Introduction
Boucher-Neuhäuser syndrome [BNS; Mendelian
Inheritance in Man (MIM) reference no. 215470; www.omim.org] is a rare autosomal recessive disorder
and is characterized by spinocerebellar ataxia, eye abnormalities
and a failure of the pituitary gland to stimulate gonadal
development during puberty (1–8).
Previously, mutations in patatin-like phospholipase domain
containing 6 (PNPLA6) have been linked to BNS (9–13).
PNPLA6 (MIM reference no. 603197; www.omim.org; 19p13.2) encodes for a neuropathy target
esterase (13,14). A total of five different
transcripts have been identified, with the longest transcript,
transcript variant 1 (GenBank accession no. NM_001166111.1;
www.ncbi.nlm.nih.gov/genbank),
encoding a protein (GenBank accession no. NP_001159583.1) of 1,375
amino acids (14). In addition to
BNS, variations in the PNPLA6 gene have been implicated in
Gordon Holmes syndrome, spastic paraplegia, photoreceptor
degeneration and pure cerebellar ataxia (12,15–17).
Hypogonadotropic hypogonadism may be identified at
the onset of puberty (18–20). Gait ataxia in BNS generally
manifests prior to early adulthood, although late-onset ataxia has
additionally been reported (2,9). In
the present study, a 39-year-old male was initially diagnosed with
hypogonadotropic hypogonadism at the age of 14 years. Subsequent
magnetic resonance imaging (MRI) and funduscopic examination
suggested that the symptoms were a result of BNS. The cause of the
symptoms was further confirmed by the presence of a compound
heterozygous PNPLA6 mutation.
Materials and methods
Patients
The proband and his parents were recruited at the
People's Hospital of Henan Province (Zhengzhou, China) in March
2016. The age range of patients was 39–70 years old. Written
informed consent was obtained. The present study was approved by
the ethics committees of Central South University and The People's
Hospital of Henan Province.
Clinical assessments
Endocrine manifestation in serum isolated from whole
blood samples (1 ml per patient) were examined using the following
ELISA kits: Human luteinizing hormone ELISA kit (cat. no. ab108651;
Abcam, Cambridge, UK), human follicle stimulating hormone ELISA kit
(cat. no. ab108641; Abcam), human estradiol E2 ELISA kit (cat. no.
ab108640; Abcam), human testosterone ELISA kit (cat. no. ab174569;
Abcam), human prolactin ELISA kit (cat. no. ab108655; Abcam), human
progesterone ELISA kit (cat. no. ab108654; Abcam), human
adrenocorticotropic hormone ELISA kit (cat. no. ENZ-KIT138-0001;
Enzo Life Sciences, Inc., Farmingdale, NY, USA), human
triiodothyronine ELISA kit (cat. no. ab108664; Abcam), human
thyroxine ELISA kit (cat. no. ab108686; Abcam), human thyroid
stimulating hormone ELISA kit (cat. no. ab108659; Abcam) and human
parathyroid hormone ELISA kit (cat. no. ab230931; Abcam). The
retina was examined using routine funduscopic examination; the
reflexes of the two legs were detected using electromyography; and
the brain structure was examined using MRI. The results of MRI were
analyzed using fluid-attenuated inversion recovery (21). And the middle cerebral artery was
examined using magnetic resonance angiography (22).
Whole exome sequencing
Genomic DNA was isolated from peripheral blood
leukocytes (1 ml per patient). The whole exome was captured using a
Sure Select Human All Exon kit (Agilent Technologies, Inc., Santa
Clara, CA, USA), and was sequenced on an Illumina HiSeq2500
instrument (Illumina, Inc., San Diego, CA, USA). The sequenced
reads were aligned to the human genome reference (University of
California Santa Cruz hg 19 version; www.epigenomebrowser.org/index.html) using the
Burrows-Wheeler Aligner (bwa-0.7.17) (23). Read qualities were recalibrated
using the Genome Analysis Toolkit (GATK-4.0.2.1; Broad Institute,
Cambridge, MA, USA). The GATK IndelRealigner was used to realign
reads around insertion/deletion sites. The single nucleotide
variants (SNVs) and small insertions and deletions (InDels) were
generated with the GATK Unified Genotyper in parallel with the
SAMtools (samtools-1.7; www.htslib.org/doc/samtools.html) pipeline. ANNOVAR
(version 2.3.1) (24) was used to
annotate the detected variations. ExAC (version 0.3.1; exac.broadinstitute.org) and 1,000 Genomes
(www.internationalgenome.org)
(25) were used to identify the
population frequencies of mutations.
Deleterious missense SNVs were predicted using the
following tools: i) SIFT [sift.jcvi.org;
a single nucleotide polymorphism (SNP) with SIFT score <0.05
predicts a negative effect on the encoded amino acid]; ii)
Polyphen2 (genetics.bwh.harvard.edu/pph2; a SNP with score
between 0.85–1.0 may be predicted to be damaging to the encoded
amino acid, while a SNP with score of 0.0–0.15 may be predicted to
be benign, and a SNP with score of 0.15–1.0 predicts possible
damage); iii) MutationTaster (www.mutationtaster.org; a score close to 1 indicates
the given variant to be disease-causing); iv) MutationAssessor
(mutationassessor.org/r3; High/Medium
means functional and Low/Neutral means non-functional); and v)
Combined Annotation Dependent Depletion (CADD; cadd.gs.washington.edu), in which a scaled CADD score
of 20 means that a variant is among the top 1% of deleterious
variants in the human genome, and a scaled CADD score of 30 means
that the variant is in the top 0.1% (26).
Polymerase chain reaction (PCR)-sanger
sequencing
PCR was used to amplify the exons containing the
PNPLA6 variants identified in the whole exome sequencing.
PCR was performed in a total volume of 20 µl with 50 ng genomic DNA
as template, 10 µl 2X Taq Mastermix (E005; Novoprotein Scientific,
Inc., Summit, NJ, USA), 1 µl forward primer (10 µmol/ml) and 1 µl
reverse primer (10 µmol/ml) under the following thermocycling
conditions: 94°C for 2 min; followed by 40 cycles of 94°C for 20
sec, 65°C for 20 sec and 72°C for 45 sec; and a final extension
step at 72°C for 5 min. The amplified products were used for Sanger
sequencing as performed by BioSune Biotechnology Co. (Shanghai,
China). The sequencing results were analyzed by using DNA
Star-Megalign software (version 3.3.8; DNASTAR, Inc., Madison, WI,
USA). The primers used for PCR were as follows:
PNPLA6-E29-forward (F), 5′-CAGGCTGTGTGTGGCGTTACGTC-3′;
PNPLA6-E29-reverse (R), 5′-GCACATGTCGCTGTCCACAGGCAC-3′;
PNPLA6-E30-F, 5′-GGACATCGCCCGCAGCATG-3′; and
PNPLA6-E30-R, 5′-ATAGATCTGGTCGAACTTCCCAAAG-3′.
Structural modeling of proteins
Structural modeling was performed using I-TASSER
(zhanglab.ccmb.med.umich.edu/I-TASSER). The models were
visualized using PyMOL (version 0.99; DeLano Scientific LLC., Palo
Alto, CA, USA).
Results and Discussion
Endocrine manifestation
The proband is a 39-year-old male (as of 2016). He
was diagnosed with hypogonadotropic hypogonadism 25 years ago at
the age of 14 years. He was subsequently treated intermittently for
2 years with testosterone, human chorionic gonadotropin and human
menopausal gonadotropin, leading to significant growth in the
testes and penis. The proband was married 6 years ago, although he
experienced poor sexual performance and the wife of the proband did
not conceive. In 2015, the proband was enrolled at The People's
Hospital of Henan Province due to infertility.
As illustrated in Table
I, the luteinizing hormone (LH), follicle-stimulating hormone
(FSH) and testosterone levels of the proband were below normal,
whereas the levels of adrenocorticotropic hormone, cortisol and
thyroid hormones were in the normal range. A gonadotropin-releasing
hormone (GnRH) stimulation test was additionally performed. As
exhibited in Table I, GnRH failed
to induce an obvious increase in FSH and LH.
 | Table I.Baseline hormonal profile and results
of gonadotrophin releasing hormone stimulation test. |
Table I.
Baseline hormonal profile and results
of gonadotrophin releasing hormone stimulation test.
|
| Time, min |
|---|
|
|
|
|---|
| Hormone | 0 | 30 | 60 | 90 | 120 | Normal range,
basal |
|---|
| LH | 0.27 | 0.59 | 0.59 | 0.48 | 0.57 | 1.2–8.6 mIU/ml |
| FSH | 1.97 | 1.07 | 1.69 | 1.87 | 1.81 | 1.3–19.3
mIU/ml |
| E2 | 38.32 |
|
|
|
| <53 pg/ml |
| T | 2.9 |
|
|
|
| 1.75–7.81
ng/ml |
| PRL | 5.41 |
|
|
|
| 2.64–13.13
ng/ml |
| P | 0.32 |
|
|
|
| 0.1–0.84 ng/ml |
| Cortisol |
|
|
|
|
|
|
| 08.00
a.m. | 8.60 |
|
|
|
| 5–25 µg/dl |
| 04.00
p.m. | 5.76 |
|
|
|
| 2.5–12.5 µg/dl |
| 12.00
a.m. | <1.0 |
|
|
|
| 2.9–13 µg/dl |
| ACTH |
|
|
|
|
|
|
| 08.00
a.m. | 29.8 |
|
|
|
| 12–46 pg/ml |
| 04.00
p.m. | 22.8 |
|
|
|
| 6–23 pg/ml |
| FT3 | 5.06 |
|
|
|
| 3.5–6.5 pmol/l |
| FT4 | 14.34 |
|
|
|
| 11.5–22.7
pmol/l |
| TSH | 1.217 |
|
|
|
| 0.55–4.78
µIU/ml |
| PTH | 71.3 |
|
|
|
| 12–88 pg/ml |
Retinal degeneration and cerebellar
atrophy
In a routine funduscopic examination, bilateral
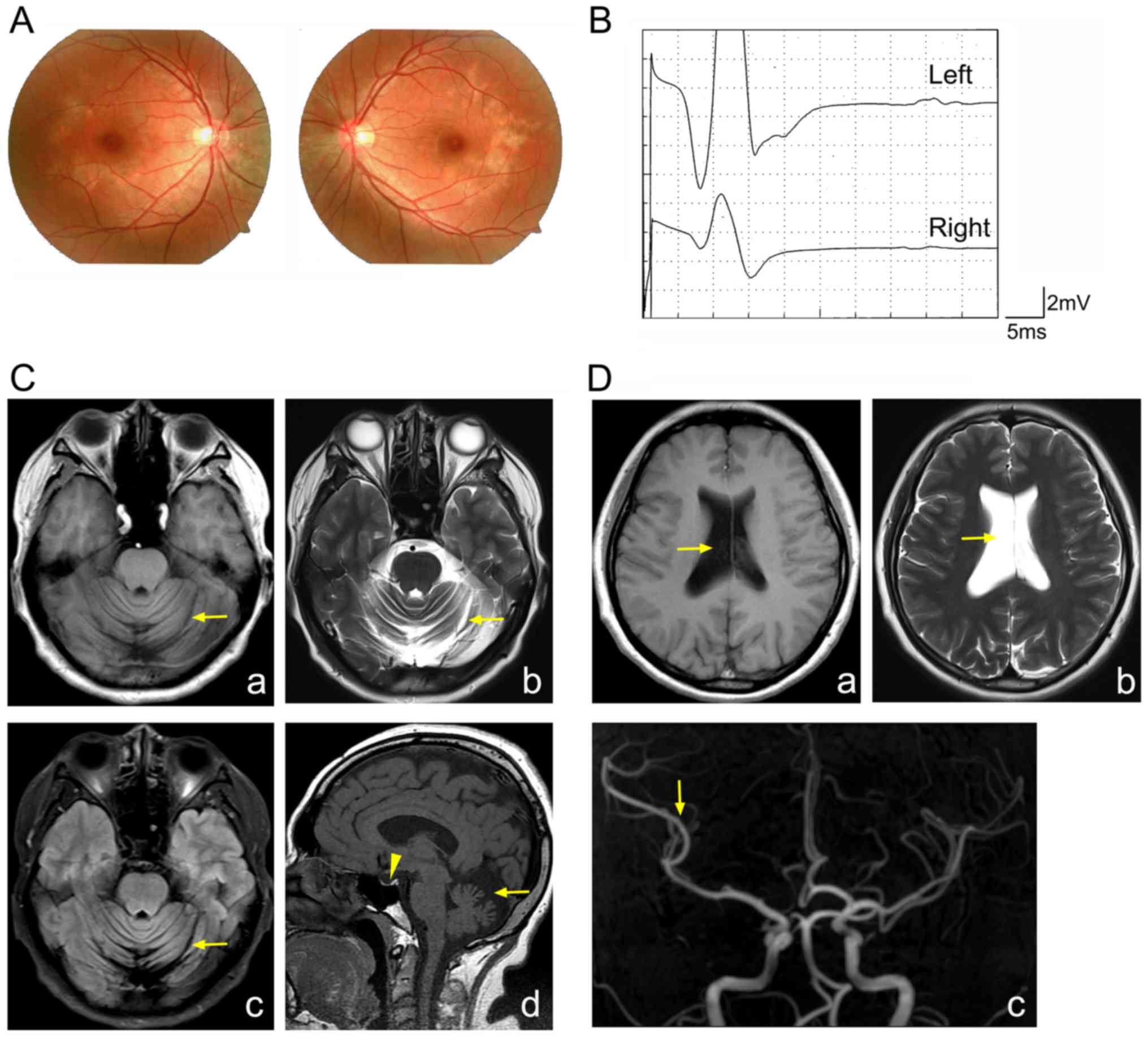
macular pigment epitheliopathy was identified in the proband
(Fig. 1A). Therefore, it was
hypothesized that the proband may be affected by BNS.
The proband did not exhibit obvious gait ataxia;
however, gait ataxia in patients with BNS may manifest as late as
the age of 50 years (9). Therefore
a thorough neurological examination was performed. No defect was
detected in electroencephalography and hearing tests (data not
shown). However, the proband presented with subtle defect in the
finger-naseversch and alternate motion tests. In addition, the
proband exhibited cavus and a reduced tendon reflex.
Electromyography demonstrated the absence of
H-reflection in both legs of the proband (Fig. 1B). MRI demonstrated marked
cerebellar atrophy, with widened cerebellar sulci and increased
cerebrospinal fluid (Fig. 1C).
Furthermore, MRI illustrated a reduction in the size the pituitary,
the expansion of the right lateral side, and a reduction in the
branches of the right middle cerebral artery (Fig. 1C and D). From these results, the
proband was diagnosed as having BNS.
Whole exome sequencing
To understand the genetic mechanism, whole exome
sequencing of the proband was performed. A total of 39.81 million
clean reads were obtained following the removal of low-quality
reads and adaptor or contaminant sequences. A total of 98.94% of
the exome regions were covered. The sample had an average of
46.8-fold coverage of the entire exon region, of which 93.9, 80.6
and 64.4% of the targeted regions were covered at least 10, 20 and
30 times, respectively. Using the GATK, a total of 21,797 coding
SNVs and 584 small indels were identified. These included 10,054
missense variants, 73 nonsense variants, 189 splice site variants
and 322 frame-shift indel mutations.
Compound heterozygous mutation in
PNPLA6
A total of two PNPLA6 mutations
(c.3386G>T, p.G1129V and c.3534G>C, p.W1178C; Fig. 2A and B) were identified. To
determine whether the two PNPLA6 mutations (c.3386G>T,
p.G1129V; c.3534G>C, p.W1178C) were located in the same or
different chromosomes, the mutations were analyzed in the parents
of the proband. As exhibited in Fig.
2A and B, the proband inherited PNPLA6 (c.3534G>C,
p.W1178C) and PNPLA6 (c.3386G>T, p.G1129V) from his
father and mother, respectively. Therefore, the PNPLA6
mutations exist in a compound heterozygous form.
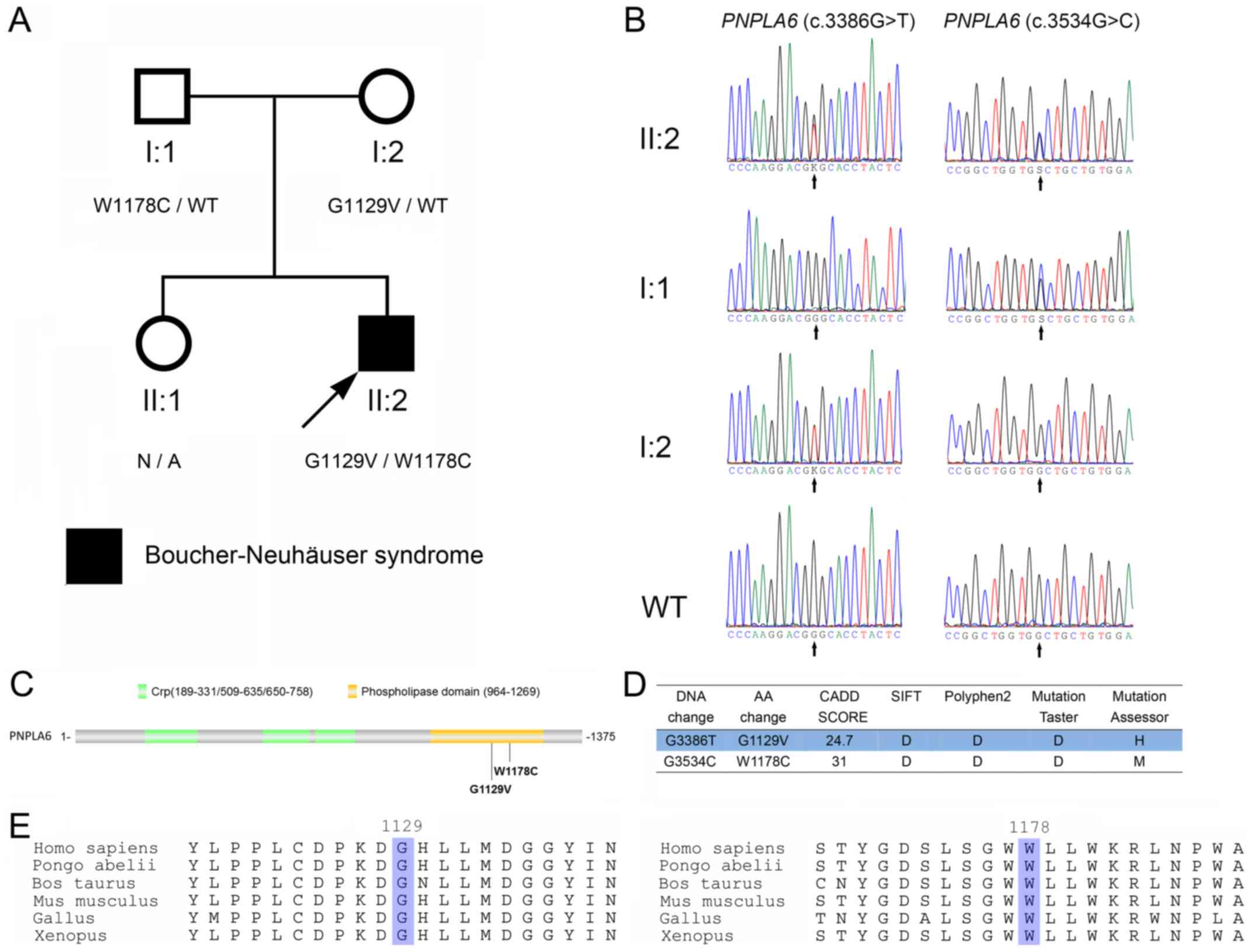 | Figure 2.Identification of PNPLA6
mutations in the patient. (A) The pedigree of the family. Subjects
in the family are identified by the Roman and Arabic numerals below
the symbol, in which the Roman numerals denote the generations.
Open symbols, unaffected; filled symbols, affected; squares, male;
circles, female; arrow, the proband. (B) Sanger sequencing of
codons 3,376–3,396 and 3,524–3,544 of the PNPLA6 genes from
the proband (II:2), his father (I:1) and mother (I:2), and a
wild-type control. K represents G or T; S represents G or C. (C)
Schematic diagram of PNPLA6 protein. The G1129V and W1178C
mutations identified in the present study are indicated with
arrows. (D) The prediction of PNPLA6 mutations using SIFT,
Polyphen2, MutationTaster, MutationAssessor and CADD. (E)
Conservative analysis of the glycine 1,129 and tryptophan
1,178-containing portions of the PNPLA6 protein. The glycine 1,129
and tryptophan 1,178 are highlighted. PNPLA6, patatin-like
phospholipase domain containing 6; Crp, cAMP-binding domain of
CRP. |
The PNPLA6 (c.3386G>T, p.G1129V) and
PNPLA6 (c.3534G>C, p.W1178C) mutations were located in
the 29th and 30th exons, respectively. The two mutations were
located in the phospholipid esterase domain of the PNPLA6 protein
(Fig. 2C), and were predicted to
be pathologically damaging using SIFT, Polyphen2, MutationTaster,
MutationAssessor and CADD (Fig.
2D). Neither PNPLA6 (c.3386G>T, p.G1129V) nor
PNPLA6 (c.3534G>C, p.W1178C) were identified in the ExAC
and 1,000 Genomes databases. Glycine 1,129 and tryptophan 1,178 are
evolutionarily conserved (Fig.
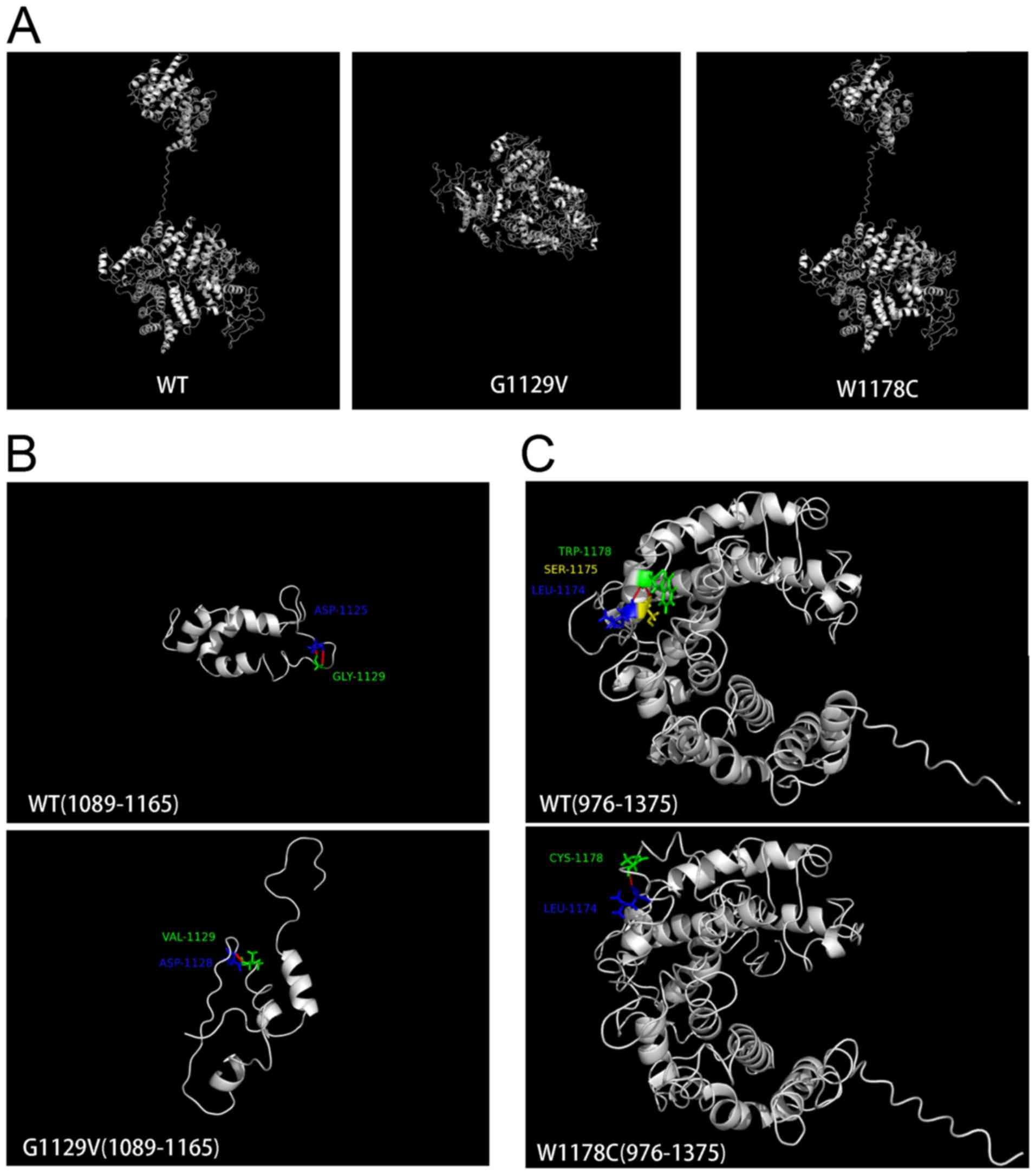
2E). Structural modeling was additionally performed using
I-TASSER (27). As exhibited in
Fig. 3, mutations led to
conformational alterations in the PNPLA6 protein. The overall
conformation of W1178C PNPLA6 only demonstrated a minor alteration
compared with wild-type (WT) PNPLA6, while the overall conformation
of G1129V PNPLA6 was markedly altered (Fig. 3A). Glycine 1,129 formed a hydrogen
bond with aspartic acid at 1,125 in WT PNPLA6; however, a G1129V
mutation led to an alternative hydrogen bond between valine 1,129
and aspartic acid at 1,128 (Fig.
3B). Tryptophan 1,178 formed hydrogen bonds with serine 1,175
and leucine 1,174 in WT PNPLA6, whereas cysteine 1,178 only formed
a hydrogen bond with leucine 1,174 in W1178C PNPLA6 (Fig. 3C).
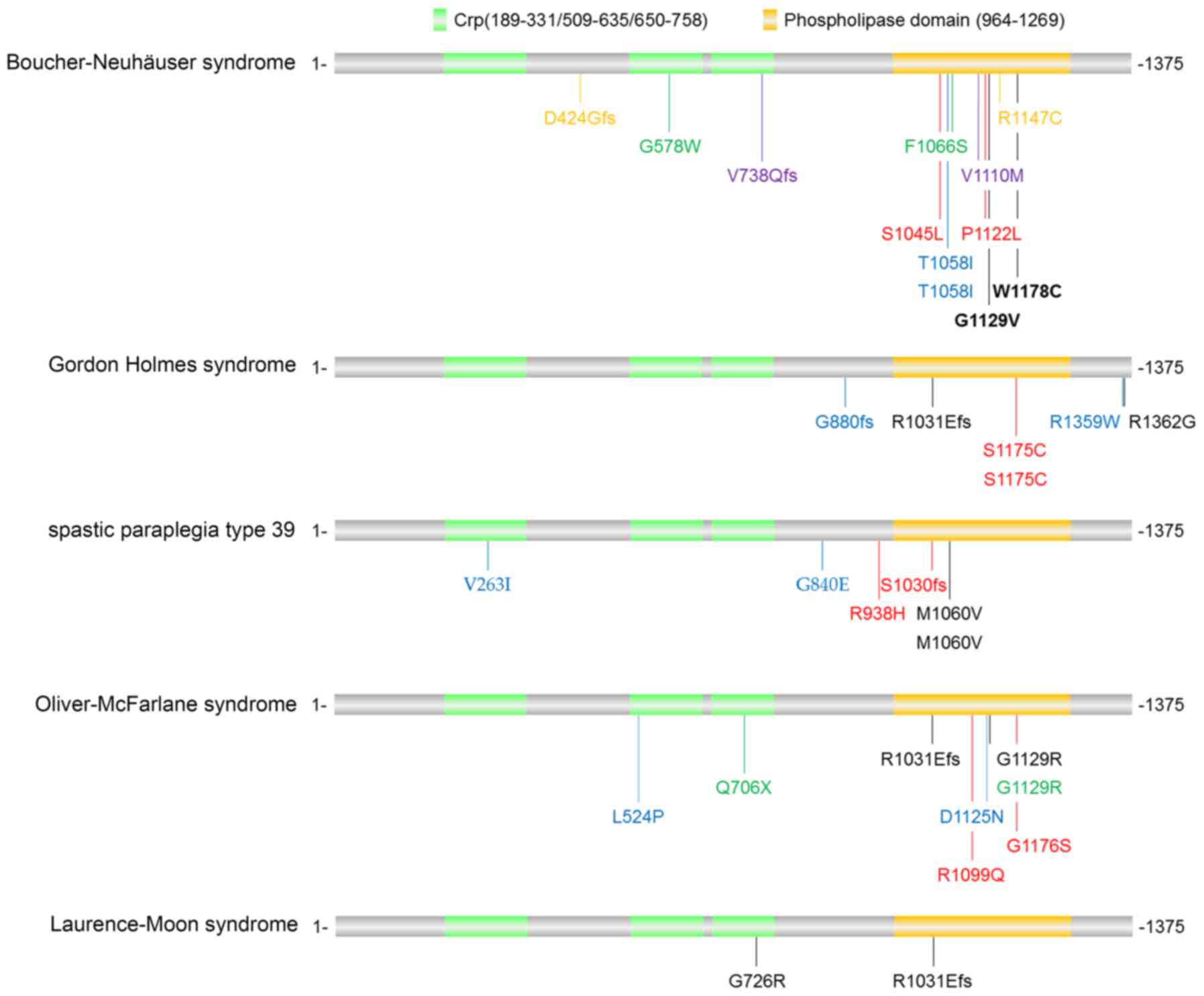
PNPLA6 mutations have been identified in a panel of
disorders, including Boucher-Neuhäuser syndrome, Gordon Holmes
syndrome, spastic paraplegia type 39, Oliver-McFarlane syndrome and
Laurence-Moon syndrome, with partly distinct and partly overlapping
clinical phenotypes. For instance, Boucher-Neuhäuser syndrome and
Gordon Holmes syndrome cause cerebellar ataxia and hypogonadotropic
hypogonadism. However, Boucher-Neuhäuser syndrome additionally
presents with chorioretinal dystrophy, whereas Gordon Holmes
syndrome leads to brisk reflexes. By contrast, spastic paraplegia
type 39 exhibits upper motor neuron involvement, peripheral
neuropathy, and sometimes reduced cognitive functioning and/or
cerebellar ataxia; Oliver-McFarlane syndrome exhibits trichomegaly,
chorioretinal dystrophy, short stature, intellectual disability and
hypopituitarism; and Laurence-Moon syndrome is characterized by
chorioretinopathy, pituitary dysfunction, childhood onset of
ataxia, spastic paraplegia and peripheral neuropathy (16,28).
PNPLA6-associated disorders are usually
diagnosed by clinical and neuroimaging results combined with
biallelic pathogenic variants in PNPLA6. The mutations
identified in patients with PNPLA6-associated disorders
(12,13,28–30)
are illustrated in Fig. 4. The
majority of the mutations were located in the phospholipid esterase
domain of the PNPLA6 protein, although there was no
disease-specific bias in these mutations. Hufnagel et al
(28) envisioned that various
PNPLA6 mutations may lead to different activity alterations
in the neuropathy target esterase, and more severe reductions may
cause more severe and early-onset presentations. However, further
studies in animal models are required to uncover why mutations in
one gene (PNPLA6) lead to various clinical presentations in
different cell types.
In conclusion, the first BNS patient from China was
identified in the present study. Although the proband did not
exhibit gait ataxia at the age of 39 years old, MRI examination
clearly indicated cerebellar atrophy. Genetic analysis confirmed
the clinical manifestation. The present study implied that whole
exome sequencing may be used for correct diagnosis and proper
genetic counseling prior to the occurrence of obvious symptoms.
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the
National High-tech R&D Program (grant no. 2015AA020502),
National Basic Research Program of China (grant no. 2012CB517904),
National Natural Science Foundation of China (grant nos. 31371187,
31501152, 31401218, 81770780 and 81728013), the Basic and Frontier
Project from the Department of Science, Technology of Henan
Province (grant no. 142300410071) and the Fundamental Research
Funds for the Central Universities of Central South University
(grant nos. 1053320170642, 1053320171214 and 1053320170804).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RZ, YZ, WHL and JDL conceived the study. RZ, YW,
JLL, ZLZ and DNC recruited the patients. YZ, JW and JDL performed
the experiments. RZ, YZ, JW, YW, JLL, ZLZ, XTZ, DNC, WHL and JDL
analyzed the data. YZ and JDL wrote the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained. The present
study was approved by the ethics committees of Central South
University and the People's Hospital of Henan Province.
Consent for publication
Patients provided written informed consent for the
use of data in this study.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Jbour AK, Mubaidin AF, Till M, El-Shanti
H, Hadidi A and Ajlouni KM: Hypogonadotrophic hypogonadism, short
stature, cerebellar ataxia, rod-cone retinal dystrophy, and
hypersegmented neutrophils: A novel disorder or a new variant of
Boucher-Neuhauser syndrome? J Med Genet. 40:e22003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kate MP, Kesavadas C, Nair M, Krishnan S,
Soman M and Singh A: Late-onset Boucher-Neuhäuser Syndrome (late
BNS) associated with white-matter changes: A report of two cases
and review of literature. J Neurol Neurosurg Psychiatry.
82:888–891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lima-Martinez MM, Gil V, Zerpa J, Rivas P,
Gomez-Perez R and Osuna J: Boucher-Neuhäuser syndrome. Endocrinol
Nutr. 60:218–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ling H, Unnwongse K and Bhidayasiri R:
Complex movement disorders in a sporadic Boucher-Neuhäuser
Syndrome: Phenotypic manifestations beyond the triad. Mov Disord.
24:2304–2306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tarnutzer AA, Gerth-Kahlert C, Timmann D,
Chang DI, Harmuth F, Bauer P, Straumann D and Synofzik M:
Boucher-Neuhäuser syndrome: Cerebellar degeneration, chorioretinal
dystrophy and hypogonadotropic hypogonadism: Two novel cases and a
review of 40 cases from the literature. J Neurol. 262:194–202.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu SI, Kim JL, Lee SG, Kim HW and Kim SJ:
Ophthalmologic findings of Boucher-Neuhäuser syndrome. Korean J
Ophthalmol. 22:263–267. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Santos AV, Saraiva PF and Breia PN:
Significance of neuroimaging in the diagnosis of Boucher-Neuhauser
syndrome. Acta Med Port. 16:193–195. 2003.(In Portuguese).
PubMed/NCBI
|
|
8
|
Makita Y: Ataxia-hypogonadism syndrome
(Boucher-Neuhäuser syndrome). Ryoikibetsu Shokogun Shirizu.
238–239. 2001.(In Japanese). PubMed/NCBI
|
|
9
|
Deik A, Johannes B, Rucker JC, Sánchez E,
Brodie SE, Deegan E, Landy K, Kajiwara Y, Scelsa S,
Saunders-Pullman R and Paisán-Ruiz C: Compound heterozygous PNPLA6
mutations cause Boucher-Neuhäuser syndrome with late-onset ataxia.
J Neurol. 261:2411–2423. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koh K, Kobayashi F, Miwa M, Shindo K,
Isozaki E, Ishiura H, Tsuji S and Takiyama Y: Novel mutations in
the PNPLA6 gene in Boucher-Neuhäuser syndrome. J Hum Genet.
60:217–220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matsuzaka Y, Ohkubo T, Kikuti YY, Mizutani
A, Tsuda M, Aoyama Y, Kakuta K, Oka A, Inoko H, Sakabe K, et al:
Association of sick building syndrome with neuropathy target
esterase (NTE) activity in Japanese. Environ Toxicol. 29:1217–1226.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Synofzik M, Gonzalez MA, Lourenco CM,
Coutelier M, Haack TB, Rebelo A, Hannequin D, Strom TM, Prokisch H,
Kernstock C, et al: PNPLA6 mutations cause Boucher-Neuhauser and
Gordon Holmes syndromes as part of a broad neurodegenerative
spectrum. Brain. 137:69–77. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Topaloglu AK, Lomniczi A, Kretzschmar D,
Dissen GA, Kotan LD, McArdle CA, Koc AF, Hamel BC, Guclu M, Papatya
ED, et al: Loss-of-function mutations in PNPLA6 encoding neuropathy
target esterase underlie pubertal failure and neurological deficits
in Gordon Holmes syndrome. J Clin Endocrinol Metab. 99:E2067–E2075.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Richardson RJ, Hein ND, Wijeyesakere SJ,
Fink JK and Makhaeva GF: Neuropathy target esterase (NTE): Overview
and future. Chem Biol Interact. 203:238–244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sogorb MA, Pamies D, Estevan C, Estévez J
and Vilanova E: Roles of NTE protein and encoding gene in
development and neurodevelopmental toxicity. Chem Biol Interact.
259:352–357. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Synofzik M, Hufnagel RB and Züchner S:
PNPLA6-related disordersGeneReviews® [Internet]. Pagon
RA, Adam MP, Ardinger HH, et al: University of Washington; Seattle,
WA: pp. 1993–2018
|
|
17
|
Wiethoff S, Bettencourt C, Paudel R, Madon
P, Liu YT, Hersheson J, Wadia N, Desai J and Houlden H: Pure
cerebellar ataxia with homozygous mutations in the PNPLA6 gene.
Cerebellum. 16:262–267. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Braslavsky D, Grinspon RP, Ballerini MG,
Bedecarrás P, Loreti N, Bastida G, Ropelato MG, Keselman A, Campo
S, Rey RA and Bergadá I: Hypogonadotropic hypogonadism in infants
with congenital hypopituitarism: A challenge to diagnose at an
early stage. Horm Res Paediatr. 84:289–297. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dwyer AA, Jayasena CN and Quinton R:
Congenital hypogonadotropic hypogonadism: Implications of absent
mini-puberty. Minerva Endocrinol. 41:188–195. 2016.PubMed/NCBI
|
|
20
|
Kim SH: Congenital hypogonadotropic
hypogonadism and kallmann syndrome: Past, present, and future.
Endocrinol Metab (Seoul). 30:456–466. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Manara R, Salvalaggio A, Favaro A, Palumbo
V, Citton V, Elefante A, Brunetti A, Di Salle F, Bonanni G and
Sinisi AA: Kallmann Syndrome Neuroradiological Study Group: Brain
changes in Kallmann syndrome. AJNR Am J Neuroradiol. 35:1700–1706.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Uchino A, Saito N, Mizukoshi W and Okada
Y: Anomalous origin of the occipital artery diagnosed by magnetic
resonance angiography. Neuroradiology. 53:853–857. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
1000 Genomes Project Consortium, . Auton
A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini
JL, McCarthy S, McVean GA and Abecasis GR: A global reference for
human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kircher M, Witten DM, Jain P, O'Roak BJ,
Cooper GM and Shendure J: A general framework for estimating the
relative pathogenicity of human genetic variants. Nat Genet.
46:310–315. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang J, Yan R, Roy A, Xu D, Poisson J and
Zhang Y: The I-TASSER suite: Protein structure and function
prediction. Nat Methods. 12:7–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hufnagel RB, Arno G, Hein ND, Hersheson J,
Prasad M, Anderson Y, Krueger LA, Gregory LC, Stoetzel C, Jaworek
TJ, et al: Neuropathy target esterase impairments cause
Oliver-McFarlane and Laurence-Moon syndromes. J Med Genet.
52:85–94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kmoch S, Majewski J, Ramamurthy V, Cao S,
Fahiminiya S, Ren H, MacDonald IM, Lopez I, Sun V, Keser V, et al:
Mutations in PNPLA6 are linked to photoreceptor degeneration and
various forms of childhood blindness. Nat Commun. 6:56142015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rainier S, Bui M, Mark E, Thomas D, Tokarz
D, Ming L, Delaney C, Richardson RJ, Albers JW, Matsunami N, et al:
Neuropathy target esterase gene mutations cause motor neuron
disease. Am J Hum Genet. 82:780–785. 2008. View Article : Google Scholar : PubMed/NCBI
|