Introduction
Heat shock proteins (HSPs) have been first
discovered as stress-inducible proteins (1,2). It
is generally recognized that HSPs intracellularly act as molecular
chaperones and regulate proteostasis under stress conditions
(1,2). Based on their molecular masses, HSPs
are currently classified into seven families, including HSPA
(HSP70), HSPB (small HSPs), HSPC (HSP90) and HSPH (HSP110)
(3). Among small HSPs with monomer
molecular mass in the range of 12–43 kDa, HSP27, an ATP-independent
molecular chaperone, is induced by the heat shock associated with
physical and chemical stresses, including radiation, oxidative
stress and various chemotherapies (1). HSP27 is able to bind to improperly
folded proteins and further transfer them to the ATP-dependent
chaperones or to the protein degradation machines including
proteasomes or autophagosomes. The functions of HSP27 are regulated
by post-translational modifications such as phosphorylation
(1). Unphosphorylated HSP27 forms
large aggregated oligomers while its phosphorylation results in the
conformational changes leading to dissociated small oligomers
(1). On the other hand, HSP90, an
ATP-dependent molecular chaperone, ubiquitously and abundantly
exist in a variety of unstressed cells, represent 1–2% of total
cellular proteins (2). In addition
to protein folding, it is well known that HSP90 regulates the
binding of glucocorticoid to its specific receptor under
physiological conditions (2).
Evidence is accumulating that HSPs are involved in a variety of
pathophysiological cell processes (1,2).
Bone metabolism is strictly regulated by two
functional cells, osteoblasts and osteoclasts (4). The former cells contribute to bone
formation, whereas the latter cells are responsible for bone
resorption. In order to maintain the stability of bone and the
integrity, bone tissue is constantly reconstructed by a sequential
process consisted of the resorption of old bone and the subsequent
formation of new bone, so called bone remodeling (5). Regarding HSP27 in osteoblasts,
down-regulation of osteoblast proliferation is reportedly
accompanied by a transient increase in the HSP27 mRNA expression
(6). In addition, it has been
shown that estrogen facilitates the induction of HSP27 stimulated
by heat (7). We have previously
reported that endothelin-1 (ET-1), a bone remodeling agent,
stimulates the induction of HSP27 in osteoblast-like MC3T3-E1 cells
and that p38 mitogen-activated protein (MAP) kinase and stress
activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK)
play as positive regulators in the HSP27 induction (8,9). In
addition, we demonstrated that the mineralization of MC3T3-E1 cells
is modulated by the expression level of HSP27 protein and its
phosphorylation status (10). On
the other hand, as for HSP90 in osteoblasts, the expression of
HSP90 protein is reportedly induced by bisphosphonate, the most
useful medicine for osteoporosis treatment (11). In addition, it has recently been
shown that low-intensity pulsed ultrasound stimulation reportedly
upregulates HSP90 level, leading to the formation of mineralized
nodule (12). However, the details
of both HSP27 and HSP90 in osteoblasts have not yet been
clarified.
In the present study, we investigated the
involvement of HSP90 in the ET-1-stimulated HSP27 induction in
these cells. We herein show that HSP90 inhibitors amplify the
ET-1-induced HSP27 protein levels in MC3T3-E1 cells and that the
up-regulating effect of HSP90 inhibitors was exerted via
SAPK/JNK.
Materials and methods
Materials
ET-1 was obtained from Peptide Institute Inc.
(Minoh, Osaka, Japan). Geldanamycin was purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The inhibitors
17-demethoxygeldanamycin (17-DMAG) and SP600125 were obtained from
EMD Millipore (Billerica, MA, USA). Onalespib was purchased from
Selleck Chemicals (Houston, TX, USA). HSP27 antibodies and
glyceraldehyde 3 phosphate dehydrogenase (GAPDH) antibodies were
obtained from Santa Cruz Biotechnology, Inc., (Dallas, TX, USA).
Phospho-specific p38 MAP kinase antibodies, p38 MAP kinase
antibodies, phospho-specific SAPK/JNK antibodies and SAPK/JNK
antibodies were purchased from Cell Signaling Technology, Inc.,
(Danvers, MA, USA). An ECL western blotting detection system was
purchased from GE Healthcare Life Sciences (Little Chalfont, UK).
Other materials and chemicals were obtained from commercial
sources. Geldanamycin, 17-DMAG, onalespib and SP600125 were
dissolved in dimethyl sulfoxide. The maximum concentration of
dimethyl sulfoxide was 0.1%, which did not affect the assay for
western blot analysis.
Cell culture
Cloned osteoblast-like cells, MC3T3-E1 cells that
have been derived from newborn mouse calvaria (13), were maintained as previously
described (14). In brief, the
cells were cultured in α-minimum essential medium (α-MEM)
containing 10% fetal bovine serum (FBS) at 37ºC in a humidified
atmosphere of 5% CO2/95% air. The cells were seeded into
90-mm diameter dishes (20×104/dish) for western blot
analysis. After 5 days, the medium was exchanged to α-MEM
containing 0.3% FBS. The cells were then used for experiments after
48 h.
Western blot analysis
Western blotting was performed as described
previously as follows (15). The
cultured cells were pretreated with various doses of geldanamycin,
17-DMAG or onalespib for 60 min, and then stimulated by 0.1 µM ET-1
or vehicle in the presence of inhibitors in α-MEM containing 0.3%
FCS for the indicated periods. The cells were washed twice with
phosphate-buffered saline and then lysed, homogenized and sonicated
in a lysis buffer containing 62.5 mM Tris/HCl, pH 6.8, 2% sodium
dodecyl sulfate (SDS), 50 mM dithiothreitol and 10% glycerol.
SDS-polyacrylamide gel electrophoresis (PAGE) was performed by the
method of Laemmli in 10% polyacrylamide gel (16). The protein was fractionated and
transferred onto an Immun-Blot polyvinylidine difluoride (PVDF)
membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
membranes were blocked with 5% fat-free dry milk in Tris-buffered
saline-Tween (TBS-T; 20 mM Tris/HCl, pH 7.6, 137 mM NaCl, 0.1%
Tween-20) for 1 h before incubation with primary antibodies.
Western blot analysis was performed using HSP27 antibodies, GAPDH
antibodies, phospho-specific p38 MAP kinase antibodies, p38 MAP
kinase antibodies, phospho-specific SAPK/JNK antibodies and
SAPK/JNK antibodies with peroxidase-labeled antibodies raised in
goat anti-rabbit IgG which were used as secondary antibodies.
Peroxidase activity on PVDF membranes was visualized on X-ray film
by means of the ECL western blotting detection system.
Densitometric analysis
A densitometric analysis of the western blots was
performed using a scanner and image analysis software program
(ImageJ v1.48; National Institutes of Health, Bethesda, MD, USA).
HSP27 protein levels or the phosphorylated protein levels were
calculated as follows: The background-subtracted intensity was
respectively normalized to GAPDH intensity or the total protein
intensity, respectively, and plotted as the fold increase in
comparison to that of the control cells without stimulation.
Statistical analysis
All data are presented as the mean ± SEM of
triplicate determinations from three independent cell preparations.
The data were analyzed by ANOVA followed by Bonferroni method for
multiple comparisons between pairs. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of geldanamycin, 17-DMAG or
onalespib on ET-1-stimulated HSP27 induction in MC3T3-E1 cells
We have previously shown that ET-1 induces the
expression of HSP27 protein in osteoblast-like MC3T3-E1 cells
(8). In the present study, in
order to clarify whether HSP90 is implicated in the ET-1-stimulated
HSP27 induction in MC3T3-E1 cells, we examined the effects of HSP90
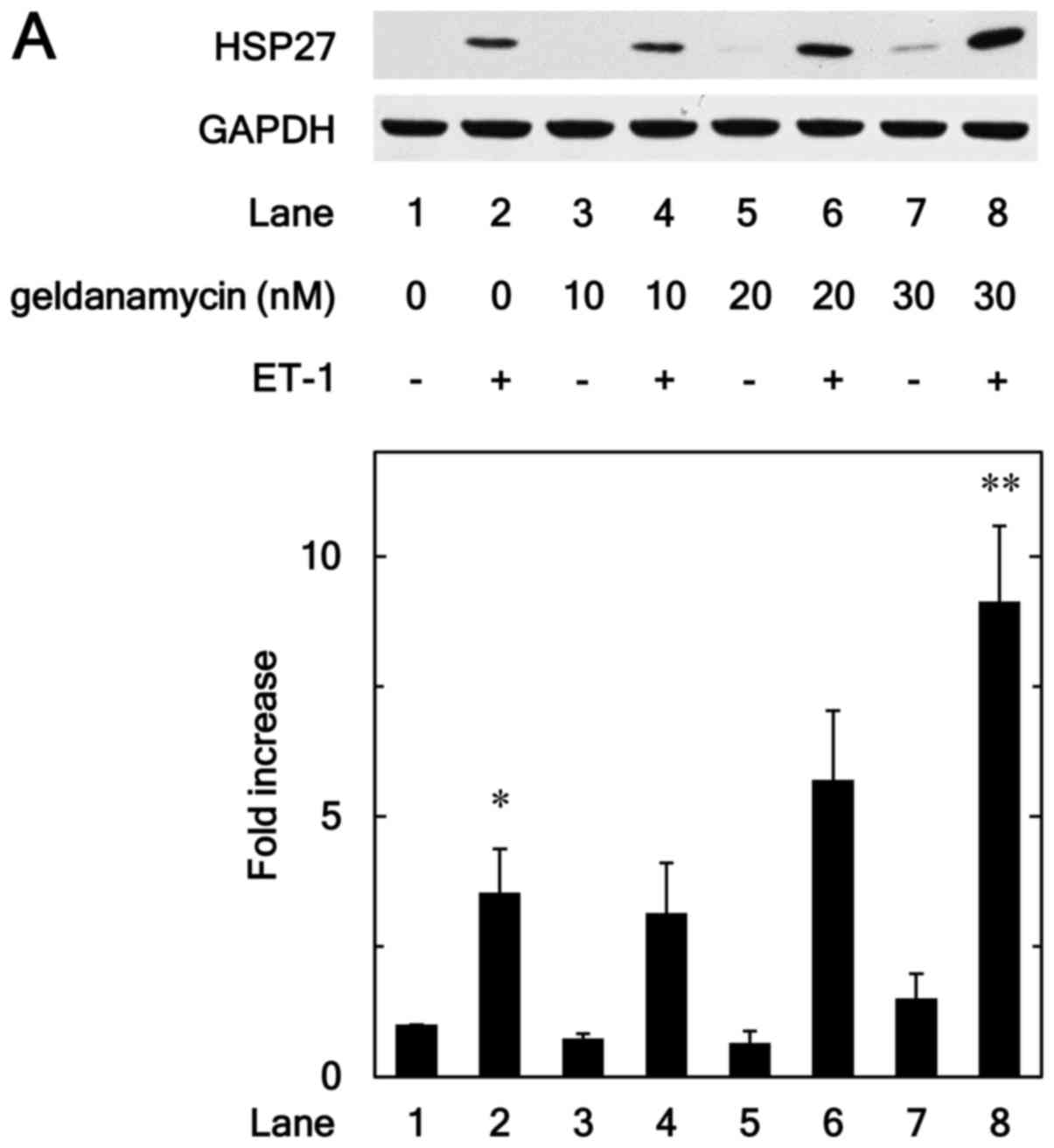
inhibitors on the HSP27 induction. Geldanamycin, an inhibitor of
HSP90 (17), significantly
enhanced the ET-1-induced levels of HSP27 protein (Fig. 1A). The effect of geldanamycin on
the HSP27 induction was dose-dependent over the range between 10
and 30 nM (Fig. 1A). We also found
that 17-DMAG, a less toxic derivative of geldanamycin (18), dose dependently augmented the
ET-1-stimulated HSP27 induction in the range between 10 and 30 nM
(Fig. 1B).
In addition, onalespib, a HSP90 inhibitor, which is
a different type from geldanamycin (19), as well as geldanamycin and its
derivative, markedly upregulated the HSP27 protein levels induced
by ET-1 (Fig. 1C).
Effects of geldanamycin, 17-DMAG or
onalespib on the ET-1-induced phosphorylation of p38 MAP kinase in
MC3T3-E1 cells
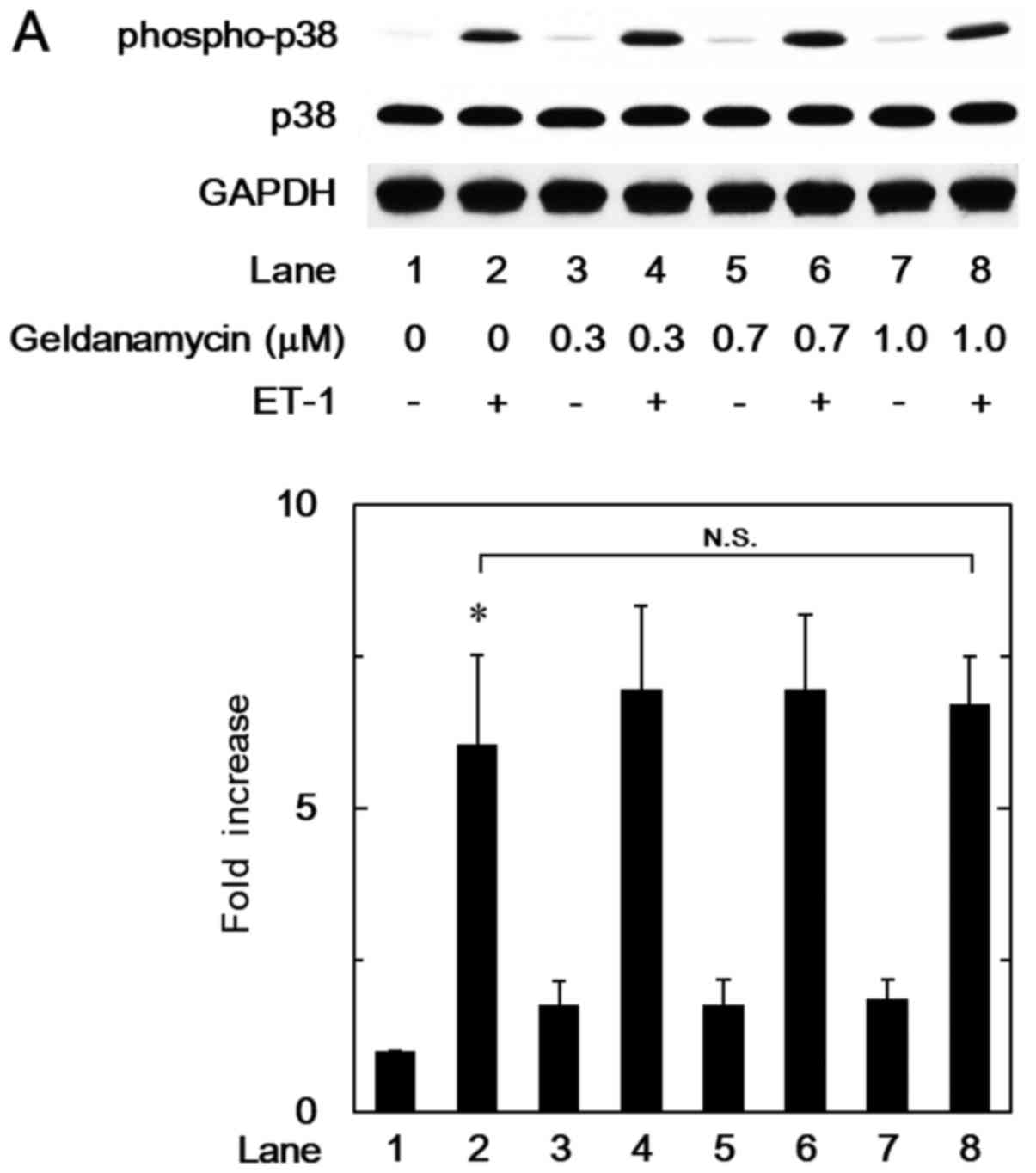
In our previous study (8), we have reported that p38 MAP kinase
acts as a positive intracellular molecule in the ET-1-stimulated
HSP27 induction in osteoblast-like MC3T3-E1 cells. In order to
investigate whether the HSP90 inhibitor-effect on the
ET-1-stimulated HSP27 induction is dependent on the activation of
p38 MAP kinase in these cells, we examined the effects of
geldanamycin, 17-DMAG or onalespib on the ET-1-induced
phosphorylation of p38 MAP kinase. However, geldanamycin (Fig. 2A), 17-DMAG (Fig. 2B) or onalespib (Fig. 2C) failed to affect the ET-1-induced
phosphorylation levels of p38 MAP kinase.
Effects of onalespib or 17-DMAG on the
ET-1-induced phosphorylation of SAPK/JNK in MC3T3-E1 cells
We have demonstrated that SAPK/JNK in addition to
p38 MAP kinase is involved in the ET-1-stimulated HSP27 induction
in MC3T3-E1 cells (9). Thus, we
next examined the effect of onalespib on the ET-1-induced
phosphorylation of SAPK/JNK. Onalespib, which alone did not affect
the SAPK/JNK phosphorylation, significantly strengthened the
ET-1-induced phosphorylation levels of SAPK/JNK (Fig. 3A). The amplifying effect of
onalespib on the SAPK/JNK phosphorylation was dose-dependent over
the range between 0.3 and 1.0 µM (Fig.
3A). Additionally, we showed that 17-DMAG remarkably enhanced
the ET-1-induced levels of phosphorylated SAPK/JNK (Fig. 3B).
Effect of SP600125 on the enhancement
by onalespib of ET-1-stimulated HSP27 induction in MC3T3-E1
cells
In order to further investigate whether HSP90
inhibitor enhances the ET-1-stimulated HSP27 induction via SAPK/JNK
activated by ET-1 in MC3T3-E1 cells, we examined effect of
SP600125, an inhibitor of SP600125 (20), on enhancement by onalespib of
ET-1-induced HSP27. SP600125 significantly reduced the
amplification by onalespib of ET-1-induced levels of HSP27 protein
(Fig. 4).
Discussion
In the present study, we showed that geldanamycin
and 17-DMAG, a geldanamycin derivative, which belong to HSP90
inhibitors (17,18), significantly potentiated the
ET-1-stimulated induction of HSP27 (HSPB1), a small HSP (HSPB), in
osteoblast-like MC3T3-E1 cells. Furthermore, onalespib (19), another HSP90 inhibitor different
from geldanamycin or its analogues, markedly increased the
ET-1-induced HSP27 protein levels. Based on our findings, it seems
likely that HSP90 plays a suppressive role in ET-1-stimulated HSP27
induction in osteoblast-like MC3T3-E1 cells.
Regarding the intracellular signaling of ET-1 in
osteoblasts, we have demonstrated that ET-1 stimulates the
induction of HSP27 via p38 MAP kinase and SAPK/JNK among the MAP
kinase superfamily (21), in
osteoblast-like MC3T3-E1 cells (8,9).
Based on our findings, we investigated whether the amplifying
effect of HSP90 inhibitors on the ET-1-stimulated HSP27 induction
is due to the modulation of these MAP kinases in MC3T3-E1 cells.
However, the HSP90 inhibitors, geldanamycin, 17-DMAG and onalespib,
did not have significant effects on the ET-1-induced levels of
phosphorylated p38 MAP kinase. Therefore, it seems unlikely that
HSP90 affects the ET-1-stimulated HSP27 induction via the signaling
pathway of p38 MAP kinase in osteoblast-like MC3T3-E1 cells. On the
other hand, we showed that onalespib significantly augmented the
ET-1-induced phosphorylated levels of SAPK/JNK. In addition, the
SAPK/JNK phosphorylation induced by ET-1 was markedly enhanced by
17-DMAG. Furthermore, we demonstrated that the enhancement by
onalespib of the ET-1-stimulated HSP27 induction was reduced by an
inhibitor of SAPK/JNK, SP600125 (20). Taking our findings into account as
a whole, it is most likely that HSP90 acts at a point upstream from
SAPK/JNK and negatively regulates the ET-1-stimulated HSP27
induction in osteoblast-like MC3T3-E1 cells. The potential
mechanism of HSP90 in the ET-1-induced HSP27 in osteoblasts shown
here is summarized in Fig. 5.
It is firmly established that HSP90, a major
molecular chaperone, plays as a central regulator of proteostasis
such as protein folding under stress conditions (2). In addition to protein folding,
accumulating evidence indicates that HSP90 is implicated in a
variety of physiological and pathological cell processes including
responses to steroid hormones, and neurodegenerative diseases
(2). Regarding HSP90 in
osteoblasts, it has been shown that bisphosphonate, the most useful
medicine for osteoporosis, stimulates expression of HSP90 in
osteoblasts (11). In addition,
low-intensity pulsed ultrasound stimulation reportedly enhances
osteoblasts proliferation and up-regulates HSP90, leading to dense
mineralization (12). We have
previously demonstrated that phosphorylated HSP27 acts as a
negative regulator of calcification in osteoblast-like MC3T3-E1
cells whereas un-phosphorylated HSP27 has a stimulatory effect on
the mineralization (10). It is
well known that the functions of small HSPs are regulated by
post-translational modifications including phosphorylation,
indicating that phosphorylated status of HSP27 has a switching role
in cellular functions (1). Our
present results show that HSP90 inhibitors limits the
ET-1-stimulated HSP27 induction in osteoblast-like MC3T3-E1 cells.
Based on these findings, it seems likely that HSP90 regulates the
mineralization of osteoblasts through modulating the HSP27 protein
levels. Our present findings regarding about the regulation by
HSP90 inhibitors of the HSP27 induction in osteoblasts might
provide a new aspect of HSP90 as a therapeutic target for metabolic
bone diseases such as osteoporosis. However, the physiological
significance of HSP27 in osteoblasts remains unclear. Further
investigations using another human osteoblast cells such as USO2 or
MG63 would be required to clarify the exact roles of HSP90 and
HSP27 in bone metabolism.
In conclusion, our results strongly suggest that
HSP90 negatively regulates ET-1-stimulated HSP27 induction at a
point upstream of SAPK/JNK in osteoblasts.
Acknowledgements
The authors would like to thank Yumiko Kurokawa for
her skillful technical assistance.
Funding
The present study was funded by Grant-in-Aid for
Scientific Research (grant nos. 26462289, 15K10487, 17K11002) from
the Ministry of Education, Culture, Sports, Science and Technology
of Japan, and the Research Funding for Longevity Sciences (grant
no. 26–12, 28-9) from National Center for Geriatrics and
Gerontology, Japan.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KF, TO and OK conceived and designed the
experiments. KF, TK, GS and RMN performed the experiments. KF, RMN,
OK and HT analyzed the data. KF, TO, OK and HT wrote the paper. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mymrikov EV, Seit-Nebi AS and Gusev NB:
Large potentials of small heat shock proteins. Physiol Rev.
91:1123–1159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schopf FH, Biebl MM and Buchner J: The
HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 18:345–360. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kampinga HH, Hageman J, Vos MJ, Kubota H,
Tanguay RM, Bruford EA, Cheetham ME, Chen B and Hightower LE:
Guidelines for the nomenclature of the human heat shock proteins.
Cell Stress Chaperones. 14:105–111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kular J, Tickner J, Chim SM and Xu J: An
overview of the regulation of bone remodelling at the cellular
level. Clin Biochem. 45:863–873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sims NA and Gooi JH: Bone remodeling:
Multiple cellular interactions required for coupling of bone
formation and resorption. Semin Cell Dev Biol. 19:444–451. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shakoori AR, Oberdorf AM, Owen TA, Weber
LA, Hickey E, Stein JL, Lian JB and Stein GS: Expression of heat
shock genes during differentiation of mammalian osteoblasts and
promyelocytic leukemia cells. J Cell Biochem. 48:277–287. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cooper LF and Uoshima K: Differential
estrogenic regulation of small M(r) heat shock protein expression
in osteoblasts. J Biol Chem. 269:7869–7873. 1994.PubMed/NCBI
|
|
8
|
Kawamura H, Otsuka T, Matsuno H, Niwa M,
Matsui N, Kato K, Uematsu T and Kozawa O: Endothelin-1 stimulates
heat shock protein 27 induction in osteoblasts: Involvement of p38
MAP kinase. Am J Physiol. 277:E1046–E1054. 1999.PubMed/NCBI
|
|
9
|
Tokuda H, Niwa M, Ito H, Oiso Y, Kato K
and Kozawa O: Involvement of stress-activated protein kinase/c-Jun
N-terminal kinase in endothelin-1-induced heat shock protein 27 in
osteoblasts. Eur J Endocrinol. 149:239–245. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kato K, Adachi S, Matsushima-Nishiwaki R,
Minamitani C, Natsume H, Katagiri Y, Hirose Y, Mizutani J, Tokuda
H, Kozawa O and Otsuka T: Regulation by heat shock protein 27 of
osteocalcin synthesis in osteoblasts. Endocrinology. 152:1872–1882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Romanello M, Bivi N, Pines A, Deganuto M,
Quadrifoglio F, Moro L and Tell G: Bisphosphonates activate
nucleotide receptors signaling and induce the expression of Hsp90
in osteoblast-like cell lines. Bone. 39:739–753. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miyasaka M, Nakata H, Hao J, Kim YK,
Kasugai S and Kuroda S: Low-intensity pulsed ultrasound stimulation
enhances heat-shock protein 90 and mineralized nodule formation in
mouse calvaria-derived osteoblasts. Tissue Eng Part A.
21:2829–2839. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sudo H, Kodama H, Amagai Y, Yamamoto S and
Kasai S: In vitro differentiation and calcification in a new clonal
osteogenic cell line derived from newborn mouse calvaria. J Cell
Biol. 96:191–198. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kozawa O, Suzuki A, Tokuda H and Uematsu
T: Prostaglandin F2alpha stimulates interleukin-6 synthesis via
activation of PKC in osteoblast-like cells. Am J Physiol.
272:E208–E211. 1997.PubMed/NCBI
|
|
15
|
Kato K, Ito H, Hasegawa K, Inaguma Y,
Kozawa O and Asano T: Modulation of the stress-induced synthesis of
hsp27 and alpha B-crystallin by cyclic AMP in C6 rat glioma cells.
J Neurochem. 66:946–950. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ochel HJ, Eichhorn K and Gademann G:
Geldanamycin: The prototype of a class of antitumor drugs targeting
the heat shock protein 90 family of molecular chaperones. Cell
Stress Chaperones. 6:105–112. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jez JM, Chen JC, Rastelli G, Stroud RM and
Santi DV: Crystal structure and molecular modeling of 17-DMAG in
complex with human Hsp90. Chem Biol. 10:361–368. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferraldeschi R, Welti J, Powers MV, Yuan
W, Smyth T, Seed G, Riisnaes R, Hedayat S, Wang H, Crespo M, et al:
Second-generation HSP90 inhibitor onalespib blocks mRNA splicing of
androgen receptor variant 7 in prostate cancer cells. Cancer Res.
76:2731–2742. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kyriakis JM and Avruch J: Mammalian MAPK
signal transduction pathways activated by stress and inflammation:
A 10-year update. Physiol Rev. 92:689–737. 2012. View Article : Google Scholar : PubMed/NCBI
|