Introduction
Oculocutaneous albinism (OCA) is a set of autosomal
recessive disorders characterized by a reduction or complete
absence of melanin in the skin, hair and eyes. The affected
individuals present with signs and symptoms including photophobia,
nystagmus, poor visual acuity and iris transillumination (1,2). No
effective therapy for OCA has been found previously. The worldwide
prevalence of all known forms of OCA is estimated to be 1:17,000
(3). In the Chinese Han population
of Shandong, the prevalence is ~1:18,000 and 3.80% of the
population are carriers (4).
To date, mutations in at least 16 genes have been
reported to be responsible for OCA (5). The nosology of OCA is based on
classification with genetic defects in molecules, comprising 12
syndromic OCA genes and seven non-syndromic OCA genes or loci,
including OCA1 (MIM 203100), OCA2 (MIM 203200), OCA3 (MIM 203290),
OCA4 (MIM 606574), OCA5 (MIM 615312), OCA6 (MIM 113750) and OCA7
(MIM 615179).
Mutations in both alleles of the tyrosinase (TYR)
gene can cause OCA1, which is characterized by a complete or
incomplete lack of melanin. OCA1 is the most predominant subtype in
the Chinese Han population (5).
OCA2, associated with mutations in the OCA2 gene, is the most
common type of albinism in the black African population (6). OCA3 is rare in Asiatic populations
associated with tyrosinase-related protein 1 (TYRP1). OCA4, OCA6
and OCA7 are caused by mutations in SLC45A2, SLC24A5 and C10 or
f11, respectively. OCA5 is a novel genetic cause mapped to
chromosome 4q24 (7).
As TYR mutations are responsible for 70.1% of cases
of OCA in the Chinese population (5), the present study performed direct
sequencing of TYR in 17 patients with OCA, which revealed 12
mutations in 10 patients, respectively, including two novel
mutations c.1198T>G (p.W400G) and c.819G>T (p.Q273H).
Patients and methods
Patients and clinical data
A total of 17 patients with OCA were enrolled in the
present study, including 6 women and 11 men, who were from 8
provinces of China (Beijing, Jiangsu, Hebei, Liaoning, Jilin,
Guangdong, Sichuan and Hunan). An additional 200 unrelated healthy
volunteers served as a control group. Routine screenings and
complete ophthalmological examinations were performed on all
participants following the provision of signed informed consent.
The typical clinical features of OCA, including hypopigmented hair,
skin and eyes, nystagmus, photophobia, poor vision and foveal
hypoplasia, were observed in all patients. None of the patients had
any other systemic diseases. The Institutional Review Board of the
Tongji Eye Institute of Tongji University School of Medicine
(Shanghai, China) approved the study, and all procedures were
performed in accordance with the tenets of the Declaration of
Helsinki.
Genetic analysis
Genomic DNA was extracted from peripheral leukocytes
using the Tiangen RelaxGene Blood DNA system (Tiangen Biotech, Co.,
Ltd., Beijing, China) according to the manufacturer's protocol. The
primers (Table I) were designed
for all five exons and splice junction sites of the TYR gene using
Primer3 software (version 0.4.0; http://bioinfo.ut.ee/primer3-0.4.0/). All exons and
flank regions of TYR were amplified using polymerase chain reaction
(PCR). A total of 25 µl PCR mixture contain 40 ng genomic DNA, 1 mM
each forward and reverse primers and 12.5 µl 2X Taq PCR MasterMix
(Tiangen Biotech Co., Ltd.). PCR was performed on C1000 Touch
Thermal Cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
using Touchdown PCR program with 35 cycles of amplification: 95°C
for 30 sec; 64–57°C for 30 sec beginning at 64°C and decreasing by
0.5°C each cycle for 14 cycles, until finishing at a final
annealing temperature of 57°C for 21 cycles and at 72°C for 40
sec.
 | Table I.Primers for amplification and sequence
analysis of human TYR. |
Table I.
Primers for amplification and sequence
analysis of human TYR.
| Primer | Primer sequence
(5′-3′) |
|---|
| TYR-EXON1A-F |
CCAGTTCCTGCAGACCTTGT |
| TYR-EXON1A-R |
TCATTTGGCCATAGGTCCCT |
| TYR-EXON1B-F |
GGGACCAAACTGCACAGAGA |
| TYR-EXON1B-R |
GGCTGCAATGAGTGTTCAGG |
| TYR-EXON2-F |
TGATGGATTTCTCAGAACATATCCCT |
| TYR-EXON2-R |
ACAACACATATTCTTGGTCAACTCA |
| TYR-EXON3-F |
TGGGATAATCACATAGGTTTTCAGT |
| TYR-EXON3-R |
GGTGACAACCTGATCACAGACA |
| TYR-EX0N4-F |
CCATGTCTCCAGATTTTAATATATGCC |
| TYR-EX0N4-R |
CACTTTCAGGATTTAAAGTGTTCAGGA |
| TYR-EXON5-F |
GCCTTCAAACCCAGGTGTCT |
| TYR-EXON5-R |
GGAACCTGGACATTACTTTGAGT |
The PCR products were sequenced using an ABI3730
automated sequencer (PE Biosystems, Foster City, CA, USA). Benign
polymorphisms were excluded using the database of the 1000 Genomes
Project (http://www.1000genomes.org/) and the
dbSNP National Center for Biotechnology Information database
(http://www.ncbi.nlm.nih.gov/projects/SNP/). The
albinism database (http://www.ifpcs.org/albinism/) and reported
literature were then used to determine whether the mutations had
been previously reported as pathogenic. To predict the
pathogenicity of TYR variants in the present study, in
silico analysis was performed with Sorting Intolerant From
Tolerant (SIFT; http://sift.jcvi.org/), Polymorphism
Phenotyping (PolyPhen)-2 (http://genetics.bwh.harvard.edu/pph2/), I-Mutant
(http://folding.biofold.org/i-mutant/i-mutant2.0.html)
and Human Splicing Finder (HSF) 3.0 (http://www.umd.be/HSF3/HSF.shtml), respectively. The
conservation of the amino acid substitutions were assessed by
multiple sequence alignment with sequences from Homo sapiens
(NP_000363.1), Canis lupus familiaris (NP_001002941.1),
Bos taurus (NP_851344.1), Mus musculus (NP_035791.1),
Rattus norvegicus (NP_001101005.1), Gallus gallus
(NP_989491.1), Danio rerio (NP_571088.1), Xeno
pustropicalis (NP_001096518.1) and Bacillus megaterium
(UniProtKB:B2ZB02). The crystal structure of protein data bank
(PDB): 5I3B (TYR from Bacillus megaterium with configuration
B of hydroquinone inhibitor in the active site) was used to predict
and improve understanding of the potential impact of the novel
mutation (8).
Results
In the present study, 17 patients were diagnosed
with non-syndromic OCA based on the routine screenings and complete
ophthalmological examination. The parents of all patients were
unaffected, which is consistent with the pattern of autosome
recessive inheritance. Using PCR combined with Sanger sequencing, a
total of 12 mutations were found in TYR, including 10 previously
reported mutations associated with OCA1 and two novel mutations
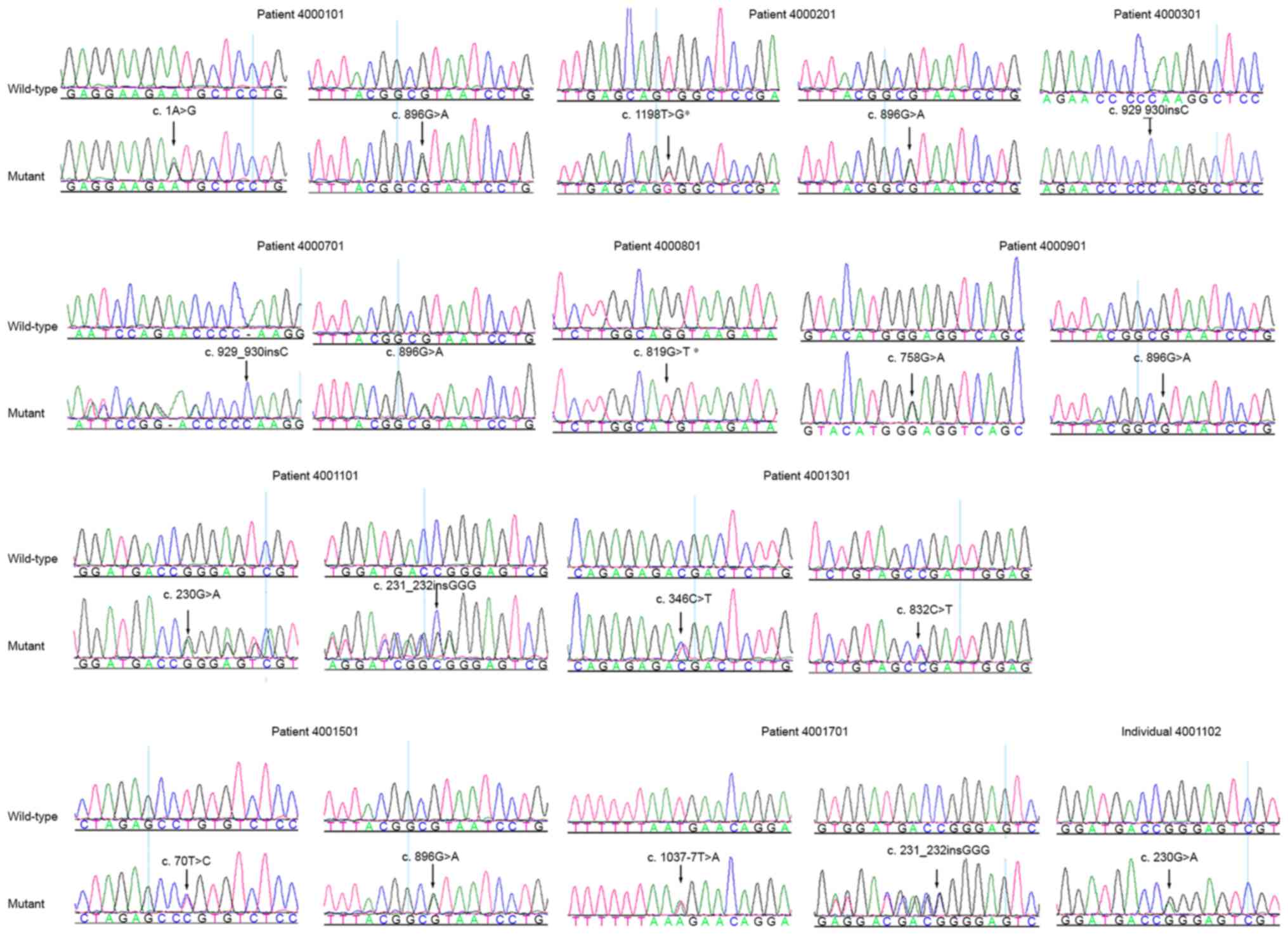
(Fig. 1 and Table II). Patient 4000101 was a compound
heterozygote for mutations c.1A>G (p.M1?) and c.896G>A
(p.R299H) in the TYR gene. Patient 4000201 was a compound
heterozygote for mutations c.1198T>G (p.W400G) and c.896G>A
(p.R299H). Patient 4000301, from a consanguineous family, was a
homozygote for mutation c.929_930insC (p.R311Kfs*7). Patient
4000701 was compound heterozygous for c.929_930insC (p.R311Kfs*7)
and c.896G>A (p.R299H). Patient 4000801 was a homozygote for
c.819G>T (p.Q273H). Patient 4000901 was a compound heterozygote
for mutations c.758G>A (p.G253E) and c.896G>A (p.R299H).
Patient 4001101 was a compound heterozygote for mutations
c.231_232insGGG (p.R77_E78insG) and c.230G>A (p.R77Q), and
c.230G>A was confirmed in the unaffected family member of
patient 4001101 (Fig. 1). Patient
4001301 carried mutations c.346C>T (p.R116*) and c.832C>T
(p.R278*), patient 4001501 carried mutations c.896G>A (p.R299H)
and c.70T>C (p.C24R), and patient 4001701 carried the mutation
c.231-232insGGG (p.R77_E78insG) and the splicing mutation
c.1037-7T>A.
| Table II.Summary of TYR mutations in the
present study. |
Table II.
Summary of TYR mutations in the
present study.
|
|
|
|
| Mutation 1 | Mutation 2 |
|
|---|
|
|
|
|
|
|
|
|
|---|
| Patient | Gender | Clinical
diagnosis | Gene | Nucleotide
change | Amino acid
change | Nucleotide
change | Amino acid
change | Province |
|---|
| 4000101 | F | OCA | TYR | c.1A>G | p.M1? | c.896G>A | p.R299H | Jilin |
| 4000201 | M | OCA | TYR | c.1198T>G | p.W400G | c.896G>A | p.R299H | Jiangsu |
| 4000301 | M | OCA | TYR | c.929_930insC | p.R311Kfs*7 | c.929_930insC | p.R311Kfs*7 | Jiangsu |
| 4000701 | M | OCA | TYR | c.929_930insC | p.R311Kfs*7 | c.896G>A | p.R299H | Guangdong |
| 4000801 | M | OCA | TYR | c.819G>T | p.Q273H | c.819G>T | p.Q273H | Guangdong |
| 4000901 | F | OCA | TYR | c.758G>A | p.G253E | c.896G>A | p.R299H | Sichuan |
| 4001101 | F | OCA | TYR |
c.231_232insGGG | p.R77_E78insG | c.230G>A | p.R77Q | Sichuan |
| 4001301 | M | OCA | TYR | c.346C>T | p.R116* | c.832C>T | p.R278* | Hunan |
| 4001501 | M | OCA | TYR | c.70T>C | p.C24R | c.896G>A | p.R299H | Beijing |
| 4001701 | M | OCA | TYR |
c.231_232insGGG | p.R77_E78insG | c.1037-7T>A | – | Jiangsu |
Among the mutations identified, two mutations
c.819G>T (p.Q273H) and c.1198T>G (p.W400G) were novel, and
their pathogenicity were predicted via in silico analysis.
The SIFT and PolyPhen-2 scores for the c.819G>T (p.Q273H)
mutation were 0.030 and 0.981, respectively, which predicted that
the mutation was ‘probably damaging’. The I-Mutant online server
predicted that the mutation may decrease protein stability with a
reliability index (RI) of 6. The c.1198T>G mutation in the TYR
gene was not detected in the 200 normal controls enrolled in the
present study, nor was it present within the dbSNP database, the
1000 Genomes Project or the albinism database (http://www.ifpcs.org/albinism/). This mutation,
resulting in the conversion from tryptophan to glycine at codon
400, was predicted to be ‘probably damaging’ with a score of 0.99
by PolyPhen2 and a score of 0 by SIFT, and it was predicted that
the protein stability may decrease with an RI of 9 by the I-Mutant
online server. Multiple sequence alignment showed that residues
W241, I237 and H245 in the TYR
protein of Bacillus megaterium are highly conserved across
species, and correspond to W400, I393 and
H404 in the human TYR protein, respectively (Fig. 2A). The crystal structure of
wild-type TYR (PDB ID: 5I3B) showed that W241 interacts
with I237 and H245, and this interaction may
be impaired when W241 is substituted by amino acid G
(Fig. 2B).
Discussion
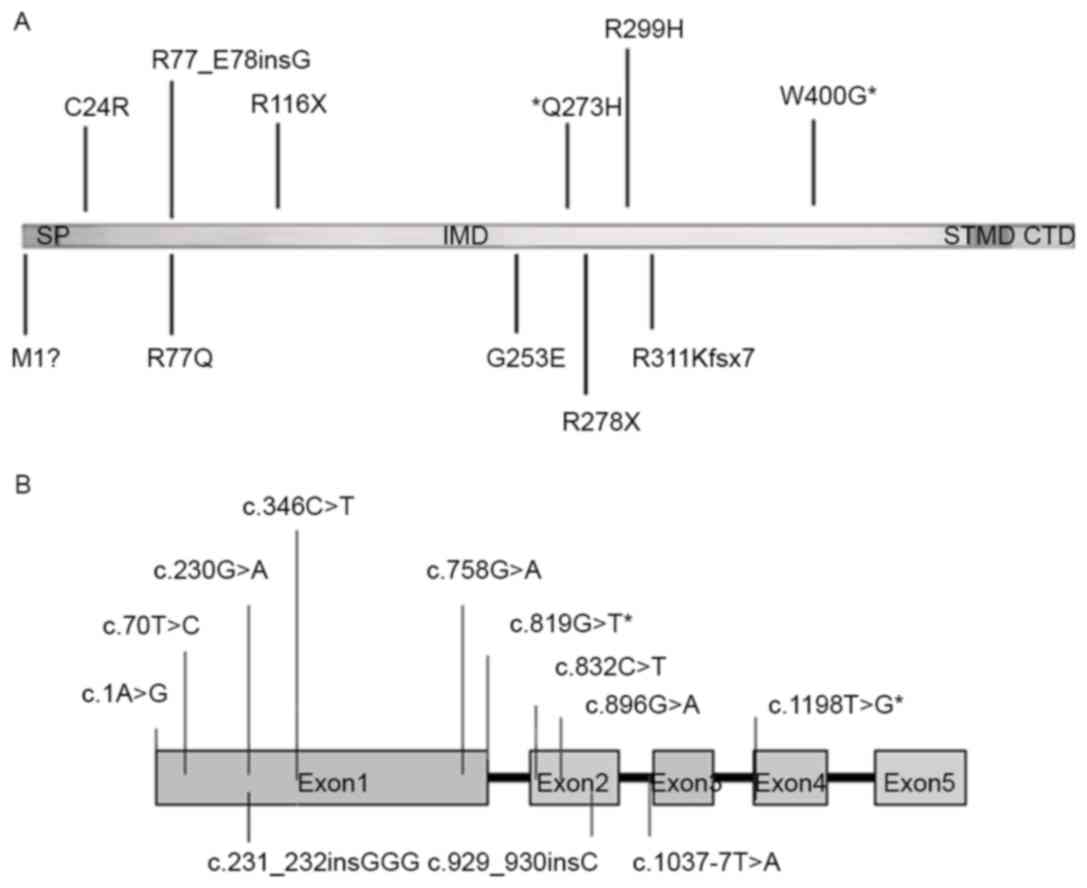
TYR is a glycoprotein containing four regions, a
signal sequence (SP; amino acid residues 1–18), an intramelanosomal
domain (IMD; residues 19–476) with a binuclear copper binding site,
a single α-helical trans-membrane domain (STMD; residues 477–497),
and a flexible C-terminal domain (CTD; residues 498–529), as shown
in Fig. 3A (9). There are three key enzymes, TYR,
TYRP1 and dopachrome tautomerase, which are important in melanin
biosynthesis (10). Among these, a
defect in TYR can lead to the complete or partial lack of melanin
as TYR catalyzes the critical first and second reactions in the
conversion of TYR to melanin (11). The human TYR gene located on
chromosome 11q14.3 is composed of five exons encoding a copper
binding protein with a molecular weight of ~75 kDa. In the present
study, mutation analysis of the TYR gene was performed in 17
patients with OCA, which identified 12 mutations as the cause of
OCA in 10 of the patients with OCA. Of these mutations, 10
mutations were clustered in exon 1 and exon 2, which are the
mutation hotspots of TYR in the Chinese Han population (5). For the additional two mutations, one
mutation c.1198T>G (p.W400 G) was located in exon 4 and another
mutation IVS2-7T>A was located in intron 2 (Fig. 3B). In Chinese patients with OCA1,
missense mutations of TYR have been found to account for almost
62.5% of all reported TYR mutations, whereas the insertion and
deletion mutations account for 25% (12).
Of the 12 mutations identified in the present study,
10 mutations were reported as causative for OCA in previous
literature. Among these 10 mutations, R299H was the most frequent
mutation in the present study (5/12). R299H is the most frequent
mutation in Chinese, Caucasian, Korean, and Christian Arab
populations, and this mutation may affect the function of TYR by
disrupting copper binding and different substitutions occurring in
R299 (5,13–15).
c.231-232insGGG is another frequent mutation in the Chinese OCA1
population, resulting in an insertion of glycine residue between
R77 and E78 (13). Mutation c.230G>A, resulting in
an amino acid change from arginine to glutamine at position 77, has
been reported in the Japanese, Korean, Chinese, German and
Pakistani populations (16). The
mutation c.929_930insC, resulting in a truncated polypeptide, is
the most frequent mutation allele in far East Asian OCA1
populations (13,17). The missense mutation c.1A>G
(p.M1?) has been found in British and Chinese patients with OCA1,
and this may alter codons initiating translation to M31, resulting
in a skipping of the putative SP (18,19).
The splicing mutation IVS2-7T>A is frequently found in patients
with OCA1 worldwide and can result in pathological splicing sites
(19–22).
It is the first time, to the best of our knowledge,
that the additional two mutations c.819G>T (p.Q273H) and
c.1198T>G (p.W400G) identified in the present study have been
associated with OCA. In the in silico analysis, c.819G>T
(p.Q273H) was predicted to be pathogenic, although it has been
reported as rs748669377 with a frequency of 1:120,054 in the Exome
Aggregation Consortium (http://exac.broadinstitute.org/). In addition, HSF 3.0
predicted that this mutation may result in disruption of the
wild-type donor site, most likely affecting splicing. However, its
exact mechanism requires further investigation for confirmation.
The other novel mutation c.1198T>G (p.W400G) occurred at the
same amino acid position as another pathogenic mutation
c.1199G>T (p.W400L). The amino acid residue W400
locating in the IMD of the human TYR protein, was found to be
highly conserved from bacteria to humans (Fig. 2A). Residues W241,
I237 and H245 in the TYR protein of
Bacillus megaterium corresponded with W400,
I393 and H404 in the human TYR protein,
respectively (Fig. 2A). Structural
analysis of wild-type TYR from Bacillus megaterium (PDB:
5I3B) showed that W241 interacts with I237
and H245, and the predicted structure of the mutated
protein shows that this substitution disrupted hydrogen bonds in
these two amino acids, impairing the spatial conformation of TYR
(Fig. 2B).
In conclusion, mutation analysis of the TYR gene in
17 patients with non-syndromic OCA revealed that 12 mutations of
TYR may have caused OCA in 10 patients. Among these mutations, two
novel mutations c.1198T>G (p.W400G) and c.819G>T (p.Q273H)
were identified, expanding on the mutation spectrum of TYR in OCA.
This may assist in the prenatal examination and genetic diagnosis
of OCA, and offers novel insight into the molecular mechanism of
OCA1.
Acknowledgements
This study was supported by the National Key Basic
Research Program of China (973 Program; grant no. 2015CB964601),
the National Natural Science Foundation of China (grant no.
81371062). Professor Jianjun Chen was supported by the Thousand
Youth Talents Program of China.
References
|
1
|
Kamaraj B and Purohit R: Mutational
analysis of oculocutaneous albinism: A compact review. Biomed Res
Int. 2014:9054722014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Summers CG: Albinism: Classification,
clinical characteristics, and recent findings. Optom Vis Sci.
86:659–662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gronskov K, Brøndum-Nielsen K, Lorenz B
and Preising MN: Clinical utility gene card for: Oculocutaneous
albinism. Eur J Hum Genet. 22:2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gong Y, Shao C, Zheng H, Chen B and Guo Y:
Study on genetic epidemiology of albinism. Yi Chuan Xue Bao.
21:169–172. 1994.(In Chinese). PubMed/NCBI
|
|
5
|
Wei A, Wang Y, Long Y, Wang Y, Guo X, Zhou
Z, Zhu W, Liu J, Bian X, Lian S and Li W: A comprehensive analysis
reveals mutational spectra and common alleles in Chinese patients
with oculocutaneous albinism. J Invest Dermatol. 130:716–724. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grønskov K, Ek J and Brondum-Nielsen K:
Oculocutaneous albinism. Orphanet J Rare Dis. 2:432007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kausar T, Bhatti MA, Ali M, Shaikh RS and
Ahmed ZM: OCA5, a novel locus for non-syndromic oculocutaneous
albinism, maps to chromosome 4q24. Clin Genet. 84:91–93. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deri B, Kanteev M, Goldfeder M, Lecina D,
Guallar V, Adir N and Fishman A: The unravelling of the complex
pattern of tyrosinase inhibition. Sci Rep. 6:349932016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lai X, Soler-Lopez M, Wichers HJ and
Dijkstra BW: Large-scale recombinant expression and purification of
human tyrosinase suitable for structural studies. PLoS One.
11:e01616972016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamaguchi Y and Hearing VJ: Melanocytes
and their diseases. Cold Spring Harb Perspect Med. 4:pii: a017046.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sánchez-Ferrer A, Rodríguez-López JN,
García-Cánovas F and García-Carmona F: Tyrosinase: A comprehensive
review of its mechanism. Biochim Biophys Acta. 1247:1–11. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu J, Choy KW, Chan LW, Leung TY, Tam PO,
Chiang SW, Lam DS, Pang CP and Lai TY: Tyrosinase gene (TYR)
mutations in Chinese patients with oculocutaneous albinism type 1.
Clin Exp Ophthalmol. 38:37–42. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsai CH, Tsai FJ, Wu JY, Lin SP, Chang JG,
Yang CF and Lee CC: Insertion/deletion mutations of type I
oculocutaneous albinism in chinese patients from Taiwan. Hum Mutat.
14:5421999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tripathi RK, Strunk KM, Giebel LB, Weleber
RG and Spritz RA: Tyrosinase gene mutations in type I
(tyrosinase-deficient) oculocutaneous albinism define two clusters
of missense substitutions. Am J Med Genet. 43:865–871. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park SK, Lee KH, Park KC, Lee JS, Spritz
RA and Lee ST: Prevalent and novel mutations of the tyrosinase gene
in Korean patients with tyrosinase-deficient oculocutaneous
albinism. Mol Cells. 7:187–191. 1997.PubMed/NCBI
|
|
16
|
Shah SA, Raheem N, Daud S, Mubeen J,
Shaikh AA, Baloch AH, Nadeem A, Tayyab M, Babar ME and Ahmad J:
Mutational spectrum of the TYR and SLC45A2 genes in Pakistani
families with oculocutaneous albinism, and potential founder effect
of missense substitution (p. Arg77Gln) of tyrosinase. Clin Exp
Dermatol. 40:774–780. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park SH, Chae H, Kim Y and Kim M:
Molecular analysis of Korean patients with oculocutaneous albinism.
Jpn J Ophthalmol. 56:98–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu N, Kong XD, Shi HR, Wu QH and Jiang M:
Tyrosinase gene mutations in the Chinese Han population with OCA1.
Genet Res (Camb). 96:e142014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Breimer LH, Winder AF, Jay B and Jay M:
Initiation codon mutation of the tyrosinase gene as a cause of
human albinism. Clin Chim Acta. 227:17–22. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ko JM, Yang JA, Jeong SY and Kim HJ:
Mutation spectrum of the TYR and SLC45A2 genes in patients with
oculocutaneous albinism. Mol Med Rep. 5:943–948. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
King RA, Pietsch J, Fryer JP, Savage S,
Brott MJ, Russell-Eggitt I, Summers CG and Oetting WS: Tyrosinase
gene mutations in oculocutaneous albinism 1 (OCA1): Definition of
the phenotype. Hum Genet. 113:502–513. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goto M, Sato-Matsumura KC, Sawamura D,
Yokota K, Nakamura H and Shimizu H: Tyrosinase gene analysis in
Japanese patients with oculocutaneous albinism. J Dermatol Sci.
35:215–220. 2004. View Article : Google Scholar : PubMed/NCBI
|