Introduction
Hepatocellular carcinoma (HCC) is one of most
frequent causes of cancer-associated mortality worldwide (1). Despite the development of specific
treatment strategies, the prognosis of HCC remains poor. The
outcomes of patients with HCC are far from satisfactory even in
patients undergoing surgical resection at early stages (2). For advanced HCC patients, sorafenib,
a multikinase inhibitor, has been demonstrated to be one of the few
effective drugs (3). Experimental
evidence has shown that it suppresses tumor growth by its capacity
to inhibit vessel formation and induces apoptosis by targeting
vascular endothelial growth factor receptor, platelet derived
growth factor-β and c-kit in a number of different cancer types
(4,5). However, it only prolonged the
survival of HCC by a few months (6). Thus, identification of drugs which
strengthen the antitumor effect of sorafenib is greatly
warranted.
Bufalin has been demonstrated to exert potent
antitumor activities in a number of human cancer types (7). It suppresses tumor cell proliferation
and angiogenesis, and induces apoptosis and differentiation in
cancer cells (8–10). In addition, it has been reported to
reverse multi-drug resistance in various types of cancers (11). Given that sorafenib and bufalin are
potent antitumor drugs, the present study speculated that greater
inhibitory effects may be generated in HCC with their combined
treatment. In a previous study, their synergistic effect has
already been confirmed by their inhibition of tumor cell
proliferation and vessel formation (12). However, whether enhanced apoptosis
would also be induced by their combined treatment requires further
exploration.
In the present study, it was shown that bufalin
promoted the inhibitory effect of sorafenib in tumor cell
proliferation. Apoptosis was also increased greatly by bufalin in
sorafenib-treated HCC cells. Furthermore, an in vivo study
was conducted using the HCC cell line SMMC-7721 as it has been
adopted to establish subcutaneous HCC tumors previously (13). It was demonstrated that the
apoptosis rate was significantly increased in mice injected with
bufalin. Ultimately, western blot analysis identified that B-cell
lymphoma 2 (Bcl-2)-associated X protein (Bax), caspase 7 and
poly-(adenosine diphosphate-ribose) polymerase (PARP) are important
molecules responsible for enhanced apoptosis. In conclusion, the
findings suggested that bufalin may promote sorafenib-induced
apoptosis in HCC. Therefore, the combination of these drugs may
have clinical utility as a favorable therapy in the treatment of
HCC.
Materials and methods
Reagents and antibodies
Sorafenib (Selleck Chemicals, Houston, TX, USA) and
bufalin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) were
dissolved in dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA) and
diluted to their working concentrations (sorafenib at
concentrations of 2.5, 5, 10 µM and bufalin at concentrations of 5,
10 and 20 nM). Antibodies against Bcl-2 (Abcam, Cambridge, UK; cat.
no. ab692), Bax (Abcam; cat. no. ab32503), caspase 7 (Bioworld
Technology, Inc., St Louis Park, MN, USA; cat. no. BS6544), caspase
8 (Bioworld Technology, Inc.; cat. no. AP0237), PARP (Bioworld
Technology, Inc.; cat. no. BS70001) and GAPDH (Bioworld Technology,
Inc.; cat. no. MB001) were also used.
Cell culture
PLC/PRF/5 and SMMC-7721 cells were purchased from
the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). Cells were cultured in high-glucose Dulbecco's
modified Eagle's medium (Hyclone; GE Healthcare, Chicago, IL, USA)
added with 10% fetal bovine serum (FBS; Hyclone; GE Healthcare) and
1% penicillin/streptomycin at 37°C containing 5% CO2.
Cells were passaged when they reached 80% confluency and used after
the third passage.
Determination of concentrations of
sorafenib and bufalin that may achieve optimal synergistic
effect
The combined index (CI) was calculated by the
CalcuSyn software. CI>1 indicated an antagonistic effect,
CI<1, indicated a synergistic effect and CI=1, indicated an
additive effect (14).
Animals
The present study was approved by Fudan University
Shanghai Cancer Center (Shanghai, China). A total of 24, 6-week old
male Balb/c nude mice weighing 20 g were used in the present study,
and were purchased from Beijing Vital River Laboratory Animal
Technology Co., Ltd. (Beijing, China). The mice were raised under
the following pathogen-free conditions: Room temperature, 20°C;
relative humidity, ~50%. The mice were given ad libitum access to
food and water and maintained under a 12-h light/dark cycle. The
mice were randomly divided into four groups: Control, sorafenib,
bufalin and the combination, with six mice per group. Animals were
raised in pathogen-free conditions and received humane care
according to the principles of animal care issued by Fudan
University (12). All experiments
conformed to the stipulations of the Animal Experimentation of
Fudan University. The mice were divided into 4 groups, those that
were subjected to daily administration of either 10 mg/kg sorafenib
(sorafenib group) via oral administration, 10 mg/kg bufalin
(bufalin group) via intraperitoneal injection, a combination of
both drugs (combination group) or saline via intragastric
administration in the vehicle (control group). The treatment lasted
for 16 days, following which the mice were sacrificed, and the
tumors were obtained.
In vivo tumorigenicity assay
The mice were not fasted prior to the following
treatments. A total of 1×107 SMMC-7721 cells in a volume
of 200 µl PBS were injected into the right flank of each mouse to
form subcutaneous tumors. When the volume of these subcutaneous
tumors reached a size of 100–300 mm3, the mice were
treated with intraperitoneal injections of 1 mg/kg bufalin (5
days/week), 30 mg/kg oral uptake of sorafenib (5 days/week), or a
combination of the two drugs (intraperitoneal injections of 1 mg/kg
bufalin combined with oral uptake of 30 mg/kg sorafenib). The
control mice were injected with saline. The tumor-bearing mice were
sacrificed following 16 days of treatments, and tumors were excised
and subjected to apoptosis assays and hematoxylin-eosin (HE)
staining. All procedures conformed to the ethical principles of
animal experimentation as stipulated by Fudan University.
HE staining
Paraffin sections were baked at 70°C for 1 h,
de-paraffinized in xylene, rehydrated in gradually varied alcohol,
and the sections were treated with 3% H2O2 to
neutralize endogenous peroxidase for 30 min. The antigen retrieval
was also conducted. Following antigen retrieval, the sections were
dipped into a Coplin jar containing Mayer's hematoxylin and
agitated for 30 sec and 1% eosin Y solution for 20 sec with
agitation at 25°C. The sections were dehydrated with two changes of
95% alcohol and two changes of 100% alcohol for 30 sec each.
Cell proliferation
Cell proliferation was determined using a Cell
Counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). HCC cells were plated in 96-well plates at a
density of 5,000 cells per well. The cells were subject to
treatment with 2.5, 5 and 10 µM sorafenib and 5, 10 and 20 nM
bufalin and the combination of both at these concentrations for 24
h at 37°C. Cell viability was measured following this incubation
with drug for 24 h using the CCK-8 kit. The absorbance was measured
at a wavelength of 450 nm with a microplate reader to determine the
cell viability rate.
Different treatments of HCC cells
HCC cells were subject to 2.5, 5, 10 µM sorafenib
and 5, 10 and 20 nM bufalin and the combination of both at these
concentrations for 24 h at 37°C.
Cell cycle assay
HCC cells were plated in 6-well plates at
2×105 cells per well and were subjected to 10 µM
sorafenib, 20 nM bufalin and the combination of 10 µM sorafenib and
20 nM bufalin for 24 h at 37°C. Then cells were trypsinized by
0.25% trypsin and then washed with PBS. A cell cycle assay was
applied (Beyotime Institute of Biotechnology; C1052). Subsequently,
cells were fixed in 70% methanol at 4°C for 2 h and stained with
propidium iodide (PI) [which consisted of 0.5 ml staining buffer,
25 µl PI staining reagent (20 X), 10 µl RNase A (50 X)] for 30 min
at 37°C. A flow cytometer was applied (FC500, Beckman Counter,
Inc., Brea, CA, USA) to detect fluorescence at excitation
wavelength of 350 nm, The multiCycle AV DNA Analysis software
(version 306; Phoenix Flow Systems, San Diego, CA, USA) was adopted
to perform analysis.
Terminal deoxynucleotidyl transferase
dUTP nick-end labelling (TUNEL) assay
Tissue apoptosis was determined with a TUNEL
Detection kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) in
compliance with the manufacturer's protocol. Tumor samples from
in vivo studies were rinsed in PBS and fixed in 10%
paraformaldehyde/PBS for 20 min at 25°C. Samples were dehydrated in
70% ethanol, paraffin embedded, and sectioned (4-µm). The slides
were rinsed twice with PBS. A total of 50 µl TUNEL reaction mixture
was added on the sample and covered with parafilm during the
incubation. The samples were incubated at 37°C for 1 h in a
humidified chamber in the dark. Finally, the samples undergoing
apoptosis were counted in three randomly chosen fields in a drop of
PBS under a fluorescence microscope.
Evaluation of apoptosis via Hochest
33258
Following treatment with sorafenib, bufalin or the
combined treatment for 24 h, a total of 1×105 HCC cells
were harvested and stained with Hochest 33258 (Beyotime Institute
of Biotechnology; C1011) for 5 min at 25°C. Apoptosis was detected
using Olympus fluorescence microscope. The excitation wavelength
was 350 nm, and the emission wavelength was 460 nm.
Evaluation of apoptosis via Annexin V
and 7-ADD
Following treatment with sorafenib, bufalin or the
combined treatment for 24 h, a total of 1×105 HCC cells
were harvested and stained with the AnnexinV-PE/7-ADD or Annexin
V-FITC/PI apoptosis detection kit (BD Biosciences). The
fluorescence intensity was detected via flow cytometry (FC500,
Beckman Counter, Inc.). Apoptosis rate was calculated by the
proportion of apoptotic cells of the total cells, using 3 randomly
chosen fields of view.
Colony formation assay
A total of 1.5×103 cells were seeded in
each well of a 6-well plate and cultured in DMEM medium containing
10% FBS. Following 10 days from seeding, the cells were stained
with crystal violet at 37°C for 20 min (Beijing Solarbio Science
& Technology Co., Ltd., Beijing, China) and counted under three
fields of view. Gelcount 1.2 software (Oxford Optronix, Abingdon,
UK) was applied to analyze the stained cells.
Western blotting
HCC cells were lysed in radioimmunoprecipitation
assay buffer containing 1% proteinase inhibitor cocktail (Beijing
Solarbio Science & Technology Co., Ltd.; R0020). The protein
concentration was determined using a bicinchoninic acid assay.
Proteins (20 µg) were loaded were separated on 10% SDS-PAGE gels
and transferred to polyvinylidene difluoride membranes 5% skim milk
was used as the blocking buffer to incubate the membrane for 1 h at
37°C. The corresponding proteins were detected with the primary
antibodies against Bcl-2 (Abcam, Cambridge, UK; cat. no. ab692;
dilution: 1:500), Bax (Abcam; cat. no. ab32503; dilution: 1:2,000),
caspase-7 (Bioworld Technology, Inc., St Louis Park, MN, USA; cat.
no. BS6544; dilution: 1:1,000), caspase-8 (Bioworld Technology,
Inc.; cat. no. AP0237; dilution: 1:1,000), PARP (Bioworld
Technology, Inc.; cat. no. BS70001; dilution: 1:1,000) and GAPDH
(Bioworld Technology, Inc.; cat. no. MB001; dilution: 1:1,000) (as
stated in Reagents and antibodies) then incubated with a
horseradish peroxide-conjugated secondary antibody (HAF008,
dilution: 1:5,000; Novus Biologicals, LLC, Littleton, CO, USA),
following which the proteins were visualized using an enhanced
chemiluminescent substrate (WBKLS0500; Merck KGaA).
Semi-quantification of the blots was conducted using ImageJ
software (version no. k 1.45; National Institutes of Health,
Bethesda, MD, USA).
Statistical analysis
Statistical analysis was performed using SPSS
software version 15.0 (SPSS, Inc., Chicago, IL, USA). Results were
presented as the mean ± standard deviation. The comparisons between
two groups were made using Student's t-test. Multi-group
comparisons of the means were made using one-way analysis of
variance with the Student-Newman-Keuls used as a post hoc test. All
experiments were repeated three times. P<0.05 was considered to
indicate a statistically significant difference.
Results
Bufalin enhances the inhibitory effect
of sorafenib on HCC cell proliferation
HCC cells were incubated with sorafenib, bufalin or
sorafenib and bufalin in combination at different concentrations.
Concentrations of 2.5, 5 and 10 µM sorafenib and 5, 10 and 20 nM
bufalin were used. The concentrations were chosen based around the
IC50 value of sorafenib and bufalin in both PLC/PRF/5 and SMMC-7721
cells. A CCK-8 assay was conducted to determine whether a
synergistic effect existed between sorafenib and bufalin. As
demonstrated in the results, the survival of HCC cells was reduced
when treated with sorafenib at concentrations ranging from 2.5 to
10 µM (Fig. 1A). Decreased
proliferation was also observed in HCC cells treated with 20 nM or
less bufalin (Fig. 1B). In
addition, combination treatment led to decreased cell proliferation
compared with either sorafenib or bufalin alone (Fig. 1C). As demonstrated in Fig. 1A and B, we have determined the
optimal concentration of sorafenib (10 µM sorafenib) and bufalin
(20 nM bufalin) that would exert the most significant synergistic
effect. Therefore, we have measured their combined effect according
to these concentrations. The inhibitory effect of the combined
drugs was more apparent as the concentration of sorafenib and
bufalin increased, as shown from the CI/fractional effect curve
(Fig. 1D and E). The combined
treatment reduced cell proliferation significantly than either of
them alone (P<0.05). For PLC/PRF/5 and SMMC-7721 cells, some
concentrations (CI<1 when combined) were demonstrated to be
synergistic whereas certain concentrations (CI>1 when combined)
were demonstrated to be antagonistic, as illustrated in Fig. 1D and E. As calculated by CI, the
combination of sorafenib (10 µM sorafenib) and bufalin (20 nM
bufalin) was demonstrated to exert the most significant synergistic
effect (P<0.05).
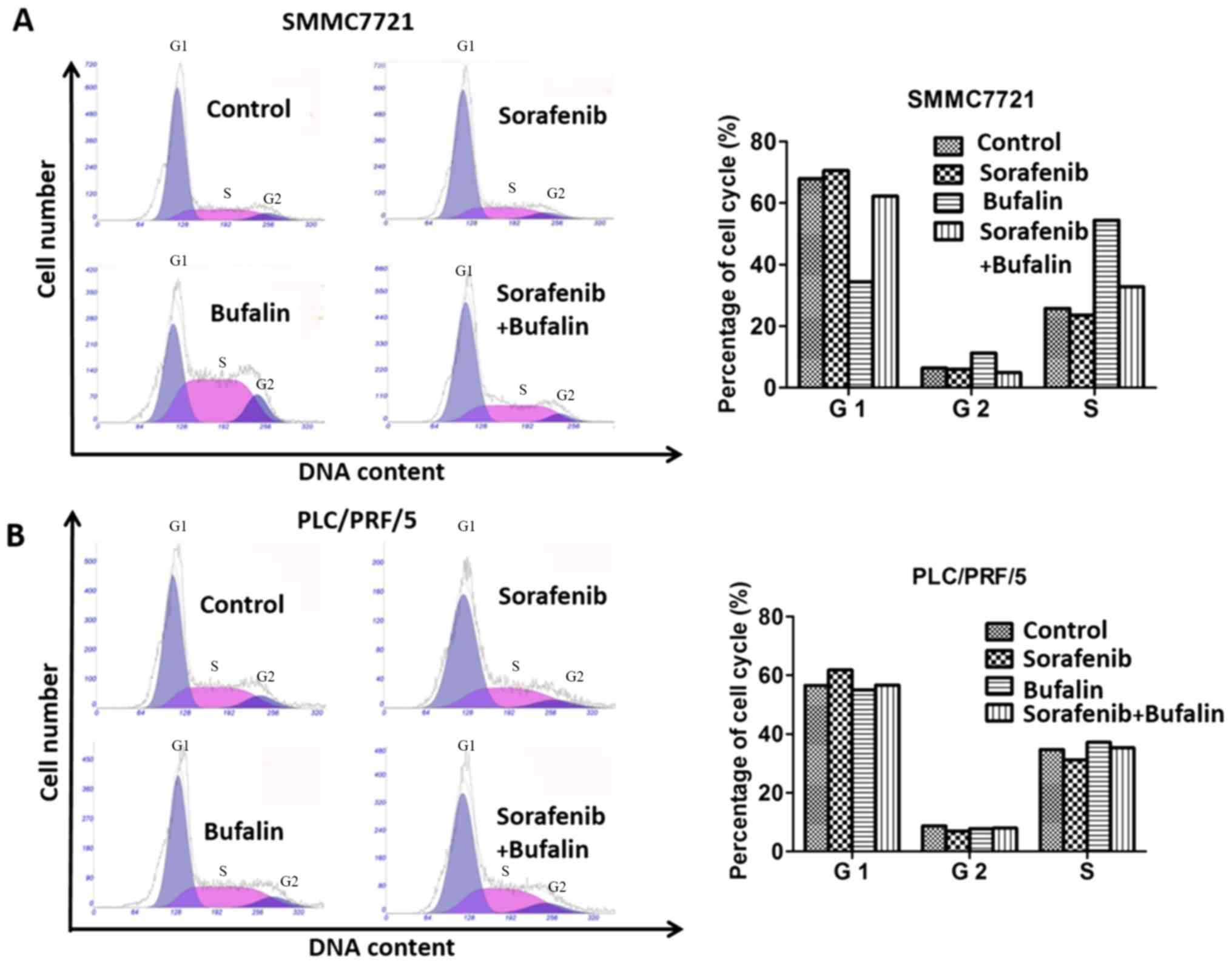
Effect of the combination treatment on
HCC cells
Cell cycle analysis of cells with different
treatments was determined by examining the cells' DNA profiles
following staining with PI. As shown from the cell cycle assay,
SMMC-7721 cells treated with sorafenib and the combination of
sorafenib+bufalin demonstrated similar proportions of cells in the
different phases of the cell cycle compared with the control group,
and the proportion of cells in the G1 phase stimulated with bufalin
was smaller compared with the control group. In the G2 phrase,
SMMC-7721 cells treated bufalin made up the largest proportion
whereas the proportion of SMMC-7721 cells with the combined
treatment was the smallest among all the groups. SMMC-7721 cells
treated with bufalin made up the largest proportion among the four
groups in S phase (Fig. 2A). With
regard to PLC/PRF/5 cells, there was no significant difference in
the proportion of cells treated with sorafenib, bufalin and the
combined drug in all phases (Fig.
2B).
Apoptosis increases in HCC cells
following a combination treatment with bufalin and sorafenib
As the present study demonstrated that the effect of
the combined treatment was more evident in inhibiting HCC
proliferation, the effect of combined treatment on apoptosis was
investigated. Concentrations of 20 nM bufalin and 10 µM sorafenib
were used to incubate HCC cells for 24 h. As shown in Fig. 3A, apoptosis was significantly
enhanced in PLC/PRF/5 cells and SMMC-7721 cells with the
combination treatment as compared with cells treated with the
control, sorafenib and bufalin alone (Fig. 3A). The Hochest 33258 staining was
used to detect apoptosis in cells with different treatments. All
the cells were stained with blue, but only the apoptotic cells were
brightly illuminated as illustrated in Fig. 3A. For SMMC-7721 cells, the controls
have the fewest cells undergoing apoptosis and the cells treated
with the combined drugs underwent the most marked level of
apoptosis. Sorafenib (10 µM) or bufalin (20 nM) promoted the
apoptosis of HCC cells, as assessed by Annexin V/PI staining, while
the combined treatment promoted HCC cell apoptosis to a greater
degree than single-agent treatment (Fig. 3B and C). These results suggested
that bufalin may promote sorafenib-induced apoptosis in HCC
cells.
Bufalin enhances the inhibitory effect
of sorafenib in tumor clone formation
The effects of sorafenib and bufalin on HCC cell
proliferation was further validated in vitro. Clone
formation analysis was conducted. The number of viable cells was
significantly fewer in the combined treatment group when compared
with the mono-drug groups, indicating the enhanced suppressive
tumor properties of the combined treatment in HCC (P<0.05;
Fig. 4A and B).
Bufalin enhances sorafenib-induced
apoptosis and necrosis in mouse HCC tissues
The nuclei of all cells were stained blue and
apoptotic nuclei were stained green. As shown in Fig. 5A, the level of apoptosis in the
tissues from mice injected with either sorafenib or bufalin was
higher than that of the control. The number of cells with
fluorescent green was the highest in mice injected with the
combined drug treatment. The apoptotic rate of each treatment group
(control, sorafenib, bufalin and the combination) was then
measured. The apoptotic rate of the sorafenib or bufalin only
treatments was significantly higher than that of the control group;
however, the highest rate was observed in mice injected with the
combination treatment (Fig. 5B).
In addition, hematoxylin and eosin (H&E) staining of tumors
from mice with different treatments was observed. The results
demonstrated that there were more areas of necrosis in tumors from
mice treated with the combined agents as compared with the other
groups (Fig. 5C).
 | Figure 5.Bufalin enhances sorafenib-induced
apoptosis in mouse HCC tissue. (A) The levels of apoptosis in the
subcutaneous tumor tissues from mice treated with sorafenib,
bufalin, the combined treatment and the control were detected. The
number of apoptotic cells in HCC tissues from mice were analyzed
via a TUNEL assay. TUNEL stained apoptotic cells were counted in
three randomly selected microscopic fields, magnification, ×10. (B)
Apoptosis rates were calculated from apoptotic cells in HCC
tissues. The corresponding P-values observed are also presented.
**P<0.01, as indicated. (C) Representative hematoxylin and eosin
images of subcutaneous tumors in mice injected with sorafenib,
bufalin, the combined treatment and the control are shown,
magnification, ×4. Three independent experiments were performed and
representative pictures are shown. HCC, hepatocellular carcinoma.
TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end
labelling. |
Effects of the combination treatment
on proteins associated with apoptosis
The expression levels of Bcl-2, Bax, caspase-7,
caspase-8 and PARP proteins were measured in HCC cells following
the different treatments. Sorafenib and bufalin treatment slightly
decreased Bcl-2 levels when compared with the control; however, the
combined treatment produced similar levels (Fig. 6). When compared with the control,
Bax expression levels in HCC cells treated with sorafenib and
bufalin did not show marked differences (Fig. 6). However, Bax expression was
upregulated in cells treated with the combination of the 2 drugs,
when compared with either drug alone (Fig. 6). Caspase-7 was upregulated in HCC
cells treated with bufalin, as compared with the control cells;
however, the difference was not statistically significant (Fig. 6). In HCC cells with the combined
treatment, the expression of caspase 7 was upregulated as compared
with the other groups (Fig. 6). In
addition, caspase-8 was slightly increased in HCC cells treated
with either bufalin or sorafenib alone, or with the combined-drug
treatment, though not significantly so (Fig. 6). Both of the two bands (35 and 20
kDa) represent caspase 7, and both of the two bands (55 and 38 kDa)
represent caspase 8. The doublet bands indicated different sized
isoforms of caspase 7 and caspase 8. Furthermore, it should be
noted that no significant difference in PARP levels were observed
between the control and sorafenib and bufalin-treated cells.
However, PARP was significantly increased in cells with the
combined drugs when compared with those treated with mono-drug, as
demonstrated in Fig. 6. The 116
kDa band is the full length PARP and the 89 kDa is the cleaved
PARP. In conclusion, the expression levels of Bax, caspase 7 and
PARP were upregulated in HCC cells with the combined treatment of
bufalin and sorafenib.
 | Figure 6.Effect of the combined treatment of
sorafenib and bufalin on the expression of proteins associated with
apoptosis. The protein expression levels of Bcl-2, Bax, caspase-7,
caspase-8 and PARP were detected in SMMC-7721 cells treated with
control, sorafenib, bufalin and combined treatment via western
blotting and analyzed by ImageJ software. All experiments were
performed three times. Representative blots and statistical
analyses are shown with the corresponding P-values. *P<0.05,
**P<0.01 and ***P<0.001, as indicated. Bcl-2, B-cell
lymphoma-2; Bax, Bcl-2 associated X protein; PARP, poly (adenosine
diphosphate-ribose) polymerase. |
Discussion
The present study demonstrated that the combination
treatment of sorafenib and bufalin was more potent in the
inhibition of HCC cell proliferation when compared with either
treatment alone. The combined drug also elicited increased cell
apoptosis. Furthermore, apoptosis-associated proteins were altered
in HCC cells with the combination treatment. Thus, the combination
of sorafenib and bufalin may lead to enhanced HCC cell death.
Sorafenib, which has been proven to suppress tumor
cell proliferation and angiogenesis, serves as the only recommended
targeted therapy for advanced HCC (3–5).
However, studies have revealed that the recurrence and progression
is still high due to the development of drug resistance (15). Therefore, drugs that may strengthen
the antitumor effect of sorafenib are warranted.
Bufalin has been widely investigated for its
antitumor effects. Despite an increasing number of studies on
bufalin, its antitumor mechanism is complex and remains to be
further explored. It has been reported to suppress tumor cell
proliferation and angiogenesis, induce apoptosis and cell
differentiation in many types of cancer (7–9).
Indeed, the effect of bufalin relies largely on its concentration.
Bufalin at low and high concentrations may exert different
functions in promoting apoptosis or inhibiting metastasis. Studies
have shown that bufalin may induce apoptosis via a number of
different mechanisms. One previous study indicated that the
intrinsic apoptotic pathway induced by bufalin is accountable for
reduced cell proliferation and tumor growth (16). In addition, the endoplasmic
reticulum (ER) stress response regulated by the inositol-requiring
enzyme signaling pathway may also contribute to bufalin-induced
apoptosis (17).
As sorafenib and bufalin are potent drugs against
HCC, the present study speculated that greater antitumor effects
may be achieved by their combined treatment. Sorafenib and bufalin
were tested to inhibit the proliferation of the PLC/PRF/5 and
SMMC-7721 cell lines via a cell viability assay. As shown in the
results, the suppressive ability of sorafenib increased when its
concentration was <10 µM. Bufalin also demonstrated its
inhibitive property against HCC cells in a dose-dependent manner.
The present study determined the optimized concentration of
sorafenib and bufalin in inhibiting HCC proliferation via CI
indexes. The most significant proliferation rate was observed in
HCC cells treated with 10 µM sorafenib and 20 nM bufalin.
Next, the present study determined the effect of the
combination treatment on cell cycle arrest. The results
demonstrated that the combination treatment (bufalin and sorafenib)
did not have a synergistic effect on cell cycle arrest, as
determined by a cell cycle assay.
Apoptosis, defined as programmed cell death, occurs
in multicellular organisms under certain physiological and
pathological circumstances, and is one of the approaches by which
organisms maintain stability (18). A number of studies have shown that
tumors have an infinite proliferative property and they also evade
apoptosis (19–21). Dysfunction of apoptosis is one of
the major mechanisms that leads to malignant tumors. Normal cells
and tissues may undergo apoptosis once their microenvironment
alters, ensuring the stability of physical activities (22,23).
Accelerated proliferation of tumor cells may be attributed to
escape from apoptosis (24).
Therefore, identification of drugs that induce apoptosis is vital
in tumor treatment.
The results presented in the present study revealed
that sorafenib combined with bufalin may lead to significantly
decreased survival in HCC cells. In addition, apoptotic properties
were observed in sorafenib and bufalin (3,4).
Therefore, it was predicted that bufalin and sorafenib may have
enhanced apoptosis in HCC cells. Next, 20 nM bufalin and 10 µM
sorafenib were adopted in the following experiments using CI index.
It was demonstrated that sorafenib combines favorably with bufalin
to induce increased apoptosis in HCC when compared with the
untreated control, and only sorafenib or bufalin-treated cancer
cells. The results showed that bufalin induced apoptosis in HCC and
also accelerated sorafenib-induced apoptosis. The levels of
apoptosis were the greatest in HCC cells that were administered the
combination treatment.
Flow cytometry analysis revealed that the
combination treatment of sorafenib and bufalin significantly
promoted apoptosis in PLC/PRF/5 and SMMC-7721 cells. Sorafenib or
bufalin induced marginal apoptosis in HCC cells, while the combined
treatment induced significant apoptosis in HCC cells when compared
with single-agent treatment. H&E staining of mouse tumors with
different treatments also showed that necrosis was the most evident
in the combined treatment group.
Apoptosis is associated with a series of signaling
pathways. Multiple mechanisms on sorafenib-induced apoptosis in HCC
cells have been reported. One previous study reported that tumors
subjected to sorafenib incubation demonstrated increased caspase-4
activation and CCAAT-enhancer-binding protein homologous protein
upregulation (25). Another study
demonstrated that sorafenib promoted apoptosis in PLC/PRF/5 and
HepG2 cells (26).
To determine the underlying mechanism by which
bufalin regulates the susceptibility of HCC cells to sorafenib, the
expression levels of anti-apoptotic and pro-apoptotic proteins were
measured in HCC cells with different treatments. The widely
accepted mechanism of apoptosis includes the mitochondrial pathway,
the death receptor pathway and the ER pathway (27–29).
All of these signaling pathways may interact with each other to
regulate apoptosis. A series of caspases are actively involved in
apoptosis. The release of caspases varies according to different
cell types and stimuli (30).
Caspase 7 and caspase 8 were measured in HCC cells administered
with different treatments in the present study. Caspase-8 is the
initiating molecule of the extrinsic apoptotic pathway; it
activates the effector caspase, which leads to cell apoptosis
(31,32). It was revealed that caspase 7 and
caspase 8 were elevated in the combination-treated HCC cells when
compared with HCC cells treated with single-agent treatments,
indicating that the combined treatment may lead to enhanced
apoptosis.
Bufalin was shown to potently promote PARP and
caspase-7 activation, while sorafenib was observed to slightly
suppress Bcl-2. Together, these results indicated that bufalin
contributed to sorafenib-induced apoptosis via the regulation of
these proteins. In the context of the present study, it is
particularly noteworthy that Bax, caspase-7 and PARP were all
upregulated in HCC cells with the combination treatment.
In the present study, sorafenib and bufalin have
been demonstrated to suppress the proliferation and promote the
apoptosis of HCC cells. Bufalin was demonstrated to synergize with
sorafenib to suppress HCC cell proliferation. The enhanced effect
of these two drugs may be attributed to bufalin's capacity to
promote the sorafenib-induced activation of Bax, PARP and
caspase-8, as shown from the western blotting results of the
present study. There are few studies exploring the effect of the
combination treatment of bufalin and sorafenib on HCC apoptosis.
Among the most common molecules participating in apoptosis, PARP
and caspase-7 were revealed to be the most significantly
altered.
A previous study demonstrated that Bax and Bcl-2,
the main Bcl-2 family members, are intimately associated with HCC
progression (33). This study
revealed that bufalin altered the levels of Bcl-2 and Bax. Bcl-2
and Bax are two of the major proteins that regulate cancer
progression. High Bcl-2 levels usually prevent tumor cells from
undergoing apoptosis, while Bax promotes apoptosis by caspase
induction via the activation of caspase-9 (34,35).
The combination of sorafenib and bufalin significantly upregulated
sorafenib-induced Bax expression; however, it only had slight
effects on Bcl-2 expression. The results of the study may provide
more of an understanding of the underlying mechanism of combined
treatment of bufalin and sorafenib against HCC.
In the present study, it was revealed that the
combination of sorafenib and bufalin was more potent in inducing
cell death, which was accompanied by the increased induction of
apoptosis, as compared with either drug alone. Thus, potentiation
of apoptosis due to the combination of the two drugs may contribute
to enhanced HCC cell death. Therefore, bufalin may also serve as an
apoptosis accelerator for sorafenib in HCC treatment. The results
suggested that the combination of sorafenib and bufalin may be a
potential therapeutic strategy for patients with advanced HCC.
However, the toxicity and clinical efficacy of this combination
therapy remain to be evaluated and therefore, further investigation
is required.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant nos. 81573753 and
81603348).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HW performed the in vivo studies and designed
the study. CZ performed the in vitro studies and wrote the
original manuscript. HC conducted the western blot experiments and
analysed the data. ZM designed the study and reviewed the
article.
Ethics approval and consent to
participate
The present study was approved by Fudan University
Shanghai Cancer Center (Shanghai, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bosch FX, Ribes J, Diaz M and Cleries R:
Primary livercancer: Worldwide incidence and trends.
Gastroenterology. 127 5 Suppl 1:S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43–9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carlomagno F, Anaganti S, Guida T,
Salvatore G, Troncone G, Wilhelm SM and Santoro M: BAY 43–9006
inhibition of oncogenic RET mutants. J Natl Cancer Inst.
98:326–334. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang YS, Adnane J, Trail PA, Levy J,
Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A,
et al: Sorafenib (BAY 43–9006) inhibits tumor growth and
vascularization and induces tumor apoptosis and hypoxia in RCC
xenograft models. Cancer Chemother Pharmacol. 59:561–574. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: SHARP investigators study group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qi F, Li A, Inagaki Y, Kokudo N, Tamura S,
Nakata M and Tang W: Antitumor activity of extracts and compounds
from the skin of the toad Bufo bufo gargarizans Cantor. Int
Immunopharmacol. 11:342–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takai N, Ueda T, Nishida M, Nasu K and
Narahara H: Bufalin induces growth inhibition, cell cycle arrest
and apoptosis in human endometrial and ovarian cancer cells. Int J
Mol Med. 21:637–643. 2008.PubMed/NCBI
|
|
9
|
Huang WW, Yang JS, Pai SJ, Wu PP, Chang
SJ, Chueh FS, Fan MJ, Chiou SM, Kuo HM, Yeh CC, et al: Bufalin
induces G0/G1 phase arrest through inhibiting the levels of cyclin
D, cyclin E, CDK2 and CDK4 and triggers apoptosis via mitochondrial
signaling pathway in T24 human bladder cancer cells. Mutat Res.
732:26–33. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yin JQ, Shen JN, Su WW, Wang J, Huang G,
Jin S, Guo QC, Zou CY, Li HM and Li FB: Bufalin induces apoptosis
in human osteosarcoma U-2OS and U-2OS methotrexate300-resistant
cell lines. Acta Pharmacol Sin. 28:712–720. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Efferth T, Davey M, Olbrich A, Rücker G,
Gebhart E and Davey R: Activity of drugs from traditional Chinese
medicine toward sensitive and MDR1- or MRP1-overexpressing
multidrug-resistant human CCRF-CEM leukemia cells. Blood Cells Mol
Dis. 28:160–168. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang H, Zhang C, Ning Z, Xu L, Zhu X and
Meng Z: Bufalin enhances anti-angiogenic effect of sorafenib via
AKT/VEGF signaling. Int J Oncol. 48:1229–1241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Zhang C, Xu L, Zang K, Ning Z,
Jiang F, Chi H, Zhu X and Meng Z: Bufalin suppresses hepatocellular
carcinoma invasion and metastasis by targeting HIF-1α via the
PI3K/AKT/mTOR pathway. Oncotarget. 7:20193–20208. 2016.PubMed/NCBI
|
|
14
|
Chen S, Wang G, Niu X, Zhao J, Tan W, Wang
H, Zhao L and Ge Y: Combination of AZD2281 (Olaparib) and GX15-070
(Obatoclax) results in synergistic antitumor activities in
preclinical models of pancreatic cancer. Cancer Lett. 348:20–28.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu J, Cui X, Qu L, Hua L, Wu M, Shen Z,
Lu C and Ni R: Overexpression of DLX2 is associated with poor
prognosis and sorafenib resistance in hepatocellular carcinoma. Exp
Mol Pathol. 101:58–65. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Chen C, Wang S, Zhang Y, Yin P,
Gao Z, Xu J, Feng D, Zuo Q, Zhao R and Chen T: Bufalin inhibits
HCT116 colon cancer cells and its orthotopic xenograft tumor in
mice model through genes related to apoptotic and PTEN/AKT
pathways. Gastroenterol Res Pract. 2015:4571932015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhai B, Hu F, Yan H, Zhao D, Jin X, Fang
T, Pan S, Sun X and Xu L: Bufalin reverses resistance to sorafenib
by inhibiting Akt activation in hepatocellular carcinoma: The role
of endoplasmic reticulum stress. PLoS One. 10:e01384852015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hajra KM and Liu JR: Apoptosome
dysfunction in human cancer. Apoptosis. 9:691–704. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, et
al: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He GW, Günther C, Thonn V, Yu YQ, Martini
E, Buchen B, Neurath MF, Stürzl M and Becker C: Regression of
apoptosis-resistant colorectal tumors by induction of necroptosis
in mice. J Exp Med. 214:1655–1662. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ucker DS and Levine JS: Exploitation of
apoptotic regulation in cancer. Front Immunol. 9:2412018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Green DR: Apoptotic pathways: Ten min to
dead. Cell. 121:671–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kwon KB, Park BH and Ryu DG: Chemotherapy
through mitochondrial apoptosis using nutritional supplements and
herbs: A brief overview. J Bioenerg Biomembr. 39:31–34. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke
AW, Wang XY, Dai Z, Peng YF, Gu CY, et al: Targeting autophagy
enhances sorafenib lethality for hepatocellular carcinoma via ER
stress-related apoptosis. Autophagy. 7:1159–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ly JD, Grubb DR and Lawen A: The
mitochondrial membrane potential (deltapsi (m)) in apoptosis; an
update. Apoptosis. 8:115–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ji BC, Hsu WH, Yang JS, Hsia TC, Lu CC,
Chiang JH, Yang JL, Lin CH, Lin JJ, Suen LJ, et al: Gallic acid
induces apoptosis via caspase-3 and mitochondrion-dependent
pathways in vitro and suppresses lung xenograft tumor growth in
vivo. J Agric Food Chem. 57:7596–7604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Logue SE and Martin SJ: Caspase activation
cascades in apoptosis. Biochem Soc Trans. 36:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang Z, Takahashi Y, Chen C, Liu Y, He H,
Tsotakos N, Serfass JM, Gebru MT, Chen H, Young MM and Wang HG:
Atg2A/B deficiency switches cytoprotective autophagy to
non-canonical caspase-8 activation and apoptosis. Cell Death
Differ. 24:2127–2138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Concha NO and Abdel-Meguid SS: Controlling
apoptosis by inhibition of caspases. Curr Med Chem. 9:713–726.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zheng Q, Wang B, Gao J, Xin N, Wang W,
Song X, Shao Y and Zhao C: CD155 knockdown promotes apoptosis via
AKT/Bcl-2/Bax in colon cancer cells. J Cell Mol Med. 22:131–140.
2018. View Article : Google Scholar : PubMed/NCBI
|