Introduction
Atherosclerosis is a complex arterial disease
characterized by lipid accumulation, inflammation and matrix
remodeling in the arterial walls. Low-density lipoprotein retention
and endothelial cell activation initiate the formation of
atherosclerotic lesions in the arterial intima (1). In the early stages of lesion
formation, monocytes are recruited to the vascular walls and
subsequently engulf lipids; after which, monocytes are transformed
to foam cells (2). Neointimal
formation is an early step in the development of atherosclerotic
plaques (3). During
atherosclerosis, vascular smooth muscle cells proliferate and
migrate to the intima, contributing to the thickening of vascular
intima associated with the pathogenesis of atherosclerosis
(4,5). In the advanced stage of lesion
formation, the accumulation of lipids, extracellular matrix and
vascular smooth muscle cells form fibroatheroma in the intima
(2). The narrowing of vessels
influences oxygen supply, thus resulting in the ischemia of
tissues, including the myocardium and brain. Atherosclerosis is one
of the major causes of heart attacks and strokes, and is
responsible for >50% of all cases of mortality in developed
countries (6).
Lysophosphatidic acid (LPA) is one of the
intermediate products of membrane phospholipid metabolism and is a
bioactive phospholipid present in almost all tissues (7). LPA has been reported to possess
various activities that affect survival, development, morphological
alterations and inflammation, via G-protein-coupled receptors
(7,8). LPA is also involved in numerous
diseases, including neurological disorders, cardiovascular
diseases, fibrosis, tumors and inflammation (8,9).
Furthermore, LPA is associated with various vessel wall cell
activities, and contributes to vasculogenesis, angiogenesis and
vascular remodeling (7,10–12).
Indirect evidence that blockade of LPA receptors reduces neointimal
hyperplasia has demonstrated that LPA may be involved in neointimal
formation following vascular injury and LPA receptor blockade
relieves atherosclerotic development (13,14);
however, the effects of LPA on neointimal formation remain
unclear.
In the present study, the effects of LPA on
neointimal formation following vascular injury were investigated.
The findings indicated that LPA may contribute to the pathology of
atherosclerosis, and may be considered a promising therapeutic
target for the treatment of atherosclerosis.
Materials and methods
Animal experimental protocol
Healthy Sprague Dawley rats (n=36; male; age, 8
weeks; weight, 240–260 g) were obtained from Liaoning Changsheng
Biotechnology Co., Ltd. (Benxi, China) and were housed in a
standard environment with controlled temperature (21–23°C),
humidity (45–55%), lighting (12 h light/dark cycle) and free access
to food and water. Rats in the vascular injury group and vascular
injury + LPA group (n=12 for each group) underwent carotid artery
balloon injury. Briefly, rats were anesthetized and fastened in a
supine position. A midline incision was performed on the skin of
the anterior neck. The left common carotid, internal and external
carotid arteries were exposed via an anterior incision of the neck.
A 2.0 F balloon catheter was introduced via the external carotid
artery and advanced towards the proximal end until it reached the
common carotid artery. The balloon was inflated with 2-fold
atmospheric pressure to obstruct the bloodstream for 30 sec. The
common carotid artery was injured by passing the inflated balloon
back and forth slowly three times. Subsequently, the catheter was
removed and the external carotid arteries and incision were closed.
Following the surgical procedure, rats received penicillin
(2×105 units; intramuscular injection; Harbin Motian
Agricultural Technology Veterinary Drug Co., Ltd., Harbin, China)
to prevent infection. Rats in the sham group received similar
operations, but no balloon-induced injury. Following
balloon-induced injury, rats in the vascular injury + LPA group
received LPA (1 mg/kg, intraperitoneal injection; Aladdin
Industrial Corporation, Shanghai, China) every 2 days for 35 days
(18 injections in total). Rats in the sham (n=12) and vascular
injury groups received an equal amount of normal saline
(intraperitoneal injection). After 35 days, the carotid artery
tissues were harvested for subsequent experiments. The present
study followed the Guide for the Care and Use of Laboratory Animals
(15) and was approved by the
Ethics Committee of The People's Hospital of China Medical
University (Liaoning, China).
Hematoxylin and eosin (HE)
staining
The carotid artery tissues were fixed in 4%
paraformaldehyde for 24 h at room temperature, dehydrated in graded
ethanol, cleared with xylene, then embedded in paraffin and cut
into 5-µm sections. The sections were deparaffinized in xylene and
rehydrated in graded ethanol series. Subsequently, the sections
were stained with hematoxylin for 5 min and eosin for 3 min at room
temperature. Images of carotid artery tissues in each group were
obtained using light microscopy with the cellSens Entry 1.9 imaging
system (Olympus Corporation, Tokyo, Japan). According to the HE
staining images, the intima, tunica media and lumen areas were
calculated.
Immunohistochemistry
Following deparaffinization and rehydration, the
paraffin-embedded sections were maintained in citrate buffer at
100°C for 10 min for antigen retrieval. Following rinsing with PBS,
the sections were incubated with 3% hydrogen peroxide at room
temperature for 15 min to inactivate the endogenous peroxidases.
Subsequently, the sections were rinsed with PBS and incubated with
normal goat serum (Beijing Solarbio Science & Technology, Co.,
Ltd., Beijing, China) at room temperature for 15 min to block
nonspecific binding sites. The sections were then incubated with a
primary antibody against proliferating cell nuclear antigen (PCNA;
1:50; Santa Cruz Biotechnology, Inc., Dallas, TX, USA; cat. no.
sc-25280) overnight at 4°C. Following rinsing with PBS, the
sections were incubated with corresponding biotin-labeled secondary
antibody (1:200; Beyotime Institute of Biotechnology, Haimen,
China; cat. nos. A0286 and A0216) for 30 min at 37°C. The sections
were then rinsed with PBS and incubated with horseradish peroxidase
(HRP)-labeled avidin (Beyotime Institute of Biotechnology) at 37°C
for 30 min. After further rinsing, the sections were visualized
with a 3,3′-diaminobenzidine (DAB) kit (Beijing Solarbio Science
& Technology, Co., Ltd.) and counterstained with hematoxylin
(Beijing Solarbio Science & Technology, Co., Ltd.) at room
temperature for 3 min. Images of each group were obtained using a
light microscope with cellSens Entry 1.9 imaging system (Olympus
Corporation). The percentage of PCNA-positive cells in neointima
was recorded.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
The paraffin-embedded sections were deparaffinized,
rehydrated and subjected to a TUNEL assay using an In situ
Cell Death Detection kit (Roche Diagnostics GmbH, Mannheim,
Germany) according to the manufacturer's protocols. Briefly, the
sections were permeabilized with 0.1% Triton X-100 for 8 min and
blocked with 3% hydrogen peroxide for 10 min. Subsequently, the
sections were incubated with a mixture of enzyme solution and label
solution (1:9) from the kit, at 37°C for 60 min in the dark.
Following rinsing with PBS, the sections were incubated with
Converter-POD at 37°C for 30 min. The sections were visualized with
a DAB kit and counterstained with hematoxylin as aforementioned.
Images were obtained using a light microscope with cellSens Entry
1.9 imaging system at ×400 magnification. The percentage of
TUNEL-positive cells in neointima was recorded.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from carotid artery tissues
using a RNApure High-purity Total RNA Rapid Extraction kit (BioTeke
Corporation, Beijing, China) and reverse transcribed to cDNA. The
reaction system included 1 µl oligo(dT)15, 1 µl random primer, 2 µl
dNTPs, 10.5 µl ddH2O, 4 µl 5× Buffer, 0.5 µl RNasin and
1 µl moloney murine leukemia virus reverse transcriptase (BioTeke
Corporation). The thermocycling conditions were: 25°C for 10 min,
followed by 42°C for 50 min. Subsequently, the mRNA expression
levels of tumor necrosis factor (TNF)-α, interleukin (IL)-10 and
IL-1β were measured via qPCR with cDNA as templates. The primer
sequences were as follows: TNF-α, forward
5′-TGGCGTGTTCATCCGTTCT-3′, reverse 5′-CCACTACTTCAGCGTCTCGT-3′;
IL-10, forward 5′-CCAGTCAGCCAGACCCACAT-3′, reverse
5′-GCATCACTTCTACCAGGTAAAAC-3′; IL-1β, forward
5′-GGGATGATGACGACCTGC-3′, reverse 5′-ACTTGTTGGCTTATGTTCTG-3′; and
β-actin, forward 5′-GGAGATTACTGCCCTGGCTCCTAGC-3′ and reverse
5′-GGCCGGACTCATCGTACTCCTGCTT-3′. RT-qPCR was performed on an
Exicycler™ 96 real-time PCR instrument (Bioneer Corporation,
Daejeon, Korea). The thermocycling conditions were as follows:
Initial denaturation at 94°C for 10 min; followed by 40 cycles of
94°C for 10 sec, 60°C for 20 sec and 72°C for 30 sec; and final
extension at 72°C for 150 sec. The relative mRNA expression levels
were normalized to β-actin and calculated using the
2−ΔΔCq method (16).
Western blot analysis
Proteins from the samples in each group were
extracted using radioimmunoprecipitation assay lysis buffer
(Beyotime Institute of Biotechnology) with 1% phenylmethanesulfonyl
fluoride (Beyotime Institute of Biotechnology). Following
determination of the protein concentration via a bichinconinic acid
(BCA) protein assay kit (Beyotime Institute of Biotechnology), the
proteins (30 µg/lane) were separated by 13% SDS-PAGE. Following
electrophoresis, the proteins were transferred onto polyvinylidene
fluoride membranes (Merck KGaA, Darmstadt, Germany). The membranes
were blocked with 5% skim milk for 1 h at room temperature and
incubated with primary antibodies against B-cell lymphoma-2 (Bcl-2;
1:400, Wuhan Boster Biological Technology, Ltd., Wuhan, China; cat.
no. BA0412), Bcl-2-associated X protein (Bax; 1:500, Sangon Biotech
Co., Ltd., Shanghai, China; cat. no. D120073), caspase-3 (1:1,000,
Abcam, Cambridge, UK; cat. no. ab2302), microtubule-associated
protein 1A/1B light chain 3 (LC3 II/I; 1:500, Cell Signaling
Technology, Inc., Danvers, MA, USA; cat. no. 2775), p62 (1:500,
Cell Signaling Technology, Inc.; cat. no. 5114) or β-actin
(1:1,000, Santa Cruz Biotechnology, Inc.; cat. no. sc-47778)
overnight at 4°C. Subsequently, the membranes were rinsed with
Tris-buffered saline with 0.15% Tween-20 (TBST) and incubated with
HRP-labeled secondary antibodies (1:5,000; Beyotime Institute of
Biotechnology; cat. nos. A0208 and A0216) at 37°C for 45 min. The
membranes were rinsed with TBST and visualized with a BeyoECL Plus
enhanced chemiluminescence reagent (Beyotime Institute of
Biotechnology). The optical densities of targeted bands were
analyzed using Gel-Pro-Analyzer software 4 (Media Cybernetics,
Rockville, MD, USA).
Measurement of malondialdehyde (MDA),
superoxide dismutase (SOD) and myeloperoxidase (MPO) levels
The carotid artery tissues were homogenized in PBS,
and underwent repeated freezing and thawing three times in liquid
nitrogen. Following centrifugation at 10,005 × g at 4°C for 10 min,
the supernatants were collected and the protein concentrations were
measured using a BCA protein assay kit. Subsequently, the levels of
MDA and SOD were measured using an MDA determination kit (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China; cat. no.
A003-1) or a SOD determination kit (WST-1 method) (Nanjing
Jiancheng Bioengineering Institute; cat. no. A001-3), according to
the manufacturer's protocols. The carotid artery tissues were
homogenized in normal saline, and the MPO levels were measured
using an MPO determination kit (Nanjing Jiancheng Bioengineering
Institute; cat. no. A044), according to the manufacturer's
protocols.
Statistical analysis
The results are presented as the means ± standard
deviation. Differences between groups were calculated using one-way
analysis of variance followed by Bonferroni's correction in
GraphPad Prism 5.0 (Graphpad Software, Inc., La Jolla, CA, USA).
Experiments were repeated three times. P<0.05 was considered to
indicate a statistically significant difference.
Results
LPA enhances vascular injury-induced
neointimal thickening
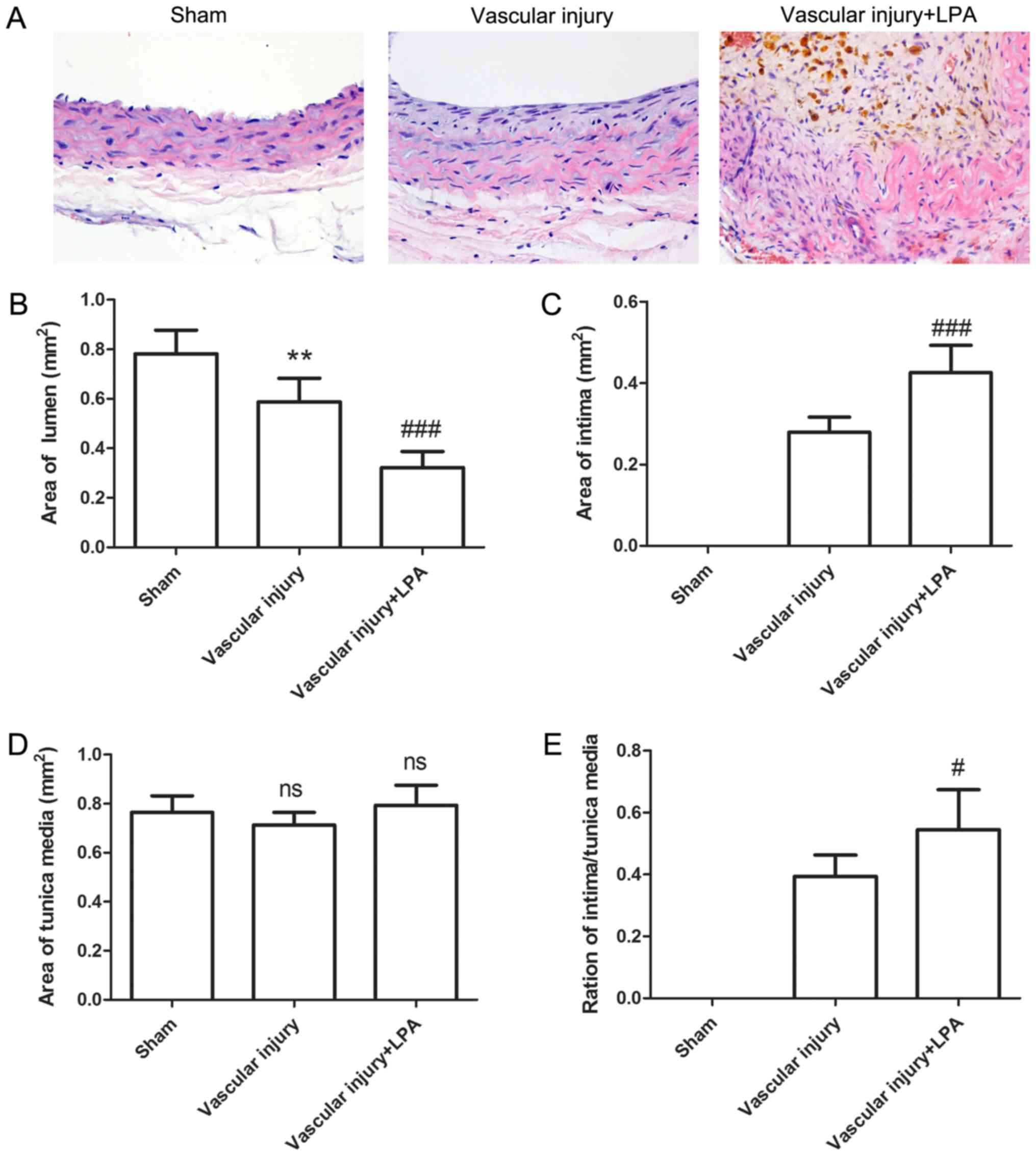
Following vascular injury, neointimal formation of
carotid artery tissues was assessed by HE staining. As presented in
Fig. 1, the carotid artery tissues
in the sham group revealed a common vascular structure with almost
nonvisible intima; however, following vascular injury, neointima
was visible, with a thickened intima. This thickening of the intima
in the injured arteries appeared to be enhanced upon LPA treatment
(Fig. 1A). Lumen, intimal and
tunica media areas were also analyzed. Compared with the sham
group, lumen area was significantly decreased in the vascular
injury group (0.781±0.096 mm2 vs. 0.588±0.095
mm2; Fig. 1B). The
lumen area further decreased following treatment with LPA (Fig. 1B). Conversely, compared with the
sham group, intimal area was significantly increased by 0.279±0.037
mm2 in the vascular injury group, and further increased
in the vascular injury + LPA group (Fig. 1C). In addition, no significant
alterations in tunica media area were observed across the three
groups (Fig. 1D). The
intima/tunica media ratio was increased by 0.394±0.069 in the
vascular injury group, and further increased in the vascular injury
+ LPA group (Fig. 1E). These
results demonstrated that intimal thickening caused by vascular
injury may be enhanced by treatment with LPA.
LPA modulates proliferation and
autophagy, but not apoptosis in neointima
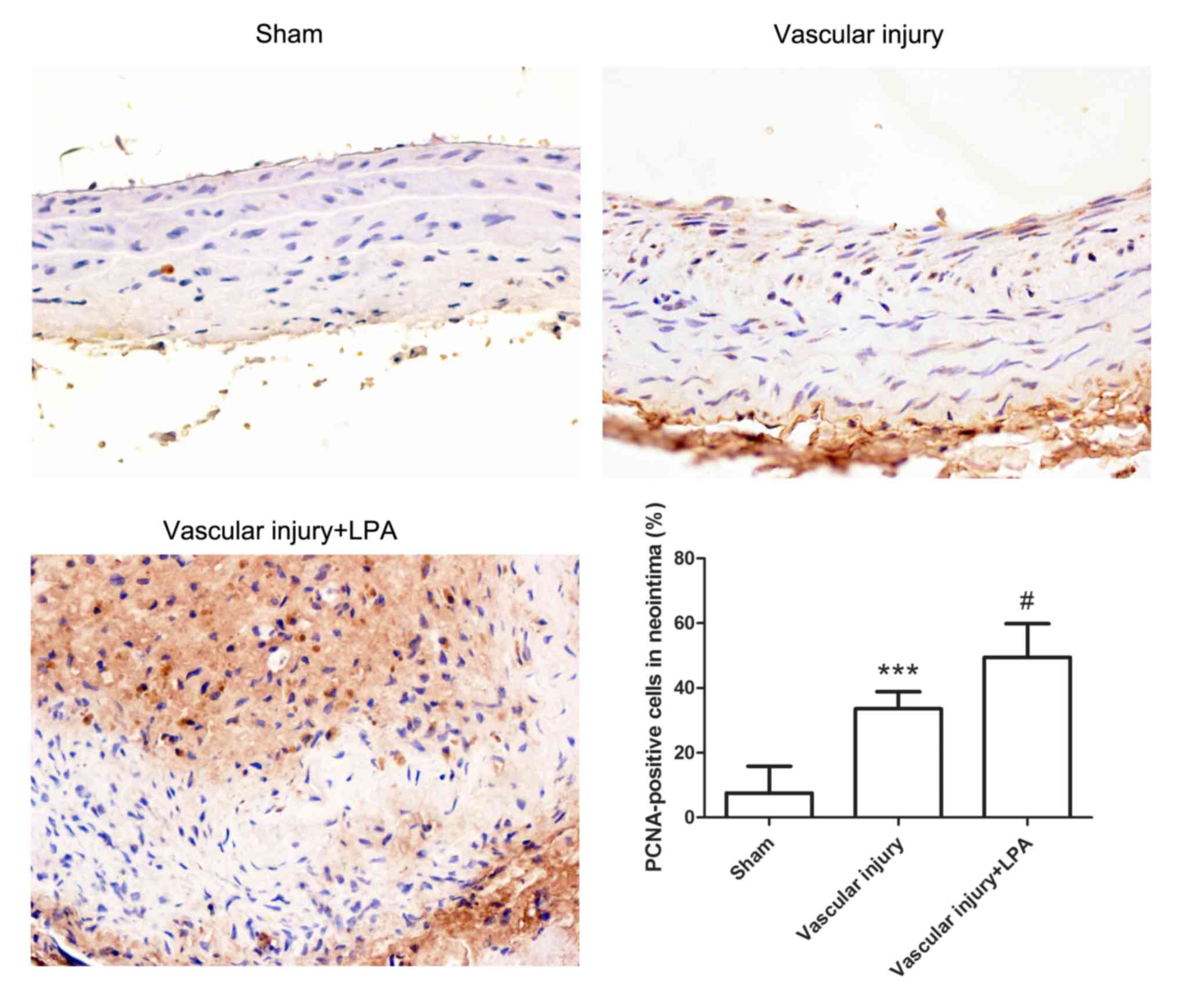
To further evaluate neointimal hyperplasia, the
expression levels of PCNA in the carotid artery tissues were
detected by immunohistochemistry. The carotid artery tissues in the
vascular injury group exhibited significantly increased PCNA
expression levels compared with in the sham group. In addition, the
carotid artery tissues in the vascular injury + LPA group
demonstrated significantly higher PCNA levels compared with in the
vascular injury group (Fig. 2).
These results indicated that LPA enhanced proliferation of
neointima.
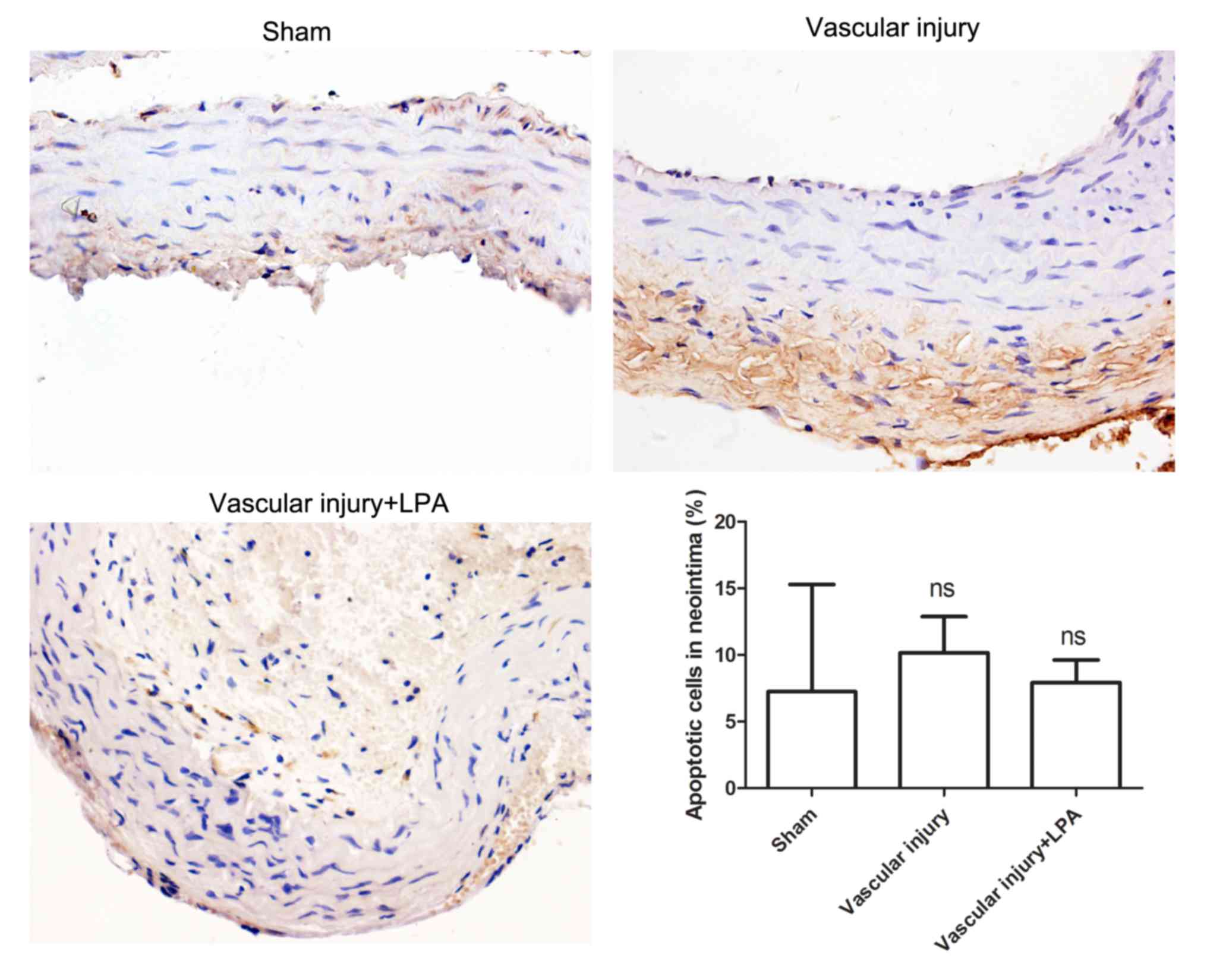
Apoptosis is very important in the formation of
neointima (17); therefore, in the
present study, the extent of apoptosis in carotid artery tissues
was investigated. The results of the TUNEL assay demonstrated that
there was no significant difference in apoptotic levels among the
various groups (Fig. 3). In
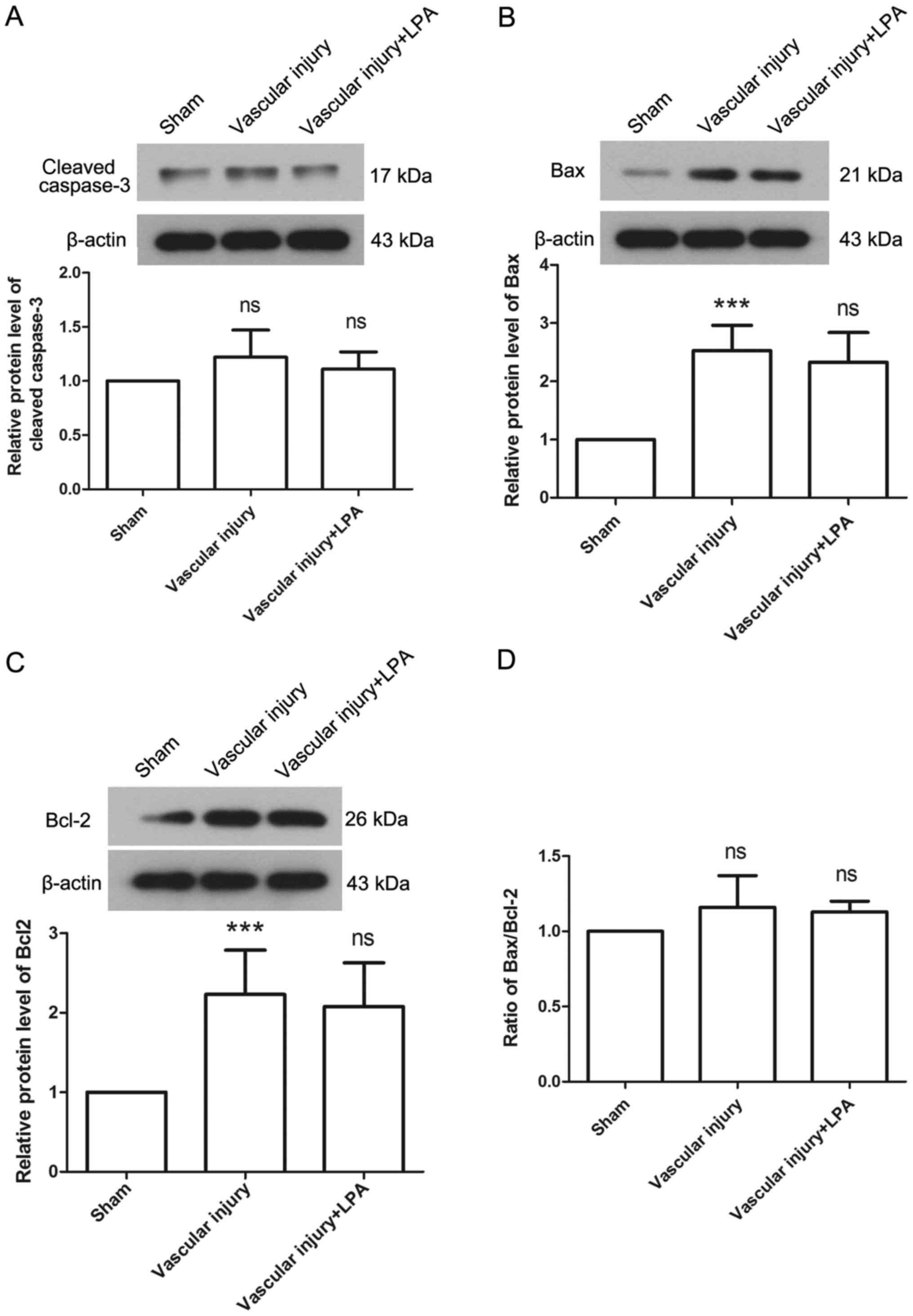
addition, the protein expression levels of caspase-3, Bax and Bcl-2
were detected to evaluate apoptosis in each group. The results
revealed that there was no significant difference in the expression
levels of caspase-3 among the groups (Fig. 4A). In the vascular injury group,
the expression levels of Bax were significantly increased compared
with the sham group (Fig. 4B). In
addition, the expression levels of Bcl-2 were significantly
increased in the vascular injury group compared with the sham group
(Fig. 4C). However, the Bax/Bcl-2
ratio exhibited no significant difference between the vascular
injury and sham groups (Fig. 4D).
Furthermore, the expression levels of Bax and Bcl-2 revealed no
significant difference between the vascular injury and vascular
injury + LPA groups (Fig. 4B-D).
These results were consistent with the results of TUNEL assay, and
demonstrated that LPA had no effect on apoptosis in vascular
injury-induced neointima.
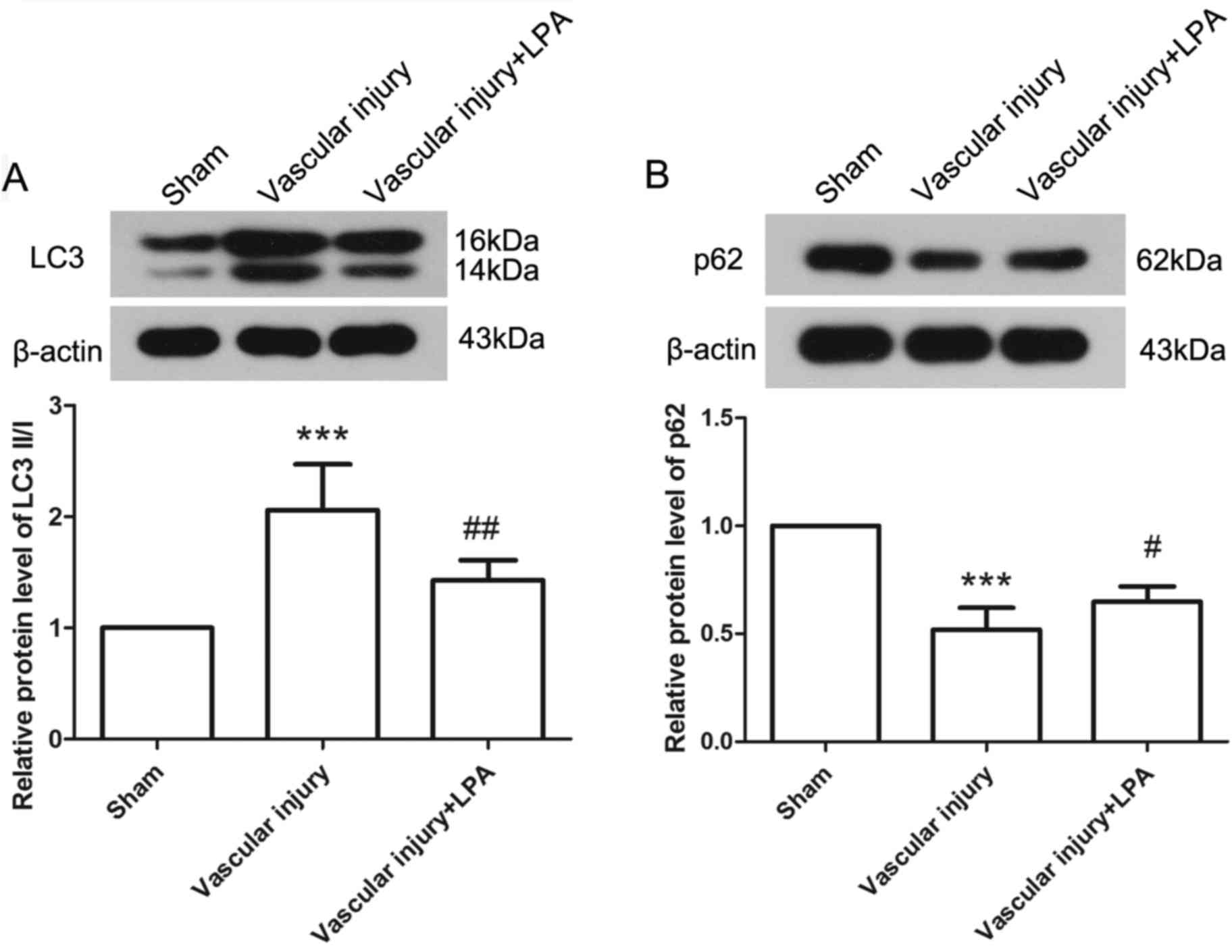
The levels of autophagy were elevated in response to
vascular injury, as determined by the expression levels of LC3 II/I
and p62 via western blotting. The results demonstrated that, in the
vascular injury group, the expression levels of LC3 II/I and p62
were significantly increased and decreased, respectively, compared
with in the sham group, thus indicating activation of autophagy.
However, upon treatment with LPA, the expression levels of LC3 II/I
were significantly decreased and those of p62 were significantly
increased compared with the vascular injury group (Fig. 5). These results demonstrated that
activation of autophagy induced by vascular injury may be inhibited
by LPA.
LPA affects inflammation and oxidative
stress induced by vascular injury
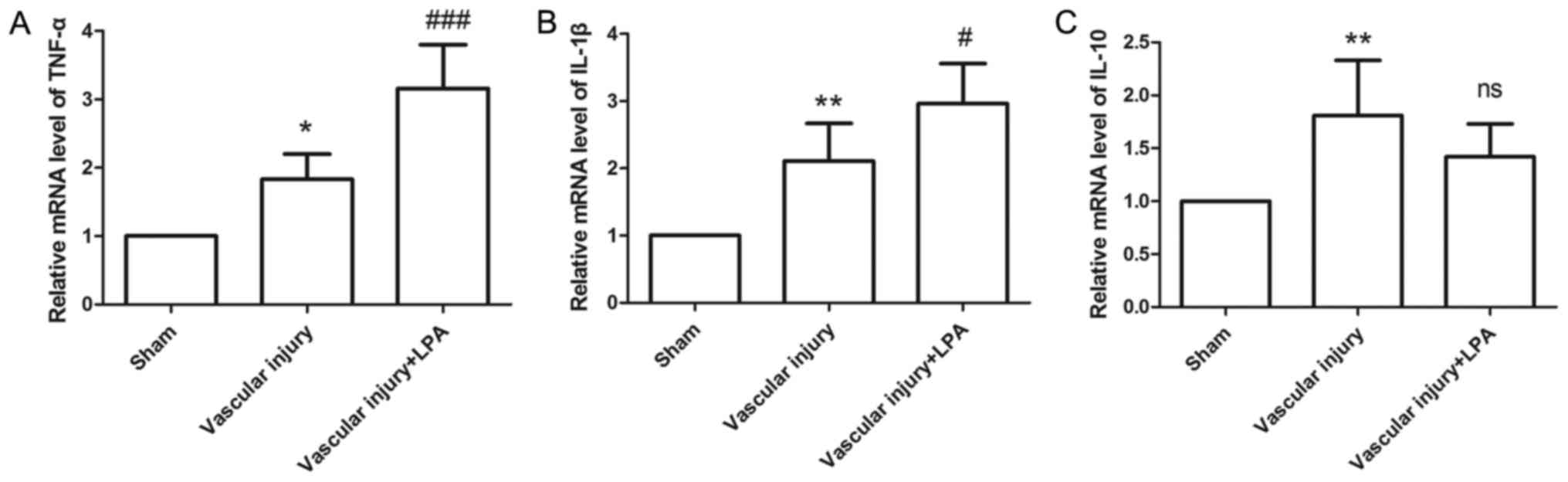
In the present study, the expression levels of
TNF-α, IL-1β and IL-10 were measured, in order to evaluate
inflammatory status. In the vascular injury group, the expression
levels of TNF-α, IL-1β and IL-10 were significantly increased
compared with the sham group (Fig.
6). In the vascular injury + LPA group, the expression levels
of TNF-α and IL-1β were significantly enhanced upon treatment with
LPA compared with the vascular injury group (Fig. 6A and B); however, the expression
levels of IL-10 in the vascular injury + LPA group exhibited a
slight, but not significant decrease following treatment with LPA
(Fig. 6C). These results suggested
that LPA may affect vascular injury-induced inflammation.
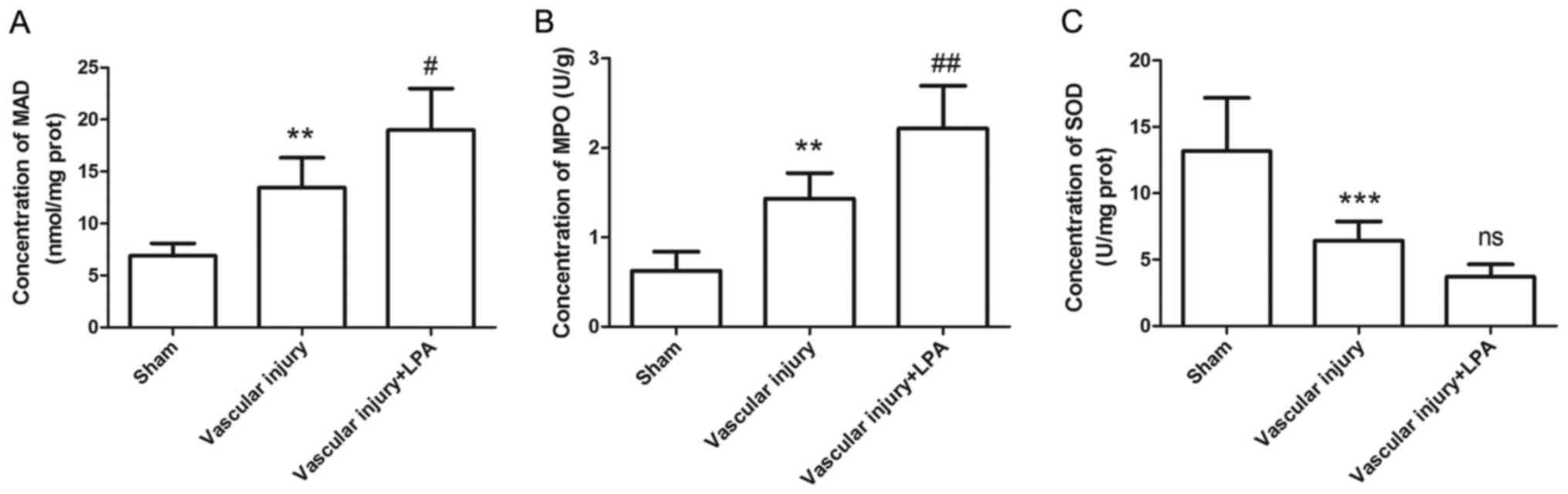
Since oxidative stress also contributes to formation
of neointima, the levels of MDA, MPO and SOD were measured using
corresponding kits. The results indicated that, compared with the
sham group, the MDA and MPO levels in the vascular injury group
were significantly increased (Fig. 7A
and B); conversely, the expression levels of SOD were
significantly decreased in the vascular injury group (Fig. 7C). In addition, in the vascular
injury + LPA group, the increased MDA and MPO levels caused by
vascular injury were significantly enhanced by LPA treatment,
whereas SOD levels were decreased; however, this was not
significant (Fig. 7). These
results indicated that LPA may enhance vascular injury-induced
oxidative stress.
Discussion
LPA serves an important role in the cardiocerebral
vascular system. In the present study, the effects of LPA on
neointimal formation following vascular injury were investigated.
The results of the present study revealed that LPA enhanced
neointimal hyperplasia in the injured carotid arteries by
modulating proliferation, autophagy, inflammation and oxidative
stress. Conversely, LPA exerted no significant effects on apoptosis
in injured carotid arteries. These results suggested that LPA may
contribute to the pathogenesis of atherosclerosis.
The expression levels of LPA are elevated in
atherosclerotic lesions (18,19),
and LPA can induce rapid activation of platelets, stimulate
angiogenesis and regulate the expression of vascular endothelial
growth factor (19–21). The present study revealed that LPA
enhanced vascular injury-induced neointimal hyperplasia via the
modulation of proliferation, autophagy, inflammation and oxidative
stress. In concordance with the present study, Zhang et al
(3) suggested that LPA may induce
neointimal formation in a rat carotid artery model by activating
peroxisome proliferator-activated-receptor γ. Subramanian et
al (13) also reported that
treatment with Ki16425, which blocks LPA receptors (LPA1 and LPA3),
inhibits neointimal formation. These findings indicated that LPA
may contribute to the pathological process of atherosclerosis. In
addition, Kritikou et al (14) suggested that the inhibition of LPA
receptors may reduce the size of atherosclerotic plaques. Since LPA
serves such a critical role in atherosclerosis, it may be
considered a promising therapeutic target for atherosclerosis.
LPA can induce DNA replication and mitosis (22,23);
excessive cell proliferation in arterial walls contributes to the
growth of plaques (5). Vascular
smooth muscle cells are the dominant type of cells that contribute
to atherosclerotic lesions. The proliferation and migration of
vascular smooth muscle cells contribute to the pathogenesis of
atherosclerosis (24). LPA has
previously been reported to serve as a mitogenic growth factor of
vascular smooth muscle cells, thus promoting their proliferation
and affecting their migration (22,25,26).
LPA also induces the proliferation and migration of vascular
endothelial cells, and activates endothelial cells to produce
adhesion molecules and secrete inflammatory cytokines, which also
contribute to the pathogenesis of atherosclerosis (27–29).
Furthermore, LPA affects the migration of fibroblasts and monocytes
(30), which are important for
neointimal formation and the pathogenesis of atherosclerosis
(31). In the present study, the
injured carotid artery tissues exhibited higher PCNA expression
levels following LPA treatment, indicating enhanced proliferation.
Therefore, LPA may contribute to the pathogenesis of
atherosclerosis.
LPA serves an indefinite role in cell apoptosis. LPA
has been reported to exert no effect on the apoptosis of colon
cancer cells, but may increase their proliferation (32). Conversely, LPA has been indicated
to induce apoptosis, but protect against cisplatin-induced
apoptosis in cervical cancer cells (33,34).
The effects of LPA have been reported to promote epithelial cell
apoptosis following lung injury, and promote the resistance of lung
fibroblasts to apoptosis (35). In
the present study, LPA enhanced neointimal hyperplasia caused by
vascular injury, but exerted no effect on the apoptosis of vascular
cells, as evidenced by TUNEL assay and unaltered caspase-3 levels
and Bax/Bcl-2 ratios. LPA also protects macrophages from apoptosis,
promoting atherosclerotic lesion formation (36).
Autophagy, which is activated by stress, nutrient
deprivation and toxic agents, is a conserved process that degrades
long-lived proteins, damaged organelles and macromolecular
aggregates for recycling (37).
Autophagy serves an important role in cholesterol metabolism and
contributes to the pathological processes of atherosclerosis
(38); however, the role of
autophagy in atherosclerosis is complex, with both detrimental and
protective effects (39). Ye et
al (40) reported that, in
injured carotid arteries, activation of autophagy influx appears in
the neointima; consistently, in the present study, autophagy was
activated in injured carotid arteries. Notably, the activation of
autophagy in injured arteries was eliminated upon treatment with
LPA in the present study. Grootaert et al (41) demonstrated that defective autophagy
may promote post-injury neointimal formation. Therefore,
LPA-induced suppression of autophagy may contribute to enhanced
neointimal hyperplasia.
Atherosclerosis is associated with chronic
inflammation. In the present study, LPA was reported to enhance the
elevated expression levels of inflammatory cytokines, TNF-α and
IL-1β, in injured carotid arteries. These results revealed that LPA
may enhance vascular injury-induced inflammation. Consistent with
the findings of the present study, previous studies demonstrated
that LPA may induce the expression of IL-1β in macrophages, thus
contributing to the development of atherosclerosis (42,43).
It has also been reported that LPA promotes the synthesis and
release of TNF-α by T lymphocytes (44). IL-10 is a well-known
anti-inflammatory cytokine, which serves a protective role against
atherosclerosis (45–47). Notably, the expression levels of
IL-10 are increased in advanced or unstable atherosclerotic plaques
(48,49). In the present study, the expression
levels of IL-10 in injured carotid arteries were elevated
post-vascular injury; however, LPA exerted no effect on the
elevated expression levels of IL-10. The recruitment of immune
cells also contributes to the pathology of atherosclerosis. LPA has
been revealed to stimulate the accumulation of macrophages, promote
the migration and adhesion of monocytes to endothelium, and
contribute to the aggravation of atherosclerotic plaques (50–52).
Atherosclerosis is also associated with oxidative
stress-induced conditions (39).
In early atherosclerotic lesions, oxidative stress is activated,
thus resulting in oxidative modification of low-density
lipoprotein, which is a pathogenic factor for atherosclerosis
(53). The present study revealed
that oxidative stress induced by vascular injury was enhanced upon
treatment with LPA, thus indicating that enhanced oxidative stress
may be associated with the neointimal-promoting effects of LPA.
According to the literature, oxidative stress is also involved in
the endothelial cytotoxicity of LPA (54).
In conclusion, the present study demonstrated that
LPA may enhance neointimal hyperplasia by modulating proliferation,
autophagy, inflammation and oxidative stress, but not apoptosis,
within injured carotid arteries. The findings of the present study
indicated that LPA may contribute to the pathogenesis of
atherosclerosis and may be considered a promising target for the
treatment of atherosclerosis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XS conceptualized the study design and wrote the
manuscript. XS, JZ, FL, TZ and TG performed the experiments and
analyzed the data.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of The People's Hospital of China Medical University
(Liaoning, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Bcl-2
|
B-cell lymphoma-2
|
|
Bax
|
Bcl-2-associated X protein
|
|
DAB
|
3,3′-diaminobenzidine
|
|
HE
|
hematoxylin and eosin
|
|
HRP
|
horseradish peroxidase
|
|
IL
|
interleukin
|
|
LPA
|
lysophosphatidic acid
|
|
MDA
|
malondialdehyde
|
|
MPO
|
myeloperoxidase
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
SOD
|
superoxide dismutase
|
|
TBST
|
Tris-buffered saline with Tween
|
|
TNF
|
tumor necrosis factor
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling
|
References
|
1
|
Weber C and Noels H: Atherosclerosis:
Current pathogenesis and therapeutic options. Nat Med.
17:1410–1422. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schober A and Siess W: Lysophosphatidic
acid in atherosclerotic diseases. Br J Pharmacol. 167:465–482.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang C, Baker DL, Yasuda S, Makarova N,
Balazs L, Johnson LR, Marathe GK, McIntyre TM, Xu Y, Prestwich GD,
et al: Lysophosphatidic acid induces neointima formation through
PPARgamma activation. J Exp Med. 199:763–774. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clowes AW, Clowes MM, Fingerle J and Reidy
MA: Regulation of smooth muscle cell growth in injured artery. J
Cardiovasc Pharmacol. 14 Suppl 6:S12–S15. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fuster JJ, Fernandez P, Gonzalez-Navarro
H, Silvestre C, Nabah YN and Andres V: Control of cell
proliferation in atherosclerosis: Insights from animal models and
human studies. Cardiovasc Res. 86:254–264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sheng X, Yung YC, Chen A and Chun J:
Lysophosphatidic acid signalling in development. Development.
142:1390–1395. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi JW, Herr DR, Noguchi K, Yung YC, Lee
CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN and Chun J: LPA
receptors: Subtypes and biological actions. Annu Rev Pharmacol
Toxicol. 50:157–186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin ME, Herr DR and Chun J:
Lysophosphatidic acid (LPA) receptors: Signaling properties and
disease relevance. Prostaglandins Other Lipid Mediat. 91:130–138.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smyth SS, Cheng HY, Miriyala S,
Panchatcharam M and Morris AJ: Roles of lysophosphatidic acid in
cardiovascular physiology and disease. Biochim Biophys Acta.
1781:563–570. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Siess W and Tigyi G: Thrombogenic and
atherogenic activities of lysophosphatidic acid. J Cell Biochem.
92:1086–1094. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshida K, Nishida W, Hayashi K, Ohkawa Y,
Ogawa A, Aoki J, Arai H and Sobue K: Vascular remodeling induced by
naturally occurring unsaturated lysophosphatidic acid in vivo.
Circulation. 108:1746–1752. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Subramanian P, Karshovska E, Reinhard P,
Megens RT, Zhou Z, Akhtar S, Schumann U, Li X, van Zandvoort M,
Ludin C, et al: Lysophosphatidic acid receptors LPA1 and LPA3
promote CXCL12-mediated smooth muscle progenitor cell recruitment
in neointima formation. Circ Res. 107:96–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kritikou E, van Puijvelde GH, van der
Heijden T, van Santbrink PJ, Swart M, Schaftenaar FH, Kröner MJ,
Kuiper J and Bot I: Inhibition of lysophosphatidic acid receptors 1
and 3 attenuates atherosclerosis development in LDL-receptor
deficient mice. Sci Rep. 6:375852016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Research NRCUIfLA, . Guide for the care
and use of laboratory animals. Washington (DC): National Academies
Press (US); 1996
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu H, Clarke MC, Figg N, Littlewood TD and
Bennett MR: Smooth muscle cell apoptosis promotes vessel remodeling
and repair via activation of cell migration, proliferation and
collagen synthesis. Arterioscler Thromb Vasc Biol. 31:2402–2409.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bot M, Bot I, Lopez-Vales R, van de Lest
CH, Saulnier-Blache JS, Helms JB, David S, van Berkel TJ and
Biessen EA: Atherosclerotic lesion progression changes
lysophosphatidic acid homeostasis to favor its accumulation. Am J
Pathol. 176:3073–3084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rother E, Brandl R, Baker DL, Goyal P,
Gebhard H, Tigyi G and Siess W: Subtype-selective antagonists of
lysophosphatidic Acid receptors inhibit platelet activation
triggered by the lipid core of atherosclerotic plaques.
Circulation. 108:741–747. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sweat RS, Azimi MS, Suarez-Martinez AD,
Katakam P and Murfee WL: Lysophosphatidic acid does not cause
blood/lymphatic vessel plasticity in the rat mesentery culture
model. Physiol Rep. 4:e128572016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rivera-Lopez CM, Tucker AL and Lynch KR:
Lysophosphatidic acid (LPA) and angiogenesis. Angiogenesis.
11:301–310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim J, Keys JR and Eckhart AD: Vascular
smooth muscle migration and proliferation in response to
lysophosphatidic acid (LPA) is mediated by LPA receptors coupling
to Gq. Cell Signal. 18:1695–1701. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu YJ, Rathi SS, Chapman DC, Arneja AS and
Dhalla NS: Mechanisms of lysophosphatidic acid-induced DNA
synthesis in vascular smooth muscle cells. J Cardiovasc Pharmacol.
41:381–387. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chappell J, Harman JL, Narasimhan VM, Yu
H, Foote K, Simons BD, Bennett MR and Jørgensen HF: Extensive
proliferation of a subset of differentiated, yet plastic, medial
vascular smooth muscle cells contributes to neointimal formation in
mouse injury and atherosclerosis models. Circ Res. 119:1313–1323.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Komachi M, Damirin A, Malchinkhuu E, Mogi
C, Tobo M, Ohta H, Sato K, Tomura H and Okajima F: Signaling
pathways involved in DNA synthesis and migration in response to
lysophosphatidic acid and low-density lipoprotein in coronary
artery smooth muscle cells. Vascul Pharmacol. 50:178–184. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Teo ST, Yung YC, Herr DR and Chun J:
Lysophosphatidic acid in vascular development and disease. IUBMB
Life. 61:791–799. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee H, Goetzl EJ and An S:
Lysophosphatidic acid and sphingosine 1-phosphate stimulate
endothelial cell wound healing. Am J Physiol Cell Physiol.
278:C612–C618. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee H, Lin CI, Liao JJ, Lee YW, Yang HY,
Lee CY, Hsu HY and Wu HL: Lysophospholipids increase ICAM-1
expression in HUVEC through a Gi- and NF-kappaB-dependent
mechanism. Am J Physiol Cell Physiol. 287:C1657–C1666. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin CI, Chen CN, Chen JH and Lee H:
Lysophospholipids increase IL-8 and MCP-1 expressions in human
umbilical cord vein endothelial cells through an IL-1-dependent
mechanism. J Cell Biochem. 99:1216–1232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pilquil C, Dewald J, Cherney A, Gorshkova
I, Tigyi G, English D, Natarajan V and Brindley DN: Lipid phosphate
phosphatase-1 regulates lysophosphatidate-induced fibroblast
migration by controlling phospholipase D2-dependent phosphatidate
generation. J Biol Chem. 281:38418–38429. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sartore S, Chiavegato A, Faggin E, Franch
R, Puato M, Ausoni S and Pauletto P: Contribution of adventitial
fibroblasts to neointima formation and vascular remodeling: From
innocent bystander to active participant. Circ Res. 89:1111–1121.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Leve F, Peres-Moreira RJ, Binato R,
Abdelhay E and Morgado-Diaz JA: LPA induces colon cancer cell
proliferation through a cooperation between the ROCK and STAT-3
pathways. PLoS One. 10:e01390942015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dong Y, Wu Y, Cui MZ and Xu X:
Lysophosphatidic acid triggers apoptosis in HeLa cells through the
upregulation of tumor necrosis factor receptor superfamily member
21. Mediators Inflamm. 2017:27547562017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sui Y, Yang Y, Wang J, Li Y, Ma H, Cai H,
Liu X, Zhang Y, Wang S, Li Z, et al: Lysophosphatidic acid inhibits
apoptosis induced by cisplatin in cervical cancer cells. Biomed Res
Int. 2015:5983862015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Funke M, Zhao Z, Xu Y, Chun J and Tager
AM: The lysophosphatidic acid receptor LPA1 promotes epithelial
cell apoptosis after lung injury. Am J Respir Cell Mol Biol.
46:355–364. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koh JS, Lieberthal W, Heydrick S and
Levine JS: Lysophosphatidic acid is a major serum noncytokine
survival factor for murine macrophages which acts via the
phosphatidylinositol 3-kinase signaling pathway. J Clin Invest.
102:716–727. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rajawat YS and Bossis I: Autophagy in
aging and in neurodegenerative disorders. Hormones (Athens).
7:46–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ouimet M, Ediriweera H, Afonso MS,
Ramkhelawon B, Singaravelu R, Liao X, Bandler RC, Rahman K, Fisher
EA, Rayner KJ, et al: microRNA-33 regulates macrophage autophagy in
atherosclerosis. Arterioscler Thromb Vasc Biol. 37:1058–1067. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Perrotta I and Aquila S: The role of
oxidative stress and autophagy in atherosclerosis. Oxid Med Cell
Longev. 2015:1303152015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ye LX, Yu J, Liang YX, Zeng JS, Huang RX
and Liao SJ: Beclin 1 knockdown retards re-endothelialization and
exacerbates neointimal formation via a crosstalk between autophagy
and apoptosis. Atherosclerosis. 237:146–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grootaert MO, da Costa Martins PA, Bitsch
N, Pintelon I, De Meyer GR, Martinet W and Schrijvers DM: Defective
autophagy in vascular smooth muscle cells accelerates senescence
and promotes neointima formation and atherogenesis. Autophagy.
11:2014–2032. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kirii H, Niwa T, Yamada Y, Wada H, Saito
K, Iwakura Y, Asano M, Moriwaki H and Seishima M: Lack of
interleukin-1beta decreases the severity of atherosclerosis in
ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 23:656–660.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang CL, Lin ME, Hsu HY, Yao CL, Hwang
SM, Pan CY, Hsu CY and Lee H: Lysophosphatidic acid-induced
interleukin-1 beta expression is mediated through Gi/Rho and the
generation of reactive oxygen species in macrophages. J Biomed Sci.
15:357–363. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang D, Zhang Y, Zhao C, Zhang W, Shao G
and Zhang H: Effect of lysophosphatidic acid on the immune
inflammatory response and the connexin 43 protein in myocardial
infarction. Exp Ther Med. 11:1617–1624. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oslund Pinderski LJ, Hedrick CC, Olvera T,
Hagenbaugh A, Territo M, Berliner JA and Fyfe AI: Interleukin-10
blocks atherosclerotic events in vitro and in vivo. Arterioscler
Thromb Vasc Biol. 19:2847–2853. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pinderski LJ, Fischbein MP, Subbanagounder
G, Fishbein MC, Kubo N, Cheroutre H, Curtiss LK, Berliner JA and
Boisvert WA: Overexpression of interleukin-10 by activated T
lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice
by altering lymphocyte and macrophage phenotypes. Circ Res.
90:1064–1071. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mallat Z, Besnard S, Duriez M, Deleuze V,
Emmanuel F, Bureau MF, Soubrier F, Esposito B, Duez H, Fievet C, et
al: Protective role of interleukin-10 in atherosclerosis. Circ Res.
85:e17–e24. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mallat Z, Heymes C, Ohan J, Faggin E,
Leseche G and Tedgui A: Expression of interleukin-10 in advanced
human atherosclerotic plaques: Relation to inducible nitric oxide
synthase expression and cell death. Arterioscler Thromb Vasc Biol.
19:611–616. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nishihira K, Imamura T, Yamashita A,
Hatakeyama K, Shibata Y, Nagatomo Y, Date H, Kita T, Eto T and
Asada Y: Increased expression of interleukin-10 in unstable plaque
obtained by directional coronary atherectomy. Eur Heart J.
27:1685–1689. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhou Z, Subramanian P, Sevilmis G, Globke
B, Soehnlein O, Karshovska E, Megens R, Heyll K, Chun J,
Saulnier-Blache JS, et al: Lipoprotein-derived lysophosphatidic
acid promotes atherosclerosis by releasing CXCL1 from the
endothelium. Cell Metab. 13:592–600. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gustin C, Van Steenbrugge M and Raes M:
LPA modulates monocyte migration directly and via LPA-stimulated
endothelial cells. Am J Physiol Cell Physiol. 295:C905–C914. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bot M, de Jager SC, MacAleese L, Lagraauw
HM, van Berkel TJ, Quax PH, Kuiper J, Heeren RM, Biessen EA and Bot
I: Lysophosphatidic acid triggers mast cell-driven atherosclerotic
plaque destabilization by increasing vascular inflammation. J Lipid
Res. 54:1265–1274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sparrow CP and Olszewski J: Cellular
oxidative modification of low density lipoprotein does not require
lipoxygenases. Proc Natl Acad Sci USA. 89:128–131. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Brault S, Gobeil F Jr, Fortier A, Honore
JC, Joyal JS, Sapieha PS, Kooli A, Martin E, Hardy P,
Ribeiro-da-Silva A, et al: Lysophosphatidic acid induces
endothelial cell death by modulating the redox environment. Am J
Physiol Regul Integr Comp Physiol. 292:R1174–R1183. 2007.
View Article : Google Scholar : PubMed/NCBI
|