized to the TCGA database. We then
further analyzed the differentially expressed RNA sequencing data
by bioinformatics analysis. The bioinformatics analysis is shown in
Fig. 1.
Functional enrichment of Gene Ontology
(GO) and pathway analysis
We analyzed the biological processes of aberrantly
expressed intersection mRNAs through the Database for Annotation,
Visualization, and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/), which used GO
database to investigate the potential functions of these aberrantly
expressed intersection mRNAs (19). The potential functions of mRNAs
participating in the pathways were then analyzed using the Kyoto
Encyclopedia of Genes and Genomes (KEGG) database.
Construction of ceRNA network
We made use of the theory in which lncRNAs regulate
miRNA by binding and sequestering them and miRNAs in turn regulate
mRNAs via lncRNA-miRNA-mRNA interactions in the competitive
endogenous RNA network (20).
Therefore, we selected the abnormally expressed lncRNA, miRNA, and
mRNA in the intersection of three groups based on fold-change
>2.0 and P<0.05. Next, we used miRanda (http://www.microrna.org) to predict the miRNA targets
and investigate lncRNA-miRNA relationships. Meanwhile, Target scan
(http://www.targetscan.org/) and miRbase
targets (http://mirdb.org) were used to predict miRNA
target genes. Finally, we combined the differentially expressed
data from TCGA with the predicted targets of miRNAs to select and
the results of miRNAs that predicted target lncRNAs and mRNAs to
select commonly regulated lncRNAs and mRNAs. In accordance with the
principle of negative regulation of ceRNA, we select the most
negative regulated miRNA, lncRNAs and mRNA to build the ceRNA
regulatory network, using Cytoscape version 3.0 to construct it
(21). Fig. 1 shows a flow chart outlining the
steps used to bulid the ceRNA network.
Association analysis between CC
specific lncRNAs and clinical features
We chose the key lncRNAs to be included in the ceRNA
network according to the comprehensively bioinformatics analysis of
the CC RNA sequencing data in TCGA. In the next step, we further
analyzed the relationships between CC-specific lncRNAs and patients
clinical features including race, pathological stage, tumor grade,
TNM stage, FIGO stage and HPV infection. Subsequently, we chose
several of the key lncRNAs in the ceRNA network and validate the
accuracy and reliability of results from the bioinformatics
analysis using RT-qPCR to analyze 31 newly diagnosed CC
patients.
Extraction of total RNA from clinical
samples and RT-qPCR verification of bioinformatics results
We random selected 17 key lncRNAs associated with CC
patients clinical features that had high association scores in the
above bioinformatics ceRNA network. Then, we utilized RT-qPCR to
analyzed the actual expression levels of these lncRNAs in 31 newly
diagnosed CC patients. We chose GAPDH as the endogenous standard to
confirm the accuracy and reliability of our bioinformatics
analysis. Total RNA were isolated from tissues specimens of the CC
patients using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to the manufacturer's
protocol, and the purity of the isolated RNA was assessed using
NanoDrop 2000 spectrometer (Thermo Fisher Scientific, Inc.).
Reverse transcription reactions and RT-qPCR were performed
according to the manufacturer's protocol, using the reverse
transcription system and qPCR Master Mix kit (Promega Corporation,
Madison, WI, USA) as well as the Step One Plus™ PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) to detect the
expression levels of lncRNAs. All the primers were produced by
Generay Biotech Co., Ltd. (Shanghai, China). The RT-qPCR results
were calculated using the 2−ΔΔCq method (22) with the formula [ΔCq=(Cq
RNAs-Cq GAPDH) and ΔΔCq=ΔCqtumor
tissues-ΔCqadjacent non-tumor tissues].
Statistical analysis
Data analysis was performed using SPSS software
version 24.0 (IBM Corp., Armonk, NY, USA). The final results were
expressed as mean ± standard deviation. Student's t-test were used
to compare the fold-change between groups of sequencing data. In
all cases, P<0.05 was considered to indicate a statistically
significant difference. In addition, we used receiver operating
characteristic (ROC) curves and the area under the curve (AUC) to
judge the diagnostic value of 6 lncRNAs in CC patients.
Results
Cancer specific lncRNAs in CC
Base on TCGA database ‘Level 3’ CESC RNA-Sequencing
(RNA-Seq) data, we observed that 71 lncRNAs were abnormality
expressed in 289 CC patients tumor tissues compared to 6 adjacent
normal cervical tissues with a fold-change >2 and P<0.05.
Subsequently, we obtained abnormally expressed lncRNAs from 68 FIGO
stage I CC tissues, 68 FIGO stage II tissues, and 71 FIGO stage
III–IV tissues when compared to adjacent normal cervical tissues.
In order to further narrow the scope of bioinformatics analysis and
improve the accuracy, we chosed 51 lncRNAs that were common to all
three groups (Fig. 2). There were
42 lncRNAs (13 upregulated; 29 downregulated; Table I) involved in the ceRNA network in
these 51 lncRNAs.
 | Table I.Differentially expressed intersection
lncRNAs between FIGO stage I/Normal, FIGO stage II/Normal and FIGO
stage III–IV/Normal. |
Table I.
Differentially expressed intersection
lncRNAs between FIGO stage I/Normal, FIGO stage II/Normal and FIGO
stage III–IV/Normal.
| Name (lncRNA) | Gene ID | Regulation | Average
fold-change | -Log (P) |
|---|
| EMX2OS | 196047 | Down | −81.30 | 5.921 |
| MIR4697HG | 283174 | Down | −24.39 | 4.096 |
| MIR100HG | 399959 | Down | −20.00 | 6.778 |
| MBNL1-AS1 | 401093 | Down | −14.78 | 4.075 |
| MEG3 | 55384 | Down | −9.46 | 3.989 |
| LINC01140 | 339524 | Down | −9.38 | 3.550 |
| A2M-AS1 | 144571 | Down | −9.09 | 3.509 |
| TPTEP1 | 387590 | Down | −8.33 | 4.281 |
| NR2F1-AS1 | 441094 | Down | −8.11 | 3.611 |
| MIR99AHG | 388815 | Down | −7.89 | 3.605 |
| LINC00341 | 161176 | Down | −7.14 | 4.617 |
| SMIM10L2B | 644596 | Down | −6.00 | 6.015 |
| LINC00663 | 284440 | Down | −5.08 | 3.868 |
| EPB41L4A-AS1 | 114915 | Down | −5.00 | 4.382 |
| LINC00312 | 29931 | Down | −5.00 | 5.436 |
| LINC00950 | 92973 | Down | −4.11 | 3.353 |
| SYS1-DBNDD2 | 767557 | Down | −3.85 | 3.970 |
| SNHG7 | 84973 | Down | −3.75 | 7.000 |
| ATP1A1-AS1 | 84852 | Down | −3.66 | 3.732 |
| RASA4CP | 401331 | Down | −3.66 | 3.974 |
| ILF3-AS1 | 147727 | Down | −3.61 | 3.879 |
| INE2 | 8551 | Down | −3.61 | 5.543 |
| FLJ10038 | 55056 | Down | −3.37 | 6.436 |
| ACVR2B-AS1 | 100128640 | Down | −3.37 | 3.619 |
| FAM66C | 440078 | Down | −3.37 | 3.522 |
| AMZ2P1 | 201283 | Down | −3.37 | 3.508 |
| LOH12CR2 | 503693 | Down | −3.33 | 3.032 |
| ZNF876P | 642280 | Down | −3.06 | 5.301 |
| FTX | 100302692 | Down | 2.40 | 4.494 |
| MIR9-3HG | 254559 | Up | 47.43 | 2.974 |
| TMPO-AS1 | 100128191 | Up | 7.15 | 4.641 |
| GOLGA2P5 | 55592 | Up | 5.93 | 6.699 |
| CDKN2B-AS1 | 100048912 | Up | 5.85 | 3.931 |
| MST1P2 | 11209 | Up | 5.49 | 3.832 |
| LINC00467 | 84791 | Up | 5.39 | 3.802 |
| DDX12P | 440081 | Up | 5.31 | 3.102 |
| ASMTL-AS1 | 80161 | Up | 4.92 | 3.468 |
| GEMIN8P4 | 492303 | Up | 4.72 | 3.610 |
| GOLGA2P10 | 80154 | Up | 4.55 | 4.017 |
| OIP5-AS1 | 729082 | Up | 3.09 | 3.046 |
| LOC146880 | 146880 | Up | 2.59 | 4.999 |
| EP400NL | 347918 | Up | 2.33 | 7.000 |
Functional enrichment analysis
The function of differentially expressed mRNAs in CC
was analyzed at the GO and KEGG pathway levels by DAVID
Bioinformatics tool. There were 2,650 differentially expressed
mRNAs between CC tumor tissues and adjacent normal cervical tissues
in FIGO stage form the TCGA. Focused on these differentially
expressed genes, there were 2,484 differentially expressed mRNAs
between CC tumor tissues and adjacent normal cervical tissues in
FIGO stage I; 2,392 differentially expressed mRNAs in FIGO stage II
and 2,650 differentially expressed mRNAs in FIGO stage III–IV. We
analyzed the enrichment of these 2,057 differentially expressed
mRNAs in the GO database (Fig. 2),
then analyzed the upregulated and downregulated mRNAs. We found
that the highest enriched GO terms were mitotic cell cycle, cell
division, DNA replication and apoptotic process in upregulated
transcripts. and cell adhesion, signal transduction, transcription
and DNA-dependent in downregulated transcripts (Fig. 3).
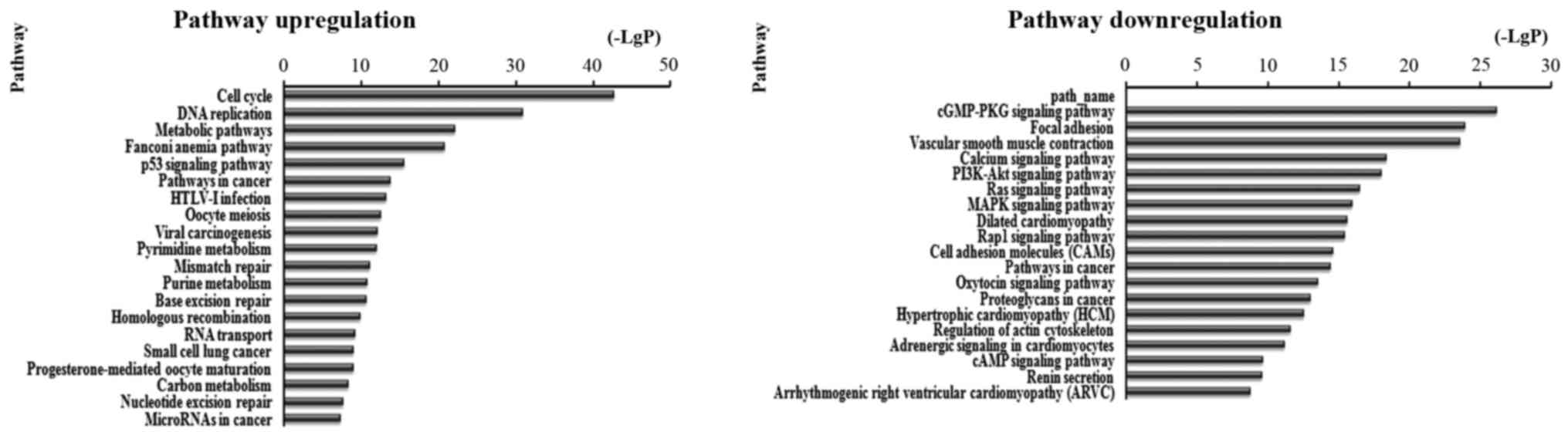
There were 87 pathways corresponded to upregulated
transcripts by pathway analysis; the main enriched pathway was the
Cell cycle. In the 109 pathways in the downregulated transcripts;
the main enriched pathway was cGMP-PKG signaling pathway. We
separately described the top 20 KEGG pathways, including
downregulated and upregulated genes (Fig. 4). Among these pathways, the p53
signaling pathway, viral carcinogenesis, PI3K-Akt signaling
pathway, Ras signaling pathway, MAPK signaling pathway, mTOR
signaling pathway and Rap1 signaling pathway may be related to
development and prognosis of cancer. In addition, other pathways
such as cGMP-PKG signaling pathway, Cell cycle and leukocyte
transendothelial migration were also associated with cancer
pathways (Table II and Fig. 4).
| Table II.KEGG pathways enriched by the coding
genes involved in the competing endogenous RNA network. |
Table II.
KEGG pathways enriched by the coding
genes involved in the competing endogenous RNA network.
| A, Upregulated
genes |
|---|
|
|---|
| KEGG pathways | Genes |
|---|
| Cancer related |
|
|
Pathways in cancer, MicroRNAs
in cancer, prostate cancer, p53 signaling pathway, PI3K-Akt
signaling pathway, small cell lung cancer, HIF-1 signaling pathway,
viral carcinogenesis | E2F3, TPM3, MYB,
NUP188, CCNE1, CHEK1, EPHA1, NUP50, SRPK1, WWC1, E2F3, EXO1, NXT2,
ACACA, CDC25A, GALNT3, SLC2A1, TCF7, XPO5, ELK4, PDE7A, PAK6, PIGA,
BCL2L11, HK2 |
| Non-cancer
related |
|
| HTLV–I
infection, cell cycle, RNA transport, renal cell carcinoma, axon
guidance |
|
|
| B, Downregulated
genes |
|
| KEGG
pathways | Genes |
|
| Cancer related |
|
|
Pathways in cancer, Rap1
signaling pathway, PI3K-Akt signaling pathway, Ras signaling
pathway, MAPK signaling pathway, prostate cancer, ErbB signaling
pathway, mTOR signaling pathway, endometrial cancer, small cell
lung cancer | CALD1, DOCK4, FLT1,
GAB1, GUCY1A3, KCNJ8, KCNMA1, NR4A3, PDGFRA, PPP1R12B, PTGER3,
RPS6KA2, S1PR1, SLC2A4, ST6GALNAC3, ST6GALNAC6, ZFPM2, HGF, PRKCA,
PTGER2, STAT5B, ZAK, AXIN2, BCL2, FGF2, INSR, MASP1, PRLR,
RAB11FIP2, RECK, SGCD, ZYX, NRXN3, ZEB1, ZEB2, CTSK, ESAM, THRA,
ACACB, GNAZ, SDC2, AKT3, SPG20, TBL1X, CALD1, ABCC9, MYLK, ST3GAL2,
KDR, MEF2D, ACTC1, CACNB2, ERG, DUSP3, GNG7, MAP3K3, NCAM1, SOX17,
ST3GAL3, ADCY5, FGFR1, FZD4, ITGA10, PRKG1, ATP2B4, HOXA11, MITF,
ST8SIA1, ENTPD1, MAGI2, MEF2C, AMPH, NEGR1 |
| Non-cancer
related |
|
|
cGMP-PKG signaling pathway,
focal adhesion, transcriptional misregulation in cancer, insulin
resistance, apoptosis, leukocyte transendothelial migration |
|
The ceRNA network
In our study, we found 72 differentially expressed
miRNAs with the fold-change >2 and P<0.05. We picked out 58
intersection miRNAs from these 72 miRNAs by bioinformatics analysis
of the FIGO stage (Fig. 2B). and
determined if these interacting miRNAs had a target relationship
with any of the 51 CC-specific lncRNAs. We predicted 56 miRNAs
targeted 49 key lncRNAs by miRcode (http://www.mircode.org/) (23) (Table
III) in the ceRNAs network. Then, mRNA targeted by miRNAs, we
found 49 specific miRNAs associated with 97 mRNAs (Tables II and IV). Some mRNAs targeted
cancer-associated genes, including BCL2, MAP3K3, AKT3, E2F3.
| Table III.miRNAs targeting specific
intersection key lncRNAs in CC. |
Table III.
miRNAs targeting specific
intersection key lncRNAs in CC.
| Key lncRNAs | miRNAs |
|---|
| A2M-AS1 | hsa-miR-183-5p,
hsa-miR-93-5p |
| ACVR2B-AS1 | hsa-miR-106b-5p,
hsa-miR-15b-5p, hsa-miR-93-5p |
| AMZ2P1 | hsa-miR-183-5p |
| ASMTL-AS1 | hsa-miR-30b-5p |
| ATP1A1-AS1 | hsa-miR-106b-5p,
hsa-miR-183-5p |
| CDKN2B-AS1 | hsa-miR-140-5p,
hsa-miR-195-5p |
| DDX12P | hsa-miR-10b-3p,
hsa-miR-139-5p, hsa-miR-140-3p, hsa-miR-145-5p, hsa-miR-497-5p |
| EMX2OS | hsa-miR-106b-5p,
hsa-miR-141-5p, hsa-miR-16-5p, hsa-miR-183-5p, hsa-miR-205-5p, |
|
| hsa-miR-21-3p,
hsa-miR-93-5p |
| EP400NL | hsa-miR-140-3p |
| EPB41L4A-AS1 | hsa-miR-141-5p,
hsa-miR-15b-5p, hsa-miR-16-5p |
| FAM66C | hsa-miR-15b-5p,
hsa-miR-16-5p, hsa-miR-185-5p |
| FLJ10038 | hsa-miR-106b-5p,
hsa-miR-183-5p, hsa-miR-200b-3p, hsa-miR-32-5p, hsa-miR-429 |
| FTX | hsa-miR-185-5p |
| GEMIN8P4 | hsa-miR-143-3p |
| GOLGA2P10 | hsa-miR-10b-5p,
hsa-miR-133a-3p, hsa-miR-140-3p, hsa-miR-195-5p, hsa-miR-320a,
hsa-miR-497-5p |
| GOLGA2P5 | hsa-miR-132-3p,
hsa-miR-133a-3p, hsa-miR-139-5p, hsa-miR-328-3p |
| ILF3-AS1 | hsa-miR-106b-5p,
hsa-miR-93-5p |
| INE2 | hsa-miR-106b-5p,
hsa-miR-93-5p |
| LINC00312 | hsa-miR-15b-5p,
hsa-miR-16-5p, hsa-miR-21-3p |
| LINC00341 | hsa-miR-200a-3p,
hsa-miR-205-5p, hsa-miR-425-5p |
| LINC00467 | hsa-miR-132-3p,
hsa-miR-133a-3p |
| LINC00663 | hsa-miR-106b-5p,
hsa-miR-141-3p, hsa-miR-15b-5p, hsa-miR-200a-3p, hsa-miR-93-5p |
| LINC00950 | hsa-miR-141-3p,
hsa-miR-141-5p, hsa-miR-200b-3p, hsa-miR-200c-3p,
hsa-miR-224-5p, |
|
| hsa-miR-142-3p,
hsa-miR-21-3p |
| LINC01140 | hsa-miR-142-3p,
hsa-miR-21-3p |
| LOC146880 | hsa-miR-145-5p |
| LOH12CR2 | hsa-miR-106b-5p,
hsa-miR-93-5p |
| MBNL1-AS1 | hsa-miR-106b-5p,
hsa-miR-141-3p, hsa-miR-183-5p, hsa-miR-200a-3p |
|
| hsa-miR-32-5p,
hsa-miR-93-5p |
| MEG3 | hsa-miR-106b-5p,
hsa-miR-22-5p, hsa-miR-429, hsa-miR-93-5p |
| MIR100HG | hsa-miR-183-5p |
| MIR4697HG | hsa-miR-141-5p,
hsa-miR-205-5p, hsa-miR-22-5p |
| MIR9-3HG | hsa-miR-10b-5p,
hsa-miR-139-5p, hsa-miR-140-5p, hsa-miR-143-5p, hsa-miR-195-5p,
hsa-miR-320a |
| MIR99AHG | hsa-miR-106b-5p,
hsa-miR-141-5p, hsa-miR-182-5p, hsa-miR-93-5p |
| MST1P2 | hsa-miR-328-3p |
| NR2F1-AS1 | hsa-miR-141-5p,
hsa-miR-15b-5p, hsa-miR-185-5p, hsa-miR-22-5p, hsa-miR-425-5p |
| OIP5-AS1 | hsa-miR-143-5p |
| RASA4CP | hsa-miR-182-5p |
| SMIM10L2B | hsa-miR-15b-5p,
hsa-miR-182-5p, hsa-miR-205-5p, hsa-miR-425-5p |
| SNHG7 | hsa-miR-182-5p,
hsa-miR-200a-5p |
| SYS1-DBNDD2 | hsa-miR-16-5p |
| TMPO-AS1 | hsa-miR-143-3p |
| TPTEP1 | hsa-miR-141-3p,
hsa-miR-142-3p, hsa-miR-16-5p |
| ZNF876P | hsa-miR-106b-5p,
hsa-miR-15b-3p, hsa-miR-93-5p |
| Table IV.miRNAs targeting CC-specific
mRNAs. |
Table IV.
miRNAs targeting CC-specific
mRNAs.
| miRNAs | mRNAs |
|---|
|
hsa-miR-106b-5p | BCL2L11, CALD1,
DOCK4, E2F2, E2F3, ELK4, ERBB3, FLT1, GAB1, GUCY1A3, KCNJ8, KCNMA1,
KPNA2, NR4A3, PDGFRA, PPP1R12B, PTGER3, RPS6KA2, RUNX1, S1PR1,
SLC2A |
| hsa-miR-10b-3p | MAGI2, MEF2D, PRLR,
RHOQ, XPO5 |
| hsa-miR-10b-5p | E2F3, NR4A3,
SHANK3 |
|
hsa-miR-125a-5p | BAK1, BCL2, CDKN2B,
DUSP3, E2F2, EIF4EBP1, ENPP1, FGFR1, LIFR, MAP3K3, MASP1, NUP210,
NUP50, PIP5K1C, PPAT, PPP1R12B, RHOQ, SCN4B, TDG |
|
hsa-miR-125b-5p | ACACB, BAK1, BCL2,
CDKN2B, E2F2, LIFR, MAP3K3, NUP210, PPAT, PPP1R12B, TDG, TSTA3 |
| hsa-miR-126-5p | PDGFRA |
| hsa-miR-132-3p | FGF7, MAP3K3,
PDE7A, PPP2CB, PRICKLE2 |
|
hsa-miR-133a-3p | AQP1, DAAM2,
GABARAPL1, SGCD, TBL1X, TPM3 |
| hsa-miR-139-5p | ANK2, DMD, FOXO1,
GALNT3, MRVI1, SOCS2, TPM3 |
| hsa-miR-140-3p | BCL2, GAB2, KCNMA1,
MYB, NUP188, VAMP2 |
| hsa-miR-140-5p | ACACA, DNM3,
PDGFRA, SLC2A1 |
| hsa-miR-141-3p | CDC25A, DUSP3,
E2F3, ERG, GNG7, HGF, MAP3K3, NCAM1, NME1, PIGW, RUNX1, SOX17,
ST3GAL3, ZEB1, ZEB2 |
| hsa-miR-141-5p | HGF, HSP90AA1,
NUP50, PRKCA, PTGER2, STAT5B, ZAK |
| hsa-miR-142-3p | PRLR |
| hsa-miR-143-3p | CACNA1C, HK2, LIFR,
NCAM1 |
| hsa-miR-143-5p | RHOQ, TCF7,
ZAK |
| hsa-miR-145-3p | DUSP3, ITGA10,
PDE7B |
| hsa-miR-145-5p | ELK4, FLI1, FLT1,
FZD4, PARVA, PTGFR, ST6GALNAC3, TGFBR2 |
| hsa-miR-15b-3p | CGN, NEGR1 |
| hsa-miR-15b-5p | ACACA, ADCY5, AKT3,
AXIN2, BCL2, CCNE1, CHEK1, E2F3, EPHA1, FGFR1, FOXO1, FZD4, INSR,
ITGA10, KDR, MASP1, MYB, NUP50, PPP1R12B, PRKG1, RAB11FIP2, RECK,
SGCD, SRPK1, WWC1, ZYX |
| hsa-miR-16-5p | AXIN2, BCL2, CCNE1,
CHEK1, E2F3, EPHA1, FGF2, FOXO1, INSR, MASP1, MYB, NUP50, PPP1R12B,
PRLR, RAB11FIP2, RECK, SGCD, UNG, WWC1, ZYX |
| hsa-miR-182-5p | BCL2, DSG2, MEF2D,
MITF, NUP50, PRLR, PTGER3, RECK, ST6GALNAC3, ST8SIA1, UCK2 |
| hsa-miR-183-5p | EZR, FOXO1, NRXN3,
TPM3, ZEB1, ZEB2, ZFPM2 |
| hsa-miR-185-5p | CTSK, ESAM, PAK6,
THRA |
| hsa-miR-195-5p | BCL2, CCNE1, CHEK1,
EPHA1, FGF2, FGF7, FOXO1, FZD4, GABARAPL1, MASP1, MYLK, NUP50,
PPP1R12B, PRLR, RAB11FIP2, SRPK1, WWC1, ZYX |
|
hsa-miR-200a-3p | B3GNT5, CDC25A,
DUSP3, E2F3, ERG, GAB1, MAP3K3, NME1, RUNX1 SOX17, ST3GAL3, ZEB1,
ZEB2 |
|
hsa-miR-200a-5p | FGFR1, POLA1 |
|
hsa-miR-200b-3p | ABCC9, DOCK4, E2F3,
ELK4, GAB1, MYLK, PPP1R12B, RAB11FIP2, RUNX1, ST3GAL2, TP73, ZEB1,
ZFPM2 |
|
hsa-miR-200c-3p | DOCK4, ELK4, KDR,
MEF2D, MYLK, PMAIP1, PPP1R12B, PRKCA, PTGER2, RAB11FIP2, RECK,
RUNX1, ST3GAL2, TP73, ZEB1, ZEB2, ZFPM2 |
| hsa-miR-205-5p | ACACB, DHCR24,
E2F1, ERBB3, TGFA |
| hsa-miR-21-3p | GNAZ, NRXN3,
RPS6KA2, SDC2, UCK2 |
| hsa-miR-218-5p | APH1B, BRCA1, ELK4,
GAB2, MTMR1, PRLR |
| hsa-miR-22-5p | ELK4, ENTPD1,
MAGI2, MEF2C, RAD54B, SDC1 |
| hsa-miR-224-5p | ATP2B4, HOXA11,
KCNMA1, LPAR5, NR4A3 |
|
hsa-miR-24-1-5p | CALD1, DNM3, E2F3,
TPM3 |
| hsa-miR-28-3p | LMO7 |
| hsa-miR-28-5p | ITPKB, MASP1, MPL,
PARVA |
| hsa-miR-30b-5p | BCL2L11, CACNA1C,
DMD, GALNT3, MEF2D, PRLR |
| hsa-miR-32-5p | ACTC1, AURKA,
BCL2L11, E2F3, ELK4, SDC2, SLX4, ZEB2 |
| hsa-miR-320a | AKT3, CACNA1C,
E2F3, EXO1, FLNC, GNAZ, GUCY1A3, NXT2, PRKG1, TPM3 |
| hsa-miR-328-3p | PAK6, PIGA,
RASGRP2, SLC2A1, ST3GAL3, ZAK |
| hsa-miR-361-5p | ERG, GTF2E1, PIGA,
PRICKLE2, ST8SIA1 |
| hsa-miR-362-5p | AKT3, ATP2B4,
KCNMA1, MRVI1, NRXN3 |
|
hsa-miR-374b-5p | FBXO32 |
| hsa-miR-381-3p | CACNA1C, ELK4,
FOXO1, GABARAPL1, ZFPM2 |
| hsa-miR-425-5p | AMPH |
| hsa-miR-429 | CACNB2, DOCK4,
E2F3, ELK4, ERG, GAB1, GTF2E1, GUCY1A3, MYB, RAB11FIP2, RUNX1,
ST3GAL2, TP73, ZEB1, ZFPM2 |
| hsa-miR-497-5p | ACACA, ADCY5, AKT3,
BCL2, CDC25A, CNTNAP1, E2F3, EPHA1, FGF2, FOXO1, FZD4, INSR,
ITGA10, KDR, MASP1, MYLK, NUP50, PTPRM, RAB11FIP2, RECK, SGCD,
SRPK1, WWC1, ZAK, ZYX |
| hsa-miR-93-5p | AKT3, BCL2L11,
E2F1, E2F2, ELK4, ERBB3, FLT1, GAB1, GUCY1A3, KCNJ8, KCNMA1, KIF23,
KPNA2, NR4A3, PGP, PPP1R12B, PTGER3, RBL1, RPS6KA2, RUNX1, SGCD,
SLC2A4, SPG20, ST6GALNAC3, ST6GALNAC6, TBL1X, THRA |
Based on our bioinformatics analysis, we investigate
the relationship between lncRNAs and mRNAs potential linked by
miRNAs that were identified in Tables
II and III, and build the
ceRNA (lncRNA-miRNA-mRNA) network. There were 72 differentially
expressed miRNAs identified in CC tissues samples, among which were
the 58 intersecting miRNAs (Fig.
2B). We then used the MREs principle to find the relationships
between these 58 miRNAs and 51 CC-specific lncRNAs, and detected
the potential MREs by starBase. The results showed that there were
49 specific miRNAs and 42 specific lncRNAs with potential
regulatory relationships. We then used Cytoscape 3.0 to build the
ceRNA network based on data from Tables III and IV. Fig.
5 shows the 42 lncRNAs, 49 miRNAs, and 72 mRNAs participating
in the lncRNA-miRNA-mRNA interaction network of CC.
Correlation analysis between CC
specific lncRNAs expression with clinical features
Using available clinical features from TCGA, such as
race, tumor grade, TNM stage, clinical stage, HPV infection, and
transfer, we further analyzed the 42 key lncRNAs from the ceRNA
network. The expression levels of the 19 key lncRNAs were obviously
different in patients with different clinical features (P<0.05;
Table V). For example two lncRNAs
(MST1P2 and FTX) were differently expressed in CC patients of
different race, five lncRNAs (LOH12CR2, GOLGA2P10, A2M-AS1,
ATP1A1-AS1 and ACVR2B-AS1) were differently expressed at different
pathological stage, ten lncRNAs (FAM66C, GOLGA2P5, ACVR2B-AS1,
ZNF876P, MIR9-3HG, EMX2OS, LINC00341, FLJ10038, ILF3-AS1 and
AMZ2P1) were expressed differently depending on the tumor TNM
stage, four lncRNAs (GOLGA2P5, ACVR2B-AS1, ZNF876P and MIR9-3HG)
were differently expressed at different clinical stage, four
lncRNAs (ILF3-AS1, GOLGA2P5, MIR9-3HG and FAM66C) were aberrantly
expressed depending on the patient outcome assessment and four
lncRNAs (SYS1-DBNDD2, MIR9-3HG, DDX12P, LINC00312) were differently
expressed in high and low risk types of HPV infection (Table V).
| Table V.The correlations between CC specific
lncRNAs from ceRNA network and clinical features. |
Table V.
The correlations between CC specific
lncRNAs from ceRNA network and clinical features.
| Comparisons | Downregulated | Upregulated |
|---|
| Race (Caucasian vs.
Asian) |
| MST1P2, FTX |
| Outcome (dead vs.
alive) | ILF3-AS1,
FAM66C | GOLGA2P5,
MIR9-3HG, |
| Transfer (N1 vs.
N0) | FAM66C, ZNF876P,
ACVR2B-AS1 | GOLGA2P5,
MIR9-3HG, |
| Classification
(stage 34 vs. stage 12) | LINC00341, EMX2OS,
FLJ10038 |
|
| Tumor pathological
stage (T34 vs. T12) | LINC00341, EMX2OS,
FLJ10038 | ILF3-AS1, AMZ2P1,
GOLGA2P5 |
| Tomor grade (g12
vs. g34) | LOH12CR2, A2M-AS1,
ATP1A1-AS1, | GOLGA2P10 |
|
| ACVR2B-AS1 |
| HPV infection
(high-risk vs. low-risk) | SYS1-DBNDD2,
LINC00312 | MIR9-3HG,
DDX12P |
RT-qPCR verification and ROC
In order to prove the reliability of the above
bioinformatics analysis results from TCGA, we random selected 11
key lncRNAs (DDX12P, GOLGA2P5, GOLGA2P10, LINC00467, MIR9-3HG,
MST1P2, TMPO-AS1, EMX2OS, LINC00663, MEG3, SYS1-DBNDD2) and
verified their actual expression levels in 31 pairs of newly
diagnosed clinical samples. The results showed that seven lncRNAs
were upregulation and four lncRNAs were downregulated in CC tumor
tissues compared to adjacent normal cervical tissues. The
validation results for these 11 key lncRNAs were with the above
TCGA bioinformatics results. This showed that our bioinformatics
analysis was accurate and reliable (Fig. 6 and Table VI).
| Table VI.Relative expression of lncRNAs in 31
pairs of cervical cancer tumor and non-tumor tissue. |
Table VI.
Relative expression of lncRNAs in 31
pairs of cervical cancer tumor and non-tumor tissue.
| Gene symbol | Type | Group | Mean ± SD of
ΔCq | ΔΔCqa (mean ± SD) |
2−ΔΔCq |
P-valueb | t-value |
|---|
| GOLGA2P10 | LncRNA | Tumor tissues | 8.033±2.855 | 0.184±2.521 | 2.651 | 0.697 | 0.393 |
|
|
| Adjacent non-tumor
tissues | 7.849±2.211 |
|
|
|
|
| MIR9-3HG | LncRNA | Tumor tissues | 11.173±2.732 | −2.008±2.602 | 13.293 | 0.001b | 3.917 |
|
|
| Adjacent non-tumor
tissues | 13.143±3.265 |
|
|
|
|
| DDX12P | LncRNA | Tumor tissues | 10.690±2.234 | −1.162±2.620 | 8.523 | 0.026b | 2.347 |
|
|
| Adjacent non-tumor
tissues | 11.853±2.488 |
|
|
|
|
| GOLGA2P5 | LncRNA | Tumor tissues | 10.164±2.348 | −0.789±3.451 | 11.794 | 0.160 | 1.451 |
|
|
| Adjacent non-tumor
tissues | 11.161±2.338 |
|
|
|
|
| LINC00467 | LncRNA | Tumor tissues | 11.645±2.066 | 0.098±2.448 | 1.461 | 0.839 | 0.205 |
|
|
| Adjacent non-tumor
tissues | 11.546±2.429 |
|
|
|
|
| MST1P2 | LncRNA | Tumor tissues | 11.717±3.025 | 0.646±2.109 | 0.287 | 0.139 | 1.531 |
|
|
| Adjacent non-tumor
tissues | 11.075±2.972 |
|
|
|
|
| TMPO-AS1 | LncRNA | Tumor tissues | 10.215±2.397 | −0.187±2.897 | 5.056 | 0.749 | 0.232 |
|
|
| Adjacent non-tumor
tissues | 10.402±2.619 |
|
|
|
|
| EMX2OS | LncRNA | Tumor tissues | 16.678±3.390 | 3.153±3.011 | −31.829 | 0.000b | 5.021 |
|
|
| Adjacent non-tumor
tissues | 13.525±3.836 |
|
|
|
|
| MEG3 | LncRNA | Tumor tissues | 10.082±2.958 | 2.047±3.143 | −29.352 | 0.001b | 3.566 |
|
|
| Adjacent non-tumor
tissues | 8.035±2.308 |
|
|
|
|
| LINC00663 | LncRNA | Tumor tissues | 19.529±2.851 | 1.506±2.993 | −12.051 | 0.015b | 2.614 |
|
|
| Adjacent non-tumor
tissues | 18.024±3.357 |
|
|
|
|
| SYS1-DBNDD2 | LncRNA | Tumor tissues | 4.566±1.748 | 1.537±2.676 | −16.796 | 0.005b | 3.039 |
|
|
| Adjacent non-tumor
tissues | 3.029±2.175 |
|
|
|
|
We assessed the diagnostic value of specific lncRNAs
and found that three out of six lncRNAs examined displayed good
diagnostic values (Fig. 7). ROC
curve analysis revealed AUC values of 0.773, 0.723 and 0.724 for
EMX20S, MEG3 and SYS1-DBNDD2, respectively (P<0.05; Fig. 7A), which suggested that these
lncRNAs may be good candidates for diagnostic biomarkers in CC
because their AUC values exceeded 0.7. ROC analysis also showed an
AUC value of 0.689 for MIR9-3HG (P<0.05; Fig. 7A), while results for DDX12P and
LINC00663 were not statistically significant (P>0.05; Fig. 7C). The AUC of these four lncRNAs
combined was 0.841, which was higher than that of the single lncRNA
(P<0.05; Fig. 7B).
Discussion
Despite improvements in treatment, early prevention
and diagnosis remains the most effective way to reduce morbidity
and mortality of CC (24). With
the extensive use of ThinPrep cytologic test (TCT) and HPV DNA
screening techniques, the incidence and mortality rates of CC have
declined over the past three decades, but the 5-year survival
percentage of patients has still remained below 40% (4), and 85% deaths have occured in
developing countries such as China (25). Therefore, the identification and
validation of biomarkers for early diagnosis and prognosis of CC is
an important goal. Many studies have reported lncRNAs related to
the biological regulatory functions in many cancers (26). Abnormal expression of lncRNAs has
also been widely detected in a variety of diseases (23,27).
Dysregulated lncRNAs have now emerged as key players in the
development of cancer. However, the expression profiles of lncRNA
in CC have been described in only a few studies involving small
sample size (28). Furthermore,
very few studies have examined the interaction between lncRNA, mRNA
and miRNA in CC. Results from the few studies performed have showed
that lncRNAs play an important function in ceRNA network, but their
relationships to specific ceRNA networks are still unclear
(29,30). Recently, a new ceRNA hypothesis was
proposed in which lncRNAs play a regulatory role through the
competitive binding of miRNAs (31,32).
Based on this mechanism, Li et al constructed a ceRNA
network related to oral squamous cell carcinoma (19). With further study of ceRNA network,
many researchers have showen that miRNAs regulated gens and
interact with lncRNAs in the ceRNA network (33).
In our study, we first screened lncRNAs, miRNAs and
mRNAs. The three types of non-coding RNA were related to FIGO
clinical stage in CC from the TCGA database. As far as we know,
this is the first time that lncRNA-miRNA-mRNA ceRNA networks have
been established in CC. Based on clinical information and RNA
sequencing profiles, we found that specific key lncRNAs from ceRNA
network were altered in different CC clinical manifestations by. We
further verified the expression level of 11 key lncRNAs in clinical
samples by RT-qPCR.
We investigated aberrantly expressed mRNAs in CC
intersection with RNAs from the three groups of RNA sequence data.
The results of GO and pathway analysis also revealed potential
regulatory relationship of mRNA related lncRNAs. The abnormal
signaling pathways may play important roles in the development and
progression of CC The GO results showed significant differences in
cellular functions and transcription process. The KEGG pathway
analysis showed that PI3K-Akt signaling pathway (34,35),
p53 signaling pathway (36), MAPK
signaling pathway, and viral carcinogenesis were particularly
important cancer-related pathways (37).
An increasing number of studies have also showed
that lncRNAs may bind to other transcription factors and are
involve in regulating the ceRNA network (14,38,39).
For example, the lncRNA MEG3 is an important gene for the
progression of many types of cancer including CC (40). MEG3 over-expression imposes another
level of post-transcriptional regulation, whereas MEG3 over
expression increase the expression of the miR-664 target gene,
ADH4, through competitive sponging of miR-664. Therefore, the
potential regulatiory function of lncRNA-miRNA-mRNA interactions
may also act during CC development. Based on the above analysis, we
built an lncRNA-miRNA-mRNA ceRNA network in CC through
bioinformatics analysis. We found that particular lncRNAs may be
associated with cancer. The lncRNAs such as MEG3, LINC00341 and
LINC00663 (41–43) may therefore acted as potential
molecular biomarker in other cancers, and may also be involved in
the initiation and progression of cancer. Based on our research
analysis, specific lncRNA was found to be indirectly related to
mRNAs signaling pathways in ceRNA network of CC. The analysis
results showed at leaet 10 pathways connected to cancer. Therefore,
it is believed that these key lncRNAs may played an important
regulatory role during CC formation.
We analyzed the association of 42 key lncRNAs from
the ceRNA network. The 19 key lncRNAs were related to clinical
features. According to recent studies, these included the lncRNAs
LINC00341 (42), FTX (44), LOH12CR2 (45) and LINC00312 (46), which have been reported to be
associated with prognosis in several cancers, while the function of
other lncRNAs have not yet been reported. These lncRNAs, which were
associated with clinical features, may have important research
values in the development and prognosis of CC. We also uesed
RT-qPCR to verify the expressions level of 11 key lncRNAs from the
31 pairs of newly obtained clinical samples. The result of RT-qPCR
were consistent with the result of TCGA bioinformatics analysis,
showing that it was basically reliable. The specificity and
sensitivity of lncRNAs as a test indicator were then determined by
ROC. Three lncRNAs (EMX20S, MEG3, SYS1-DBNDD2) had significant
single diagnostic values, but more important, the AUC of the
combined four lncRNAs (EMX20S, MEG3, SYS1-DBNDD2, MIR9-3HG) was
0.841 (P<0.05), which was greater than that any single lncRNA,
suggested that the combined diagnosis could improve the diagnostic
efficacy of CC.
In conclusion, we screened for key lncRNAs which
related to CC from the large number of candidate lncrRNAs in the
TCGA database by bioinformatics analysis and found differentially
expressed lncRNAs associated with different clinical features.
Importantly, we have constructed a ceRNA network which encompassed
the lncRNA-miRNA-mRNA interactions in CC, and investigated the CC
related key lncRNAs for their potential regulatory role. We also
validated key lncRNAs expression levels by RT-qPCR and thus
demonstrated the reliability and validity our bioinformatics
analysis. Furthermore, we explored the diagnostic value of some
these key lncRNAs. Our results suggested that these key lncRNAs may
be new candidate biomarkers for the clinical diagnosis,
classification and prognosis of CC. Due to sample size limitations
of TCGA database. Preliminary analysis and screening was only a
reference and exploration, our research focused on the follow-up
study for the enlarged sample size of Chinese population. Future
research studies will require molecular investigations and more
clinical samples to verify the function and mechanism of these
lncRNAs.
Acknowledgements
The authors would like to thank Mr. Donglin Cheng
for his technical support.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81673132, 81673130 and
81472939), the 333 Project of Jiangsu Province, the Fundamental
Research Funds for the Central Universities and the Innovative
Research Project for Postgraduates in Colleges of Jiangsu
Province.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
WJW and GYL conceived and designed the study. WJW
and CYL performed the experiments. WJW, CYL, JS, SY, SYX and MZ
analyzed and interpreted the results. YS performed the cervical
cancer patients' tissue sample collection and quality control. LHY
and YPP assisted with study design and provided advice throughout.
WJW performed analysis and quality control and was a major
contributor in writing the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Zhongda Hospital of Southeast University (Nanjing,
China). All patients provided written informed consent to
participate in the present study.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Sankaranarayanan R and Ferlay J: Worldwide
burden of gynaecological cancer: The size of the problem. Best
Pract Res Clin Obstet Gynaecol. 20:207–225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim HJ, Lee DW, Yim GW, Nam EJ, Kim S, Kim
SW and Kim YT: Long non-coding RNA HOTAIR is associated with human
cervical cancer progression. Int J Oncol. 46:521–530. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bray F, Ren JS, Masuyer E and Ferlay J:
Global estimates of cancer prevalence for 27 sites in the adult
population in 2008. Int J Cancer. 132:1133–1145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Mara TA, Zhao M and Spurdle AB:
Meta-analysis of gene expression studies in endometrial cancer
identifies gene expression profiles associated with aggressive
disease and patient outcome. Sci Rep. 6:366772016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dey BK, Mueller AC and Dutta A: Long
non-coding RNAs as emerging regulators of differentiation,
development and disease. Transcription. 5:e9440142014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mercer TR and Mattick JS: Structure and
function of long noncoding RNAs in epigenetic regulation. Nat
Struct Mol Biol. 20:300–307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng L, Yuan XQ, Liu ZY, Li WL, Zhang CY,
Zhang YQ, Pan X, Chen J, Li YH and Li GC: High lncRNA H19
expression as prognostic indicator: data mining in female cancers
and polling analysis in non-female cancers. Oncotarget.
8:1655–1667. 2017.PubMed/NCBI
|
|
8
|
Li CY, Liang GY, Yao WZ, Sui J, Shen X,
Zhang YQ, Peng H, Hong WW, Ye YC, Zhang ZY, et al: Identification
and functional characterization of microRNAs reveal a potential
role in gastric cancer progression. Clin Transl Oncol. 19:162–172.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu J, Liu L, Wan JX and Song Y: Long
noncoding RNA SNHG20 promotes gastric cancer progression by
inhibiting p21 expression and regulating the GSK-3β/β-catenin
signaling pathway. Oncotarget. 8:80700–80708. 2017.PubMed/NCBI
|
|
10
|
Peng Z, Wang J, Shan B, Yuan F, Li B, Dong
Y, Peng W, Shi W, Cheng Y, Gao Y, et al: Genome-wide analyses of
long noncoding RNA expression profiles in lung adenocarcinoma. Sci
Rep. 7:153312017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu Y, Biglia N, Wang Z, Shen Y, Risch HA,
Lu L, Canuto EM, Jia W, Katsaros D and Yu H: Long non-coding RNAs,
ASAP1-IT1, FAM215A and LINC00472, in epithelial ovarian cancer.
Gynecol Oncol. 143:642–649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun R, Qin C, Jiang B, Fang S, Pan X, Peng
L, Liu Z, Li W, Li Y and Li G: Down-regulation of MALAT1 inhibits
cervical cancer cell invasion and metastasis by inhibition of
epithelial-mesenchymal transition. Mol Biosyst. 12:952–962. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Z, Chen Y, Hu S, Zhang J, Wu J, Ren W,
Shao N and Ying X: Integrative analysis of protein-coding and
non-coding RNAs identifies clinically relevant subtypes of clear
cell renal cell carcinoma. Oncotarget. 7:82671–82685.
2016.PubMed/NCBI
|
|
14
|
Song X, Cao G, Jing L, Lin S, Wang X,
Zhang J, Wang M, Liu W and Lv C: Analysing the relationship between
lncRNA and protein-coding gene and the role of lncRNA as ceRNA in
pulmonary fibrosis. J Cell Mol Med. 18:991–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martin CM, Astbury K, Mcevoy L, O'Toole S,
Sheils O and O'Leary JJ: Gene expression profiling in cervical
cancer: Identification of novel markers for disease diagnosis and
therapy. Methods Mol Biol. 511:333–359. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goldfeder RL, Wall DP, Khoury MJ,
Ioannidis JPA and Ashley EA: Human genome sequencing at the
population scale: A primer on high-throughput DNA sequencing and
analysis. Am J Epidemiol. 186:1000–1009. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shukla HD: Comprehensive analysis of
cancer-proteogenome to identify biomarkers for the early diagnosis
and prognosis of cancer. Proteomes. 5:E282017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li S, Chen X, Liu X, Yu Y, Pan H, Haak R,
Schmidt J, Ziebolz D and Schmalz G: Complex integrated analysis of
lncRNAs-miRNAs-mRNAs in oral squamous cell carcinoma. Oral Oncol.
73:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo LL, Song CH, Wang P, Dai LP, Zhang JY
and Wang KJ: Competing endogenous RNA networks and gastric cancer.
World J Gastroenterol. 21:11680–11687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu H, Shi B, Wu G, Zhang Y, Zhu X, Zhang
Z, Liu C, Zhao Y, Wu T, Wang J and Chen R: Integrated analysis of
multiple data sources reveals modular structure of biological
networks. Biochem Biophys Res Commun. 345:302–309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng Y, Wei L, Guo L and Yang X: The
expression level of miR-203 in patients with gastric cancer and its
clinical significance. Pathol Res Pract. 13:1515–1518. 2017.
View Article : Google Scholar
|
|
23
|
Miao Y, Xu SY, Chen LS, Liang GY, Pu YPG
and Yin LH: Trends of long noncoding RNA research from 2007 to
2016: A bibliometric analysis. Oncotarget. 8:83114–83127. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
World Health Organization (WHO): New WHO
guide to prevent and control cervical cancer. http://www.who.int/mediacentre/news/releases/2014/preventing-cervical-cancer/en/December
3–2014
|
|
26
|
Moran VA, Perera RJ and Khalil AM:
Emerging functional and mechanistic paradigms of mammalian long
non-coding RNAs. Nucleic Acids Res. 40:6391–6400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu X, Sood AK, Dang CV and Zhang L: The
role of long noncoding RNAs in cancer: The dark matter matters.
Curr Opin Genet Dev. 48:8–15. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang L, Yi K, Wang H, Zhao Y and Xi M:
Comprehensive analysis of lncRNAs microarray profile and
mRNA-lncRNA co-expression in oncogenic HPV-positive cervical cancer
cell lines. Oncotarget. 7:49917–49929. 2016.PubMed/NCBI
|
|
29
|
Wei G: Bioinformatics analysis of microRNA
comprehensive regulatory network in congenital microtia. Int J
Pediatr Otorhinolaryngol. 79:1727–1731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qu J, Li M, Zhong W and Hu C: Competing
endogenous RNA in cancer: A new pattern of gene expression
regulation. Int J Clin Exp Med. 8:17110–17116. 2015.PubMed/NCBI
|
|
31
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thomson DW and Dinger ME: Endogenous
microRNA sponges: Evidence and controversy. Nat Rev Genet.
17:272–283. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karreth FA and Pandolfi PP: ceRNA
cross-talk in cancer: When ce-bling rivalries go awry. Cancer
Discov. 3:1113–11121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang L, Wu J, Ling MT, Zhao L and Zhao
KN: The role of the PI3K/Akt/mTOR signalling pathway in human
cancers induced by infection with human papillomaviruses. Mol
Cancer. 14:12015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang D, Sun G, Zhang H, Tian J and Li Y:
Long non-coding RNA ANRIL indicates a poor prognosis of cervical
cancer and promotes carcinogenesis via PI3K/Akt pathways. Biomed
Pharmacother. 85:511–516. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rong Z, Lu H, Lyu YY, Yang XM, Zhu LY,
Yang GD, Jiang PC, Re Y, Song WW, Wang JH, et al:
E6/E7-P53-POU2F1-CTHRC1 axis promotes cervical cancer metastasis
and activates Wnt/PCP pathway. Sci Rep. 7:447442017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ling W, Zhigang H, Tian H, Bin Z, Xiaolin
X and Hongxiu Z: HPV 16 infection up-regulates Piwil2, which
affects cell proliferation and invasion in cervical cancer by
regulating MMP-9 via the MAPK pathway. Eur J Gynaecol Oncol.
36:647–654. 2015.PubMed/NCBI
|
|
38
|
Wu Q, Guo L, Jiang F, Li L, Li Z and Chen
F: Analysis of the miRNA-mRNA-lncRNA networks in ER+ and ER-breast
cancer cell lines. J Cell Mol Med. 19:2874–2887. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li CY, Liang GY, Yao WZ, Sui J, Shen X,
Zhang YQ, Peng H, Hong WW, Ye YC, Zhang ZY, et al: Integrated
analysis of long non-coding RNA competing interactions reveals the
potential role in progression of human gastric cancer. Int J Oncol.
48:1965–1976. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He JH, Han ZP, Liu JM, Zhou JB, Zou MX, Lv
YB, Li YG and Cao MR: Overexpression of long non-coding RNA MEG3
inhibits proliferation of hepatocellular carcinoma Huh7 cells via
negative modulation of miRNA-664. J Cell Biochem. 118:3713–3721.
2015. View Article : Google Scholar
|
|
41
|
Tong GF, Qin N, Sun LW and Xu XL: Long
Noncoding RNA MEG3 suppresses glioma cell proliferation, migration
and invasion by acting as competing endogenous RNA of MiR-19a.
Oncol Res. 25:1471–1478. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liao M, Li B, Zhang S, Liu Q, Liao W, Xie
W and Zhang Y: Relationship between LINC00341 expression and cancer
prognosis. Oncotarget. 8:15283–15293. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bozgeyik E, Igci YZ, Jacksi MF, Arman K,
Gurses SA, Bozgeyik I, Pala E, Yumrutas O, Temiz E and Igci M: A
novel variable exonic region and differential expression of
LINC00663 non-coding RNA in various cancer cell lines and normal
human tissue samples. Tumor Biol. 37:8791–8798. 2016. View Article : Google Scholar
|
|
44
|
Liu F, Yuan JH, Huang JF, Yang F, Wang TT,
Ma JZ, Zhang L, Zhou CC, Wang F, Yu J, et al: Long noncoding RNA
FTX inhibits hepatocellular carcinoma proliferation and metastasis
by binding MCM2 and miR-374a. Oncogene. 35:5422–5434. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Montpetit A, Larose J, Boily G, Langlois
S, Trudel N and Sinnett D: Mutational and expression analysis of
the chromosome 12p candidate tumor suppressor genes in pre-B acute
lymphoblastic leukemia. Leukemia. 18:1499–1504. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tian Z, Wen S, Zhang Y, Shi X, Zhu Y, Xu
Y, Lv H and Wang G: Identification of dysregulated long non-coding
RNAs/microRNAs/mRNAs in TNM I stage lung adenocarcinoma.
Oncotarget. 8:51703–51718. 2017. View Article : Google Scholar : PubMed/NCBI
|