Introduction
Gynecological malignancies, particularly ovarian
cancer, cervical cancer and endometrial cancer, are serious medical
conditions in women and have been leading causes of cancer
mortality in recent years. However, the use of cancer markers for
early and progressive detection remain lacking (1). In addition, research has demonstrated
that there are close associations across the three aforementioned
types of cancer. It has been demonstrated that the progress and the
development of the three aforementioned types of cancer are
similar, which may be useful when diagnosing any one of these three
cancer types. In the case of endometrial cancer, prior to the
development of endometrial carcinoma, the endometrium undergoes
progressive neoplastic alterations in a parallel fashion to the
premalignant alterations observed in the cervix prior to the
development of cervical carcinoma (2). The rationale of oophorectomy in
surgical management is that endometrial cancer may metastasize to
the ovary, in which women with endometrial cancer are at risk for
synchronous and metachronous ovarian cancer, and the source of
estrogen may be eliminated by oophorectomy (3,4). In
cancer cells, oncogenic transformation is associated with major
alterations in gene expression (5). With the advent of large-scale
screening of cancer genomes, hundreds of genes with alterations in
different types of tumors from patients with cancer have been
identified (6–10), which revealed that cancer is a
complex disease caused by genetic alterations in multiple genes
(11,12). In order to elucidate the cancer
marker genes and biological processes associated with each type of
gynecological tumor, and the potential underlying mechanism of
associations among gynecological tumors, the contribution of
identified differentially expressed genes (DEGs) to the
pathogenesis of gynecological tumors must be understood.
To analyze different DEGs, high-throughput
experimental methods, including microarray analysis, have been
widely used in a number of studies (13,14).
A vast quantity of microarray data has been produced and deposited
in publicly-available data repositories, including the Gene
Expression Omnibus (GEO) (15).
With the methods of integrated bioinformatics analysis, researchers
have been able to advance the identification of genetic signatures.
This may provide insights into the underlying biological mechanisms
of the development of gynecological tumors.
Chung et al (16) revealed that microRNA (miRNA)-200b/a
is a direct transcriptional target of grainyhead like transcription
factor 2, which is associated with development and overall survival
in epithelial ovarian cancer. Halabi et al (17) demonstrated that 41 genes, including
matrix metalloproteinase (MMP)7 and tumor protein 53, were involved
in the potential underlying mechanisms of ovarian cancer. Espinosa
et al (18) revealed that
six genes encoding cyclin B2, cell-division cycle protein 20,
protein regulator of cytokinesis 1, synaptonemal complex protein 2,
nucleolar and spindle associated protein 1 and cyclin-dependent
kinase inhibitor 2 belonging to the mitosis pathway, were potential
markers for screening or therapeutic targets of cervical cancer.
However, biomarkers which were identified in this way have had poor
translation into actual clinical practices. Results have been
non-concordant among studies due to small sample sizes. In
addition, the studies into the associations of biomarker genes
(driver genes) remain lacking among the different types of
gynecological tumors.
A robust driver gene biomarker signature may be
beneficial for the diagnosis and targeted treatment of
gynecological tumors. In the present study, in order to identify a
driver gene biomarker signature for the three types of
gynecological tumors, data from the Metabolic Gene Rapid Visualizer
database (MERAV, which is derived from GEO) was used (19). In MERAV, microarrays were
normalized together to eliminate systematic errors caused by
different batch experiments.
The present study devised a target network for
ovarian cancer, cervical cancer and endometrial cancer using the
selected driver genes, and further investigated the identified DEGs
via functional enrichment analysis, pathway enrichment analysis and
protein-protein interaction (PPI) networks. In addition, the
present study extracted clinical information of ovarian cancer,
cervical cancer and endometrial cancer from The Cancer Genome Atlas
(TCGA) data portal. Subsequently, driver genes in each type of
cancer were analyzed. It was important to investigate the
underlying mechanism of each gynecological tumor and whether the
identified driver genes contributed to these diseases.
Subsequently, a network was generated between the miRNAs and the
identified driver genes, using the method of mining the Mir2
disease and Tarbase databases which provide information on miRNAs,
diseases and the interactions between miRNAs and genes. Finally,
the present study determined hub-genes and hub-miRNAs across the
gynecological tumors to study the potential underlying mechanisms
of the developments of gynecological tumors, which may shed light
on different strategies for the design of biological targets for
cancer therapies.
Materials and methods
Identification of gene expression
datasets
In the present study, DEGs were identified between
normal tissues and tumors extracted from the MERAV database from
the National Center for Biotechnology Information GEO database
(MERAV, http://merav.wi.mit.edu). The
experimental samples for the present study are presented in
Tables I and II. The following information was
extracted from each identified study: GEO accession number, sample
type, number of cases and controls, and gene expression data.
Studies in which the microarray data were uncertain were excluded.
The experimental protocol for the present study is presented in
Fig. 1.
 | Table I.Datasets from the Metabolic Gene
Rapid Visualizer database (cervix). |
Table I.
Datasets from the Metabolic Gene
Rapid Visualizer database (cervix).
| Tissue type | Datasets |
|---|
| Normal, n=4 | GSM176135,
GSM175833, GSM176130, GSM176140 |
| Tumor |
|
|
Squamous cell carcinoma,
n=5 | GSM152635,
GSM277702, GSM46919, GSM102527, GSM152587 |
|
Squamous cell carcinoma
non-keratinizing, n=5 | GSM179907,
GSM46942, GSM76614, GSM152580, GSM203742 |
|
Squamous cell carcinoma
keratinizing, n=3 | GSM117576,
GSM152723, GSM152751 |
|
Adenoma, n=6 | GSM179956,
GSM152667, GSM152719, GSM179853, GSM325835, GSM203622 |
| Table II.Datasets from the Metabolic Gene
Rapid Visualizer database (ovary and endometrium). |
Table II.
Datasets from the Metabolic Gene
Rapid Visualizer database (ovary and endometrium).
|
| Datasets |
|---|
|
|
|
|---|
| Tissue type | Ovary, n=4 | Endometrium,
n=22 |
|---|
| Normal tissues | GSM175789 | GSM175777,
GSM175778, |
|
| GSM176131 | GSM175779,
GSM175780, |
|
| GSM176136 | GSM175781,
GSM175783, |
|
| GSM176318 | GSM175784,
GSM175785, |
|
|
| GSM176039,
GSM176040, |
|
|
| GSM176041,
GSM176043, |
|
|
| GSM176093,
GSM176099, |
|
|
| GSM176127,
GSM176137, |
|
|
| GSM176141,
GSM176142, |
|
|
| GSM176144,
GSM176146, |
|
|
| GSM176143,
GSM176145, |
|
| Tissue type | Ovary serous
adenocarcinoma, n=11 | Endometrioid
carcinoma, n=12 |
|
| Tumors | GSM8897, GSM203626,
GSM15267, | GSM102425,
GSM117582, |
|
| GSM102445,
GSM46831, GSM152577, | GSM117586,
GSM117590, |
|
| GSM88973,
GSM152581, GSM27769, | GSM88952,
GSM88966, |
|
| GSM277737,
GSM301703 | GSM102469,
GSM102492, |
|
|
| GSM53058,
GSM88978, |
|
|
| GSM46923,
GSM46937 |
Integrated analysis of DEGs identified
in the extracted databases
Information was extracted from the microarray
datasets in MERAV which are presented in Tables I and II, respectively. Following the
intersection of the microarray datasets, the DEGs were established
between the normal and cancer tissues. In the present study, the
degree of differential gene expression was measured by fold-change
based on the Student's t-test. A fold-change value >2 or <0.5
and t-test P<0.01 for a gene was considered to be significant.
The differential expression analysis was conducted using the Linear
Models for Microarray Data package in R (20).
Protein interaction network
The DEGs were subsequently applied to the Human
Protein Reference Database (21)
(HPRD, www.hprd.org), to identify the more
complex functional interactive driver genes of separate cancer
types. Genes with interactions with each other were extracted from
the DEGs as mentioned above (presented in Tables III–X). The PPI network is a useful research
tool for investigating the cellular networks of protein
interactions, and was downloaded from the HPRD. Cancer-associated
gene-gene interaction networks were constructed by mapping the DEGs
into the HPRD PPI network for each cancer (cervix tumor, ovarian
tumor and endometrium tumor). To make it easier to identify the
driver genes, the present study calculated the lines attached to
each node, which was defined as the degree of the node. The nodes
that exhibited degrees ≥4 were defined as driver genes. The nodes
whose degree was ≥4 were considered to serve more complex roles in
the development of the diseases of interest. These nodes were then
extracted for the PPI network (Fig.
2). The present study constructed a connected network which
contained the driver genes across the three cancer types. Through
this method, it was determined whether the driver genes of the
separate cancer types had any interaction with each other. The
networks were constructed using Cytoscape version 3.3.0 (www.cytoscape.org).
| Table III.Driver genes identified by integrated
analysis of the microarray datasets (cervical squamous cell
carcinoma). |
Table III.
Driver genes identified by integrated
analysis of the microarray datasets (cervical squamous cell
carcinoma).
| Gene |
|---|
| RB1 | HTRA1 | MTOR | CLDN5 | NARF | PURA |
| MCM7 | KPNA2 | PLSCR4 | CYBA | NCAPD2 | RBM8A |
| MCM2 | LMNB1 | PRKD1 | DCUN1D1 | NCF4 | RECK |
| PLK1 | MEIS1 | PSMA5 | DDAH2 | NME4 | REV3L |
| AR | NCOA1 | PSMB10 | DMPK | NPLOC4 | RFC3 |
| PPP1CA | PBX1 | PSMB9 | EPS8 | NR2F1 | RNF126 |
| ABL1 | PIAS3 | PSMD2 | EXOSC5 | NR2F2 | RPA3 |
| LMNA | POLA2 | RACGAP1 | GABBR1 | NRAS | RRM1 |
| PTN | PPP1R14A | RTN3 | GAS6 | NTF3 | RRM2 |
| TRIP13 | AXL | SNRPB | GCH1 | NTRK2 | SAT2 |
| CAV1 | BUB1B | TOR1AIP1 | GCHFR | NUB1 | SDC2 |
| CDC20 | CCL14 | TUBA4A | GLRX3 | NUP210 | SEC24A |
| CDC6 | CCR5 | UBTF | GMFB | NUP50 | SELENBP1 |
| FLNA | COL4A5 | USP6NL | GOLGA2 | PAFAH1B3 | SERBP1 |
| FXR2 | CSNK1D | UTP3 | HOXD13 | PAK2 | SH3BP5 |
| ZHX1 | DBF4 | ACTN4 | ILK | PAM | SMC4 |
| CCNA2 | DVL3 | ADAM10 | KANK1 | PCGF2 | SNRPD1 |
| DGKZ | EFEMP2 | ANTXR2 | LAPTM5 | PHACTR4 | SNTB2 |
| MCM10 | EIF4EBP1 | ARHGAP17 | LDB2 | PLK2 | SNX27 |
| MCM6 | EZH2 | ASPM | LDOC1 | PNO1 | SPIN1 |
| PCNA | FAM46A | BID | LMO4 | PNP | SSSCA1 |
| RBPMS | HOXD10 | BMP4 | LRP1 | PPIA | STXBP2 |
| RPS6KA1 | HSPA4 | BNIP2 | LRP6 | PPIH | SUB1 |
| SAT1 | ITGB3BP | C1QA | LRRC41 | PRPF18 | TALDO1 |
| BUB1 | KLF6 | CBX4 | LZTS2 | PSMA6 | TGFBR3 |
| CSNK1E | MAD2L2 | CCNE1 | MAGEH1 | PSMB7 | TNFRSF1A |
| DCN | MAP2K4 | CCR1 | MELK | PSMD4 | UFD1L |
| FGFR1 | MAPK10 | CDC42BPA | MPDZ | PSME3 | WSB2 |
| FXYD1 | MCM5 | CENPE | MTA1 | PSMF1 | XPNPEP1 |
| GMNN | MITF | CHFR | MYCBP | PSTPIP1 | YLPM1 |
| HOXA10 | MMP9 | CIB1 | MYL9 | PTTG1 | ZMIZ1 |
| Table X.Driver genes identified by the
integrated analysis of the microarray datasets (endometrial
carcinoma). |
Table X.
Driver genes identified by the
integrated analysis of the microarray datasets (endometrial
carcinoma).
| Gene |
|---|
| EP300 | CDKN2A | F2R | AMFR | EPN3 | MMP11 |
| JUN | COL3A1 | FZD5 | AXL | EPR1 | MMP26 |
| CAV1 | EGR1 | HLA-DMB | BCL11A | FOSB | MYO5B |
| CTNNB1 | ERBB4 | HOXA10 | BCL2A1 | GALNT10 | NRG2 |
| ABL1 | FBLN1 | ID1 | BIK | GAS6 | NRXN2 |
| AR | FBN1 | ID4 | BLNK | GATA2 | PCOLCE |
| TCF4 | FLNA | IDE | C1R | GCH1 | PDGFRB |
| THBS1 | FOXO1 | INADL | C1S | GCHFR | PKD2 |
| TUBA4A | HLA-DRA | JUND | C3AR1 | GPI | PNP |
| ATXN1 | ID3 | LMO4 | CCND2 | GPRASP1 | PPP1R14A |
| COL1A1 | IGFBP5 | LNX1 | CDH11 | HLA-DQB1 | PRDM1 |
| DCN | LAMB3 | NCALD | CDKN1A | HLA-DRB1 | PSTPIP2 |
| LRP1 | MITF | NCF2 | CDKN2C | HLF | PTGDS |
| C3 | MYC | NR2F2 | CFB | HOXA9 | PTGS2 |
| COL7A1 | PLAT | PDGFRA | CGN | ID2 | R3HDM2 |
| FBLN2 | RUNX1T1 | PLEKHF2 | CLEC3B | IGFBP4 | RAB25 |
| FOS | S100A8 | PTPN13 | CLK1 | IGFBP6 | RAB3IP |
| GNAI2 | SERPINA1 | RAB8B | CXADR | IL33 | RAPGEF6 |
| IGF1 | SYK | RABAC1 | CXCL10 | IRS1 | S100A9 |
| LAMC2 | TGFB1I1 | ROR2 | DNM1 | KLF5 | SCRIB |
| MUC1 | CD14 | SFN | DPYSL2 | LAPTM5 | SEC24D |
| NID1 | COL5A1 | SFRP1 | ECM1 | LDB2 | SNTB2 |
| PRKD1 | CRMP1 | TFAP2A | EDNRA | LUC7L3 | SOX9 |
| PTPN12 | DBP | TJP2 | EFEMP2 | MAFB | SPINT1 |
| VCAN | DDR2 | TRPC1 | EFS | MAL2 | SPP1 |
| CD74 | F10 | WNT5A | ENO2 | MAPK10 | ST14 |
| SYTL1 | TJP3 | TLR3 | TRO | WASF2 | WNT4 |
| TBL1X | TLR2 | TPD52 | USP54 | WNT2 | ZEB1 |
miRNAs regulating gene network
construction
The present study analyzed the association between
miRNAs and the identified driver genes (Fig. 3). This process was performed by
extracting a list of miRNAs which were associated with the type of
cancer (cervical tumor, ovarian tumor or endometrial tumor) from
the Mir2 Disease database (www.mir2disease.org) (22). Following this step, a network was
created regarding the regulatory associations between the miRNA and
the specific driver gene of each type of cancer in order to
identify the hub-miRNAs of the gynecological tumors. The
associations of the regulation were extracted from Tarbase
(diana.cslab.ece.ntua.gr/tarbase) (23).
Functional and pathway enrichment
analysis
In order to assess the functional relevance of the
aforementioned DEGs, a pathway analysis was created based on the
Database for Annotation, Visualization and Integrated Discovery
(DAVID) (24). DAVID provides a
useful tool to analyze large gene lists, including gene ontology
(GO) and pathway analysis. DEGs in different diseases were applied
to this database in order to detect potentially represented
functions. GO-categories were organized based on the GO database
(25) (www.geneontology.org). In addition, pathway analysis
was based on the Kyoto Encyclopedia of Genes and Genomes (KEGG)
database (26) (genome.jp/kegg).
Significant categories were identified by expression analysis
systematic explorer scores, a modified Fisher's exact P-value. The
threshold for significance for a category was considered to be
P<0.01, with >4 genes for the corresponding term.
Survival analysis
The present study used TCGA database to extract
clinical information and gene expression profile information. At
the start of the analysis, the expression values of each driver
gene were listed, which were identified via the PPI network. To
find the median level of gene expression, the samples were divided
into two groups by median of expression (high expression group and
low expression group). Additionally, the corresponding clinical
information of each sample was extracted. Survival data
representing time between initial diagnosis and mortality were
downloaded directly from TCGA data portal (tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp)
(27). With this information, the
present study was able to estimate the association between the
identified driver genes of the three types of cancer mentioned
above and the survival rates of patients. All analyses were
conducted using custom-written code in R (www.r-project.org).
Results
Integrated analysis of multiple
studies to establish the driver genes in cancer
There are multiple genes that contribute to the
cause of the aforementioned cancer types and, therefore, no single
gene is a determining factor in diagnosis. It was identified that
each type of cancer was driven by different variations of genes
that serve key roles during the development of pathology. However,
no single gene may explain the heterogeneity of each type of
cancer. In the case of cervical cancer, 186 genes in squamous cell
carcinoma of the cervix (Table
III), 107 genes in keratinized squamous cell carcinoma of the
cervix (Table IV), 96 genes in
cervical adenocarcinoma Grade 3 (Table
V), 133 genes in non-keratinized squamous cell carcinoma of the
cervix (Table VI) and 203 genes
in cervical adenocarcinoma Grade 2 (Table VII) were identified to be
important. In addition, 120 genes and 76 genes were established,
respectively, in adenocarcinoma of the ovary Grade 2 and Grade 3
(Tables VIII and IX). A total of 168 genes were
established in endometrial carcinoma (Table X).
| Table IV.Driver genes identified by integrated
analysis of the microarray datasets (cervical keratinized squamous
cell carcinoma). |
Table IV.
Driver genes identified by integrated
analysis of the microarray datasets (cervical keratinized squamous
cell carcinoma).
| Gene |
|---|
| FYN | ADAM10 | ARHGAP17 | HSPB2 | PHF1 | TMOD1 |
| ZHX1 | ADAM17 | ARMCX2 | ID4 | PIK3C2B | TMSB10 |
| ABL1 | ANXA6 | BIN1 | LDB2 | PIP5K1C | TPD52 |
| BCL2L1 | AXL | CBX3 | LDOC1 | PNP | UBTF |
| FXR2 | BCL11A | CLDN5 | LMO4 | PSME3 | ZHX2 |
| TBP | CSNK1E | CNN3 | LRP1 | PSMF1 | ZMIZ1 |
| AR | DMPK | CNNM3 | LRP6 | PTOV1 | ZNF76 |
| BARD1 | ITGB3BP | CNTNAP1 | LSM5 | PTPN12 |
|
| BID | KPNA6 | CRYAB | MAGI2 | RAE1 |
|
| DDX24 | MAD2L2 | CSE1L | MAPK10 | REV3L |
|
| NCOA1 | MCL1 | CSTF1 | MIS12 | RUNX1T1 |
|
| PDGFRB | NR2F1 | EFEMP2 | MPDZ | SDC2 |
|
| PRKD1 | NTRK2 | EXOSC5 | MTA1 | SFRP1 |
|
| PSEN1 | PPP1R14A | FGFR1 | MYCBP | SH3BP5 |
|
| RBPMS | PTN | FXYD1 | NPDC1 | TAF9 |
|
| SPTAN1 | RTN3 | FZD6 | NR2F2 | TCF7L2 |
|
| TCF4 | SYK | GAS6 | NTF3 | TERF1 |
|
| TGFA | VIM | GDI1 | NUDT21 | TFDP1 |
|
| A2M | ANTXR2 | GTF3C3 | PBX1 | TGFBR3 |
|
| ACP1 | AQP1 | HOXA10 | PDGFD | TLN2 |
|
| Table V.Driver genes identified by integrated
analysis of the microarray datasets (cervical adenocarcinoma
G3). |
Table V.
Driver genes identified by integrated
analysis of the microarray datasets (cervical adenocarcinoma
G3).
| Gene |
|---|
| AR | BAD | PLD2 | CIB1 | MAPK10 | SERPINA1 |
|---|
| CAV1 | BAHD1 | PPA1 | CLDN5 | MED14 | SF1 |
| FLNA | C1QBP | PRKD1 | CUL4B | MPDZ | SMO |
| PPP1CA | CPSF6 | SAT1 | DMPK | MYL9 | SPINT2 |
| NCK2 | CSNK1D | SMAD1 | EFNB1 | NR2F1 | SSBP3 |
| PLSCR1 | DOCK1 | SNAP23 | F3 | NTF3 | SSR1 |
| SUMO4 | DVL2 | TAF1D | GDI1 | PCGF2 | STAM |
| LMNA | FXR2 | TAF9 | HOXA10 | PDPK1 | SYNE1 |
| LRP1 | FXYD1 | TCF4 | HOXD10 | PHACTR4 | TCF7L2 |
| PSEN1 | ILK | WIPI1 | HOXD13 | PHYHIP | TGFBR3 |
| PTN | LDB1 | ACVR2A | HSPA1B | PLSCR4 | TMF1 |
| CSNK1E | LMO4 | ANTXR2 | HSPBAP1 | PNPLA2 | UBTF |
| DVL3 | MAP2K4 | ATG12 | KANK1 | PPP1R10 | VAMP8 |
| MMP14 | NCOA1 | CD82 | KPNA6 | PTCH2 | WASF1 |
| PPP1R14A | NTRK2 | CDC42BPA | LDB2 | RNF138 | WASF2 |
| ALDOA | PBX1 | CDC42EP1 | LRP6 | RUNX1T1 | ZHX1 |
| Table VI.Driver genes identified by integrated
analysis of the microarray datasets (cervical non-keratinized
squamous cell carcinoma). |
Table VI.
Driver genes identified by integrated
analysis of the microarray datasets (cervical non-keratinized
squamous cell carcinoma).
| Gene |
|---|
| AR | FXR2 | FOXO1 | TLR2 | FBN2 | NTF3 |
| ABL1 | ILK | GMNN | TXNDC9 | FGR | NTRK2 |
| CAV1 | LMNA | HOXD10 | XRCC4 | FXYD1 | NUBP1 |
| CHD3 | MEIS1 | ICAM3 | YAP1 | GDI1 | PALLD |
| HIF1A | NCOA1 | ITGB2 | ADCY6 | HCLS1 | PDPK1 |
| PTPN6 | PAG1 | LCP2 | ADI1 | HLA-DMB | PGK1 |
| SAT1 | PBX1 | LRP1 | AGTPBP1 | HLA-DRA | PGLS |
| FLNA | PIAS1 | MAFG | ANTXR2 | HOXD13 | PIK3R3 |
| HOXA10 | PSEN1 | MPDZ | ANXA6 | HSPB2 | PLTP |
| PLSCR1 | PTN | NDN | ARHGDIB | LCP1 | PNP |
| RAF1 | WASF2 | NR2F2 | CDC37 | LDOC1 | PRRX1 |
| DCN | ZHX1 | PAICS | CITED2 | LILRB2 | RAB11FIP2 |
| EZR | ACTR3 | PLSCR4 | CLDN5 | LRP6 | RAB18 |
| MMP14 | BIN1 | PPP1R14A | CNN3 | MAPK10 | RFXANK |
| PDGFRB | C1QB | PPP2R1A | COL4A5 | MED14 | RUNX1T1 |
| ABCA1 | C1QC | PRDX2 | DOCK1 | MTA1 | SAT2 |
| C1QA | CSNK1D | SNTB2 | DVL2 | MYO5B | SEPHS1 |
| CSNK1E | DGKZ | SSSCA1 | ENO1 | NARF | SF1 |
| DMPK | DVL3 | TCF4 | FAM46A | NISCH |
|
| ELN | EFEMP2 | TLR1 | FBLN1 | TICAM1 |
|
| SNX2 | SYNE1 | TCF7L2 | VTA1 | TRAP1 |
|
| TMEM8B | TMOD1 | TMSB10 | TPD52 | SH3BP5 |
|
| WASF3 | ZNF76 | TEAD3 | TIMP2 | NR2F1 |
|
| Table VII.Driver genes identified by integrated
analysis of the microarray datasets (cervical adenocarcinoma
G2). |
Table VII.
Driver genes identified by integrated
analysis of the microarray datasets (cervical adenocarcinoma
G2).
| Gene |
|---|
| ABL1 | HSPA5 | ASAP1 | PSMF1 | ASS1 | EHD2 |
| AR | HTRA1 | AXL | QKI | ATRX | ENAH |
| CAV1 | LMNA | BCR | RAB4A | AURKA | ENO1 |
| PPP1CA | MEIS1 | BGN | RNF138 | AURKB | ERBB3 |
| FLNA | NTRK2 | BMP4 | SDC2 | BIN1 | FBLN1 |
| FYN | PRNP | BRCA2 | SMARCE1 | BIRC5 | GAS6 |
| MMP2 | PTPN12 | CDKN2A | SNAP29 | CAPZB | GLRX3 |
| SMAD1 | SMAD5 | CSNK1E | TAF7 | CAV2 | GOLGA2 |
| NCK2 | TAF9 | DMPK | TCF4 | CBX4 | GTF2I |
| RB1 | TTF2 | DOCK1 | TGFBR3 | CD81 | HAT1 |
| PTN | DVL2 | DR1 | THBS2 | CDT1 | HOXD10 |
| PTPN6 | EFEMP2 | FGFR1 | TIFA | CEP76 | HSPA1B |
| SMAD7 | FXR2 | FXYD1 | TIMP2 | CLDN5 | HSPB2 |
| SUMO4 | HOXA10 | GDF5 | TNFRSF1A | CLU | IDE |
| A2M | HOXD13 | GNA12 | ZHX1 | CNN3 | IFI35 |
| AP1M1 | LRP1 | KIDINS220 | ADI1 | CNTNAP1 | IFNAR1 |
| CDC5L | NCOA1 | LDOC1 | AHNAK | COL4A5 | ILK |
| EZR | NOTCH2 | LRP6 | ALDOA | COL6A3 | IQGAP1 |
| MMP14 | PBX1 | MAFG | ANTXR1 | COX5A | JAG1 |
| PIAS1 | PDGFRB | MAP2K4 | ANTXR2 | CUL4B | KANK1 |
| CD2AP | PRKD1 | MAPK10 | ANXA6 | CXCL12 | KDM2A |
| CDH1 | SAT1 | MEF2C | AQP1 | DCLRE1A | KPNA6 |
| DCN | WASF2 | POLE3 | ARHGAP17 | DDX24 | LCAT |
| DRAP1 | YAP1 | PPP1R14A | ARHGEF6 | EFNB1 | MAD2L1BP |
| ELN | ACVR2A | PRRX1 | ASH1L | EFS | MAP3K3 |
| MCM4 | NR2F2 | PLSCR4 | RUNX1T1 | SYNE1 | WNK1 |
| MED14 | NTF3 | PPA1 | SALL2 | TEAD3 | YLPM1 |
| MPDZ | NUDT21 | PPP1R10 | SAT2 | TERF1 | ZMIZ1 |
| MSN | PALB2 | PPP2R1A | SETD7 | THBS3 |
|
| MYCBP2 | PALLD | PSMB10 | SH3BP5 | TMEM8B |
|
| MYO5B | PBX3 | PURA | SH3KBP1 | TSPAN4 |
|
| NFE2L1 | PDGFD | RAB11FIP1 | SKAP1 | TWIST2 |
|
| NMI | PHACTR4 | RAB11FIP2 | SPARCL1 | UBTF |
|
| NPHS2 | PIP4K2B | RBPJ | STX3 | VGLL4 |
|
| NR2F1 | PKD2 | REPS2 | STX7 | WFDC2 |
|
| Table VIII.Driver genes identified by integrated
analysis of the microarray datasets (adenocarcinoma of the ovary
Grade 2). |
Table VIII.
Driver genes identified by integrated
analysis of the microarray datasets (adenocarcinoma of the ovary
Grade 2).
| Gene |
|---|
| JUN | MEF2C | HSPA1A | CNNM3 | GNE | PHF1 |
| FXR2 | NCOA2 | HTRA1 | COX5A | GNG4 | PKD2 |
| RAF1 | NIF3L1 | IKZF4 | CRY2 | GPRASP1 | PLA2G16 |
| RBPMS | PCBD1 | LIFR | CTF1 | HMGA1 | PLK1 |
| ZBTB16 | PDGFRA | MAPK10 | CTSD | HSPA2 | PTPN13 |
| PRKACA | PRTFDC1 | MYO15A | DCN | ICAM3 | RBBP8 |
| CAV1 | STAT5A | NFE2L1 | DST | IGFBP4 | RBP1 |
| MAP3K3 | APBB1 | NR2F6 | ELF3 | IRS1 | SDC2 |
| MAP3K5 | C1R | PER1 | ELK1 | KIAA1217 | SGK1 |
| NCOA1 | C1S | PTPN6 | ENAH | MAFG | SH3BP5 |
| PDGFRB | CALCOCO2 | SERPING1 | ENG | MRAS | SMC3 |
| SIN3A | CD2AP | SIN3B | EPS8 | NBL1 | SNCA |
| ABLIM1 | DCTN1 | TGFBR3 | ETV6 | NFATC4 | SNRNP70 |
| DDX17 | DMPK | TSC22D3 | EYA2 | NINL | SPOP |
| FEZ1 | DVL2 | UBQLN1 | FLAD1 | NR2F2 | SPTBN1 |
| GATA4 | FHL2 | ACTA2 | FOXO1 | OLFML3 | SPTBN2 |
| GOLGA2 | FLNA | BEGAIN | FOXO3 | PAICS | ST13 |
| LRP1 | FXYD1 | CCT5 | FTH1 | PDGFD | STRBP |
| TCF4 | THRA | TPM2 | TXN | USP13 | ZC3H10 |
| TEAD1 | TOP2A | TRIM21 | TXNDC9 | WTIP | ZFPM2 |
| Table IX.Driver genes identified by integrated
analysis of the microarray datasets (adenocarcinoma of the ovary
Grade 3). |
Table IX.
Driver genes identified by integrated
analysis of the microarray datasets (adenocarcinoma of the ovary
Grade 3).
| Gene |
|---|
| CDK1 | HLA-DRA | CD14 | FCGR2B | PDGFD | NR2F2 |
| AURKB | ICAM3 | CDC20 | FOS | SLPI |
|
| CAV1 | KRT7 | CDH1 | GCA | SMC4 |
|
| PTPN6 | MAD2L1 | CDKN2A | GNE | SOX9 |
|
| ZBTB16 | MAL2 | CEBPG | GPRASP1 | SPINT1 |
|
| BCL2L1 | MAP3K5 | CENPA | HLA-DMB | ST14 |
|
| HSPA1A | PDGFRA | CKS2 | HLA-DRB1 | STRBP |
|
| IRS1 | PDGFRB | CLDN1 | LAPTM5 | TACC1 |
|
| ITGB2 | PMAIP1 | CLDN3 | LCP1 | TOP2A |
|
| MCM2 | RACGAP1 | CRIP1 | LRP1 | TRIP13 |
|
| NDC80 | RBPMS | CTSS | MSLN | TYROBP |
|
| SYK | TPD52 | CXCR4 | MUC1 | ZWINT |
|
| TPD52L1 | ALOX5 | DBF4 | MUC16 | ECT2 |
|
| BCL11A | ALOX5AP | DSC2 | NCAPD2 | CCNB1 |
|
| CCNB2 | BIK | DSG2 | NR2F1 | ERBB3 |
|
Integrated PPI (protein-protein
interactions) network construction
Based on the HPRD, the interaction network of the
identified driver genes was constructed, which consisted of 101
nodes (genes that form associations) and 185 edges (biological
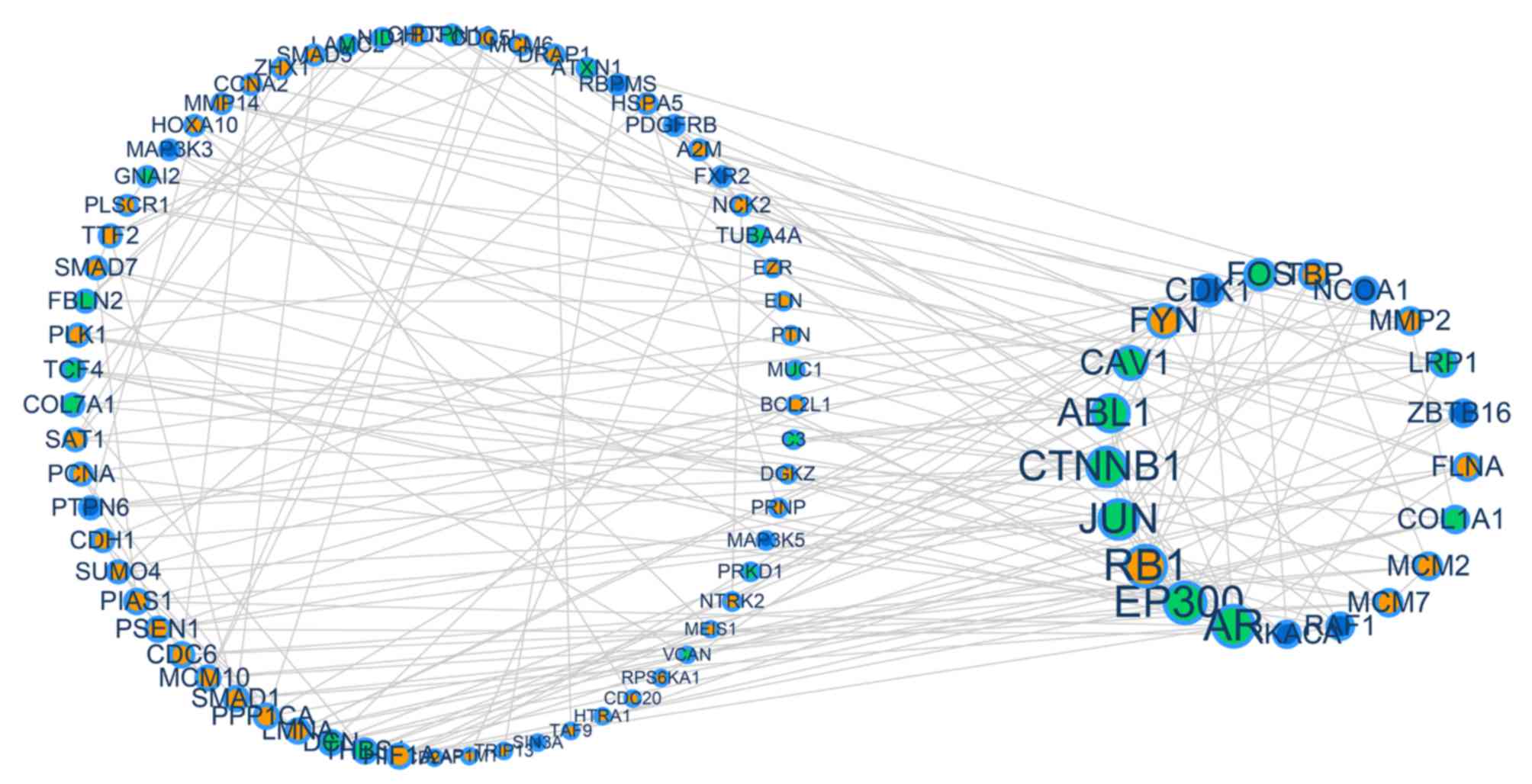
association) (Fig. 2). Genes with
a higher degree of association (degree ≥4) were observed to be
larger in size, and included the genes CDK1, CAV1, ZBTB16, Jun
proto-oncogene AP-1 transcription factor subunit (JUN), RAF1, RB1,
minichromosome maintenance complex component 2 (MCM2), AR, ABL1,
LMNA, FLNA, DCN, FYN, SMAD1, LRP1, PSEN1, EP300, CTNNB1, collagen
type I α1 chain (COL1A1) and FOS. Through this method, it was
identified that driver genes in each gynecological cancer have
contact interactions.
Comprehensive analysis of miRNA
regulation and the selected driver genes
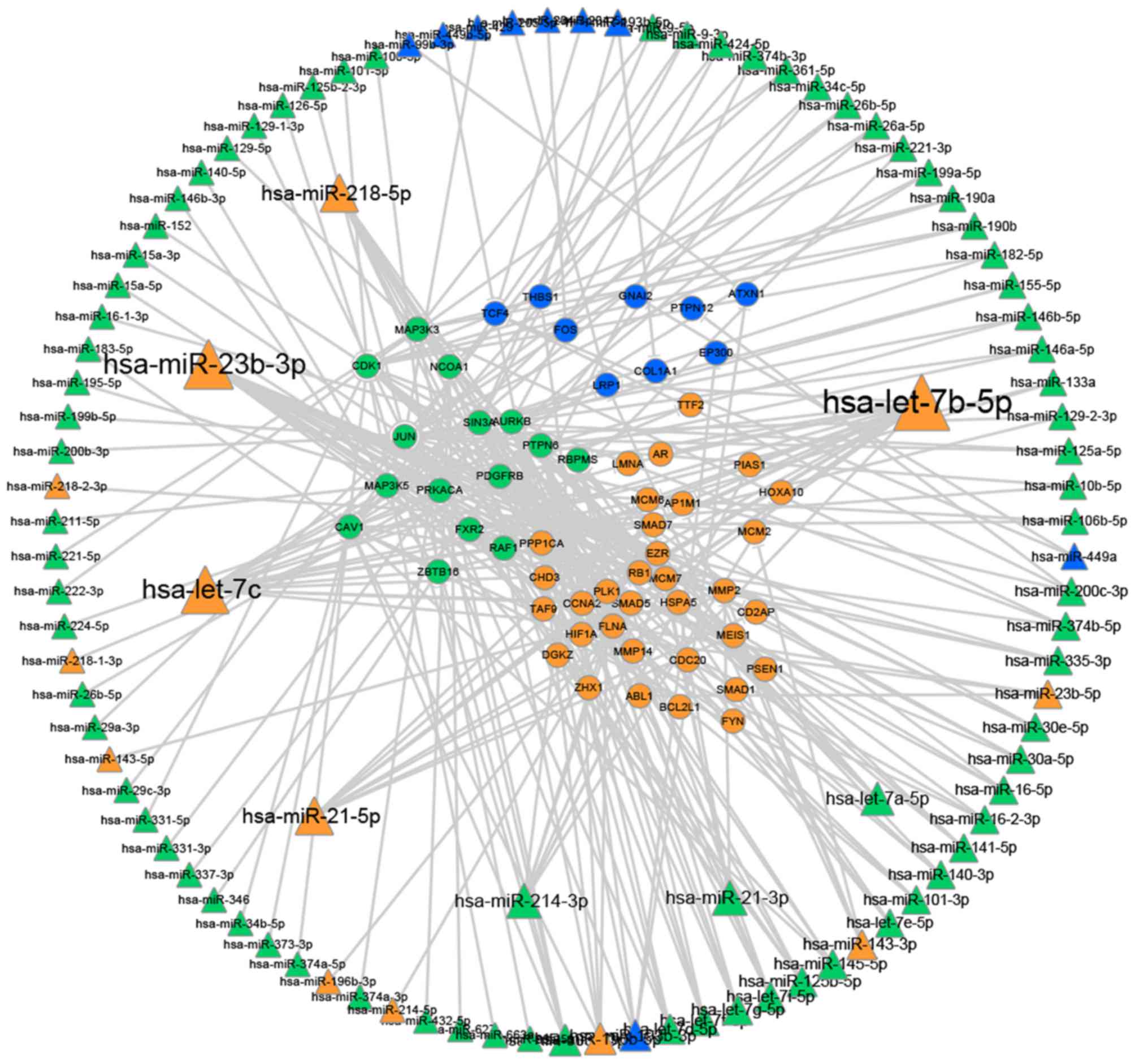
Fig. 3 illustrates
that certain miRNAs serve important roles in regulating the driver
genes. In the present study, it was demonstrated that a number of
miRNAs regulate separate networks [for example the let7 family,
miRNA (miR)-23b, miR-21, miR-214 and miR-218]. miRNAs that were
confirmed to be significant in cervical cancer, including let7c and
let7b, are also found to be associated with the other two cancers
in this study. This information may be important in establishing
the connections between the three gynecological cancer types, which
may be used in the development of targets for further research and
diagnosis.
Functional and pathway enrichment
analysis
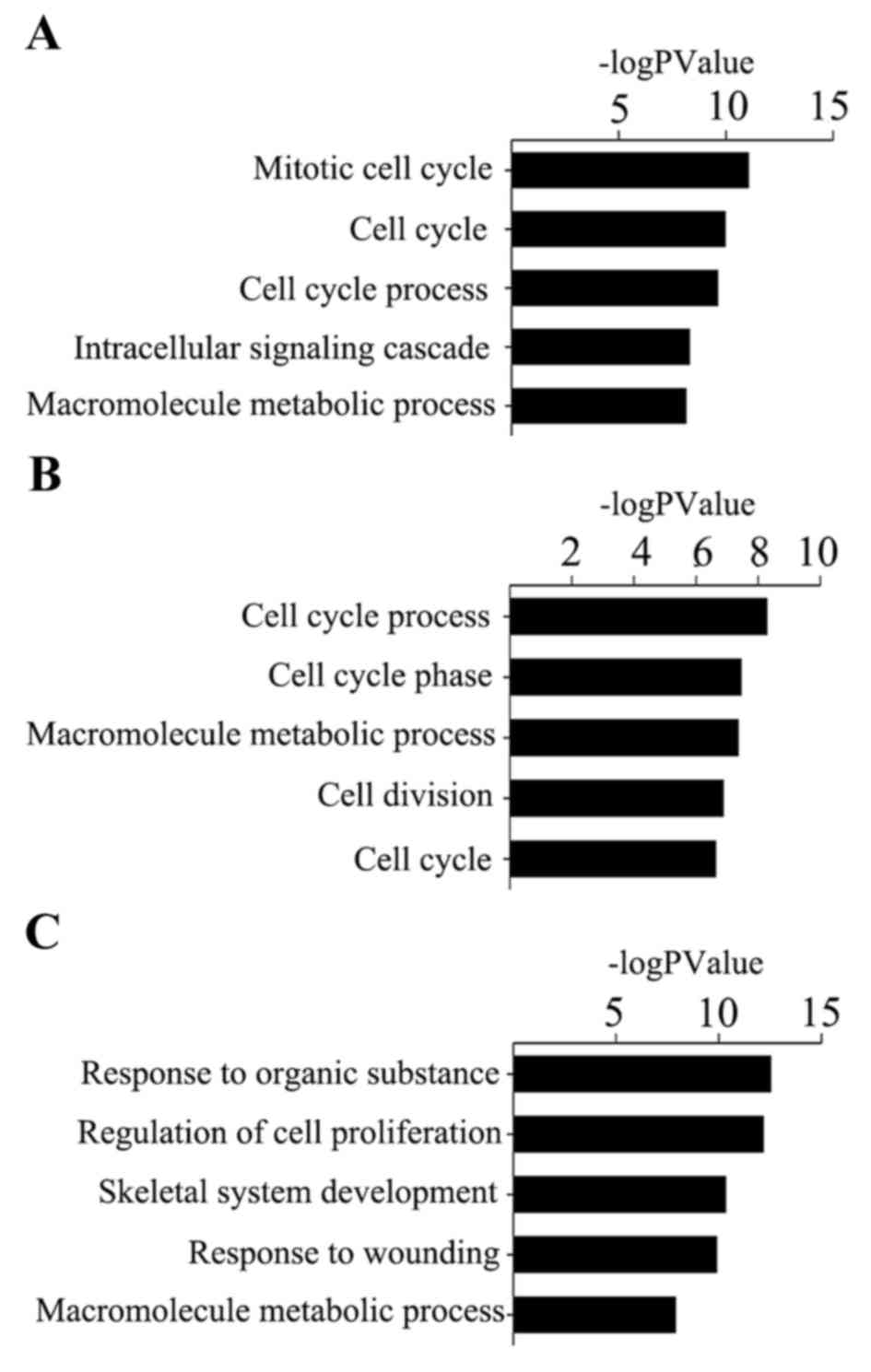
GO analysis revealed that the identified genes of
cervical tumors, ovarian tumors and endometrial tumors were
predominantly involved in the illustrated biological processes
(Fig. 4). The top three
significant biological processes of cervical cancer were ‘mitotic
cell cycle’, ‘cell cycle’ and ‘cell cycle process’, while for
ovarian cancer, the biological processes consisted of ‘cell cycle
process’, ‘cell cycle phase’ and ‘macromolecule metabolic process’.
For the progression of endometrial cancer, the top three biological
processes observed to be at fault for cancer progression were
‘response to organic substance’, ‘regulation of cell proliferation’
and ‘skeletal system development’.
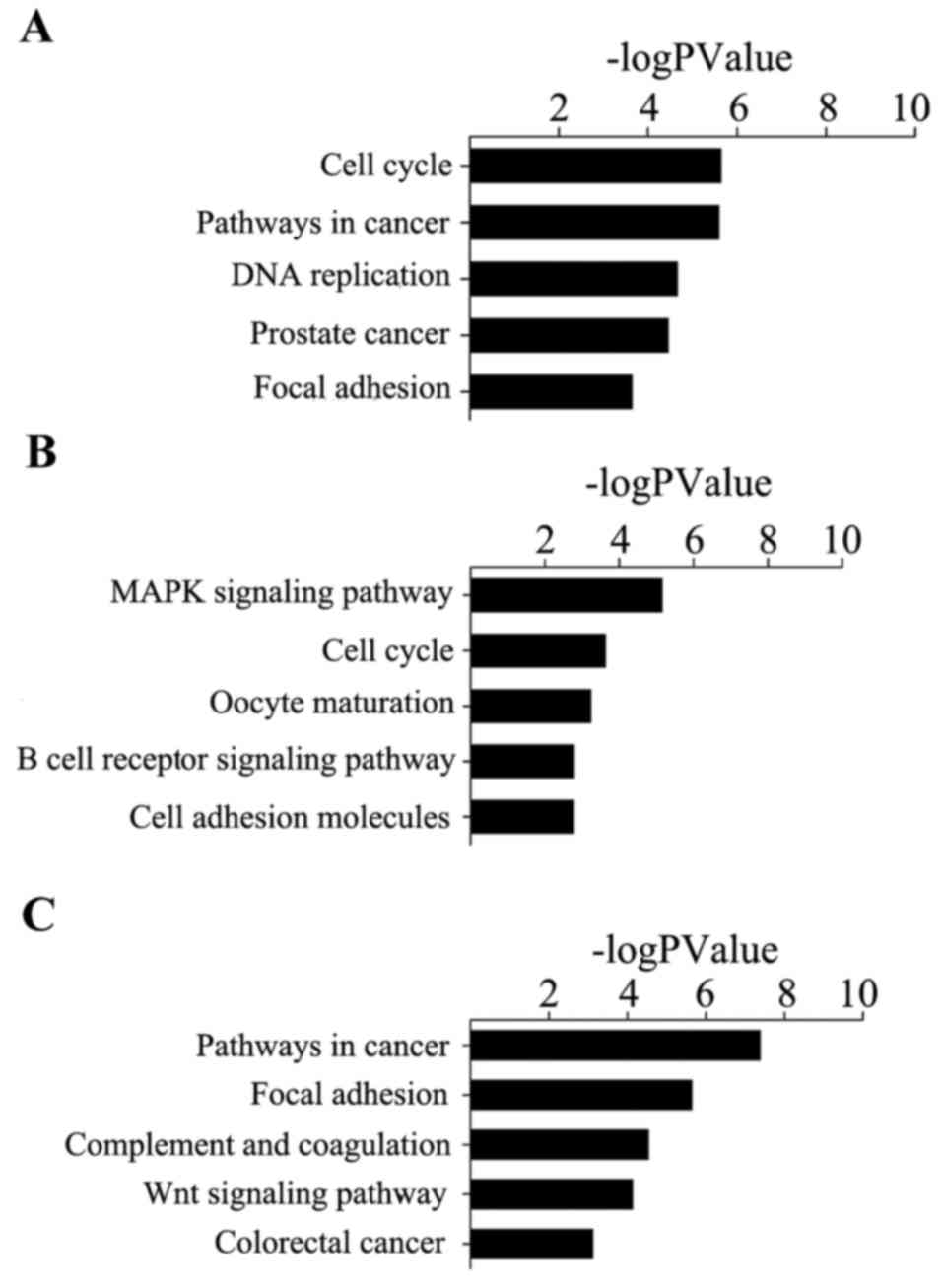
Using the method of pathway analysis, it was
revealed that genes in cervical cancer were significantly enriched
in ‘cell cycle’, ‘pathways in cancer’ and ‘DNA replication’.
Ovarian cancer was observed to be significantly enriched in ‘MAPK
signaling pathway’, ‘cell cycle’ and ‘oocyte maturation’.
Endometrial cancer was observed to be significantly enriched in
‘pathways in cancer’, ‘focal adhesion’ and ‘complement and
coagulation cascades’ (Fig.
5).
Survival analysis of patients with
gynecological tumor
Fig. 6 illustrates
the association between survival time and survival rate in the high
and low expression groups. The genes MCM2, MMP2, COL1A1 and JUN are
presented in the figure, and it was observed that the driver genes
of the expression groups were able to divide each of the target
cancer types into two groups, one of which contained the high
expression group with the other containing the low expression
group. Therefore, in order to determine whether the driver genes
had a key role in the development of gynecological tumors and the
connective function of separate cancer types, the present study
aimed to identify the association between the target cancer driver
genes and other types of gynecological cancer.
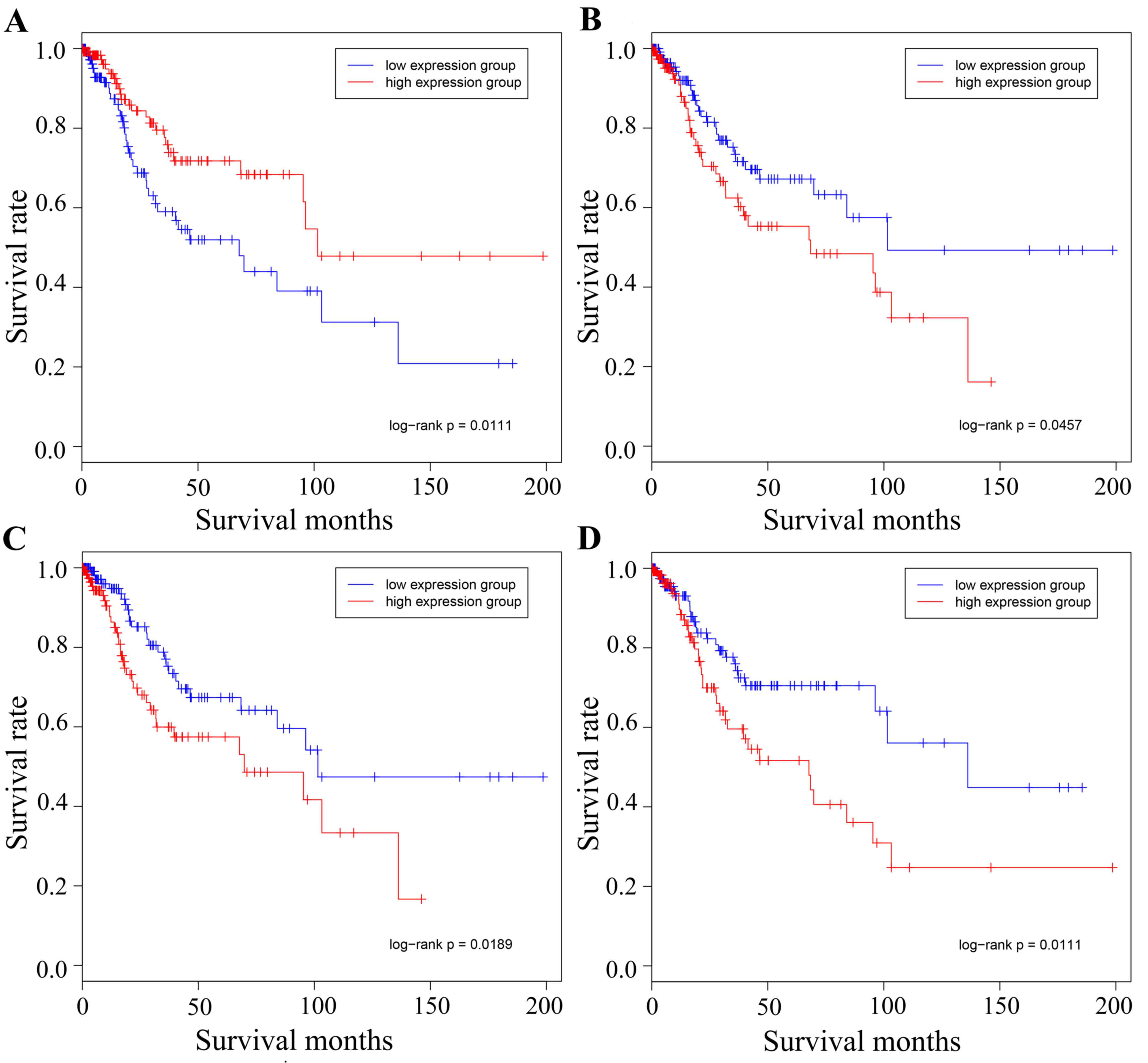 | Figure 6.Survival analysis of the different
cancer types using the representative driver genes. Survival data
representing time between initial diagnosis and mortality were
downloaded directly from TCGA data portal. The red line represents
the high expression group and the blue line represents the low
expression group. (A) Cervical hub-gene MCM2 in cervical cancer.
high and low expression of MCM2 divided the samples into two
groups, with 133 and 144 samples in each group, respectively. (B)
Cervical hub-gene MMP2 in cervical cancer, whose high and low
expression divided the group into two, with 142 and 142 samples in
each group, respectively. (C) Ovarian hub-gene COL1A1 in cervical
cancer, whose high and low expression divided the group into two,
with 143 and 141 samples in each group, respectively. (D) Ovarian
hub-gene JUN in cervical cancer, whose high and low expression
divided the group into two, with 141 and 144 samples in each group,
respectively. MCM2, minichromosome maintenance complex component 2;
MMP2, matrix metalloproteinase 2; COL1A1, collagen type I α1 chain;
TCGA, The Cancer Genome Atlas. |
Discussion
The principal challenge of high-throughput cancer
genomics is to identify specific driver genes and the underlying
mechanisms of carcinogenesis, apart from the vast quantity of
heterogeneous genomic alteration data. Numerous studies have
focused on identifying individual functional modules or pathways
involved in cancer (28–30). Based on this methodology, the
analysis of the present study focused specifically on DEGs in order
to reveal the transcriptional responses of gynecological tumors.
The results of this analysis suggested that the common biological
processes of cancer of the cervix, ovary and endometrium were those
involved in the cell cycle and the regulation of macromolecule
metabolism.
The cell cycle is the progression of biochemical and
morphological phases and events that occur in a cell during
successive cell replication or nuclear replication. Research has
shown that interference with cell cycle components may lead to
tumor formation (31). Certain
cell cycle inhibitors, including retinoblastoma protein and tumor
protein 53 may mutate during replication, causing the cell to
proliferate uncontrollably, ultimately resulting in a tumor.
Furthermore, the proportion of active cell division in tumors is
much higher compared with the rate in normal tissue.
To clarify the hub genes in ovarian cancer, cervical
cancer and endometrial cancer, DEGs were predicted to be biomarkers
for each cancer using PPI networks. It is considered that hub nodes
are genes that are highly connected with other genes and have been
predicted to serve key roles in numerous networks. In addition,
highly connected hub genes were proposed to have a considerable
role in biological development. Hub nodes have more complex
interactions compared with those of other nodes, which indicates
that they have pivotal roles in the underlying mechanisms of
disease. In addition, certain identified biomarkers of each type of
cancer were extracted from each network and these driver genes were
placed into one PPI network with the duplication hub genes
eliminated. Therefore, the particular hub genes of each
gynecological cancer and the connection nodes across the three
types of cancers may be identified. Accordingly, the identification
of hub genes and hub connected genes involved in each gynecological
cancer may lead to the discovery of the association across ovarian
cancer, cervical cancer and endometrial cancer, and may lead to the
development of effective diagnostic and therapeutic approaches.
In order to ascertain a causal association across
the three types of gynecological cancer, the present study
extracted clinical information and gene expression profile
information from TCGA database, and used the hub connected genes
identified in the PPI network to perform survival analysis. In the
present study, four noteworthy genes were identified, including
MCM2, MMP2, COL1A1 and JUN.
The present study demonstrated that MCM2 may serve a
key role in cervical cancer. A poor prognosis was associated with
lower expression. Furthermore, MCM2 was highly connected with
ovarian cancer and endometrial cancer. The results suggested that
MCM2 is a component of the DNA replication licensing complex, with
a rich binding surface that directs multiple regulatory
interactions of cancer significance, marking DNA replication
origins during the G1 phase of the cell cycle for use in the
subsequent S-phase. A deficiency of MCM2 results in death or
morbidity in the absence of an overt tumor (32). These processes of DNA replication
have been studied and used as therapeutic targets. Simon and
Schwacha (33) suggested that MCM2
was a promising target for blocking the proliferation of cancerous
and precancerous cells.
In the present study, MMP2 was identified to be
essential in causing cervical cancer. MMPs are zinc-containing
endopeptidases with an extensive range of substrate specificities.
These enzymes are able to degrade various components of
extracellular matrix (ECM) proteins. In photocarcinogenesis,
degradation of the ECM is the initial step towards tumor cell
invasion, to intrude in the basement membrane and the surrounding
stroma that primarily comprises fibrillary collagens. Additionally,
MMP2 is involved in angiogenesis, which promotes cancer cell growth
and migration (34).
COL1A1 and COL1A2 encode the α1 and α2 chains of
type I collagen, respectively (35). The primary constituents of the ECM
are collagens, adhesive glycoproteins and proteoglycans (36). Specific interactions between cells
and ECM-mediated cell-surface-associated components and
transmembrane molecules result in the control of cellular
activities, including adhesion and migration (37). Collagen is the primary component of
the ECM, which serves pivotal roles in maintaining skin and vessel
elasticity, and increasing cartilage lubricity (38). Upregulation of type II collagen
expression may contribute to ovarian cancer metastasis and
biological processes, including cell proliferation, invasion and
migration (39). The oncogene JUN
is the putative transforming gene of avian sarcoma virus 17, which
is the most extensively studied protein of the activator protein-1
complex and is involved in numerous cell activities, including
proliferation, apoptosis, survival, tumorigenesis and tissue
morphogenesis. The present study identified that COL1A1 was
important in ovarian cancer, which was highly connected with
cervical and endometrial cancer. Therefore, COL1A1 and JUN may be
potentially important associated genes of the three types of
gynecological malignancies.
miRNAs are small noncoding regulatory RNAs that
downregulate transcription by targeting specific mRNAs.
Furthermore, the present study identified that certain miRNAs were
highly associated with hub connected genes, including let7, which
is one of the founding members of the miRNA family. This miRNA was
first identified in Caenorhabditis elegans. Lee and Dutta
(40) identified six functional
let7 target sites in the 3′-untranslated region of high mobility
group AT-hook 2 (HMGA2), which reduced HMGA2 expression and cell
proliferation in a lung cancer cell line. Using genome-wide mRNA
expression analysis, Mi et al (41) identified that miRNA let7B was
downregulated in acute lymphoblastic leukemia (ALL) compared with
acute myeloid leukemia (AML). Quantitative polymerase chain
reaction analysis confirmed the downregulation of let7B in ALL
samples compared with AML samples and normal controls.
The present study identified that let7a, let7b and
let7c had strong connections with the hub genes and that these
miRNAs may serve an important part of the potential mechanism,
which may explain the connections across the hub genes.
Overall, the present study identified a number of
DEGs associated with gynecological cancer, in addition to the
functions and signaling pathways in which these genes were
involved. Comprehensive network analyses of the dysregulated gene
expression in gynecological cancers identified a series of hub
genes and the connection genes across ovarian cancer, cervical
cancer and endometrial cancer in a PPI network. Subsequently, this
study confirmed the driver genes by survival analysis using the
TCGA database. Comprehensive network analyses of miRNAs and
connection driver genes identified certain miRNAs which may be
potential therapeutic and prevention targets of gynecological
cancer. In addition, the present study demonstrated the
associations across the different gynecological cancers, which may
be useful for identifying potential useful diagnostic markers and
novel therapeutic targets. The results of this study may provide an
insight into the underlying mechanism of the aforementioned
gynecological cancers and may lead to further improvement in
diagnosis and treatment of them.
Acknowledgements
The authors would like to thank Professor Yunyan Gu
(College of Bioinformatics Science and Technology, Harbin Medical
University, Harbin, China.) for technical support and critically
reviewing the manuscript.
Funding
The present study was supported by grant no.
RC2013QN004112 from Harbin Science and Technology Innovation
Talents, China.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MY and JW conceived and designed the study; MY, LL
and JL performed the experiments and analyzed the data. MY wrote
the paper, and JW revised the manuscript and gave final approval of
the version to be published.
Ethics approval and consent to
participate
The present study was approved by the Clinical
Research Ethics Committee of the Affiliated Zhuzhou Hospital
Xiangya Medical College CSU (Zhuzhou, China), and written informed
consent was obtained from all participants.
Consent for publication
Written informed consent was obtained from all
volunteers for the publication of any associated data.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
miRNAs
|
microRNAs
|
|
GO
|
gene ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
PPI
|
Protein-protein Interaction
|
|
DEGs
|
differentially expressed genes
|
|
GEO
|
Gene Expression Omnibus
|
|
MERAV
|
Metabolic Gene Rapid Visualizer
Database
|
|
HPRD
|
Human Protein Reference Database
|
|
MMPs
|
matrix metalloproteinases
|
|
ECM
|
extracellular matrix
|
References
|
1
|
Li XY and Wang X: The role of human
cervical cancer oncogene in cancer progression. Int J Clin Exp Med.
8:8363–8368. 2015.PubMed/NCBI
|
|
2
|
Nisker JA: Screening for endometrial
cancer. Can Fam Physician. 29:961–965. 1983.PubMed/NCBI
|
|
3
|
Setiawan VW, Yang HP, Pike MC, McCann SE,
Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, et al:
Type I and II endometrial cancers: Have they different risk
factors? J Clin Oncol. 31:2607–2618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wright JD: Take 'em or leave 'em:
Management of the ovaries in young women with endometrial cancer.
Gynecol Oncol. 131:287–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lukk M, Kapushesky M, Nikkilä J, Parkinson
H, Goncalves A, Hube W, Ukkonen E and Brazma A: A global map of
human gene expression. Nat Biotechnol. 28:322–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network, .
McLendon R, Friedman A, Bigner D, Van Meir EG, Brat DJ,
Mastrogianakis GM, Olson JJ, Mikkelsen T, Lehman N, et al:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cancer Genome Atlas Research Network:
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu P, Morrison C, Wang L, Xiong D, Vedell
P, Cui P, Hua X, Ding F, Lu Y, James M, et al: Identification of
somatic mutations in non-small cell lung carcinomas using
whole-exome sequencing. Carcinogenesis. 33:1270–1276. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nik-Zainal S, Alexandrov LB, Wedge DC, Van
Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J,
Stebbings LA, et al: Mutational processes molding the genomes of 21
breast cancers. Cell. 149:979–993. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baudot A, Real FX, Izarzugaza JM and
Valencia A: From cancer genomes to cancer models: Bridging the
gaps. EMBO Rep. 10:359–366. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kreeger PK and Lauffenburger DA: Cancer
systems biology: A network modeling perspective. Carcinogenesis.
31:2–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee S, Stewart S, Nagtegaal I, Luo J, Wu
Y, Colditz G, Medina D and Allred DC: Differentially expressed
genes regulating the progression of ductal carcinoma in situ to
invasive breast cancer. Cancer Res. 72:4574–4586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wray CJ, Ko TC and Tan FK: Secondary use
of existing public microarray data to predict outcome for
hepatocellular carcinoma. J Surg Res. 188:137–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barrett T, Troup DB, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, et al: NCBI GEO: Archive for functional genomics data
sets-10 years on. Nucleic Acids Res. 39:(Database Issue).
D1005–D1010. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chung VY, Tan TZ, Tan M, Wong MK, Kuay KT,
Yang Z, Ye J, Muller J, Koh CM, Guccione E, et al:
GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian
cancer through transcriptional regulation and histone modification.
Sci Rep. 6:199432016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Halabi NM, Martinez A, Al-Farsi H, Mery E,
Puydenus L, Pujol P, Khalak HG, McLurcan C, Ferron G, Querleu D, et
al: Preferential allele expression analysis identifies shared
germline and somatic driver genes in advanced ovarian cancer. PLoS
Genet. 12:e10058922016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Espinosa AM, Alfaro A, Roman-Basaure E,
Guardado-Estrada M, Palma Í, Serralde C, Medina I, Juárez E,
Bermúdez M, Márquez E, et al: Mitosis is a source of potential
markers for screening and survival and therapeutic targets in
cervical cancer. PLoS One. 8:e559752013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shaul YD, Yuan B, Thiru P, Nutter-Upham A,
McCallum S, Lanzkron C, Bell GW and Sabatini DM: MERAV: A tool for
comparing gene expression across human tissues and cell types.
Nucleic Acids Res. 44:D560–D566. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
R Development Core Team, . R: A language
and environment for statistical computingR Foundation for
Statistical Computing. Vienna, Austria: 2017
|
|
21
|
Prasad Keshava TS, Goel R, Kandasamy K,
Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R,
Shafreen B, Venugopal A, et al: Human protein reference
database-2009 update. Nucleic Acids Res. 37:(Database Issue).
D767–D772. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2Disease: A manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res. 37:(Database Issue). D98–D104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vlachos IS, Paraskevopoulou MD, Karagkouni
D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL,
Maniou S, Karathanou K, Kalfakakou D, et al: DIANA-TarBase v7.0:
Indexing more than half a million experimentally supported
miRNA:mRNA interactions. Nucleic Acids Res. 43:(Database Issue).
D153–D159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaimal V, Bardes EE, Tabar SC, Jegga AG
and Aronow BJ: ToppCluster: A multiple gene list feature analyzer
for comparative enrichment clustering and network-based dissection
of biological systems. Nucleic Acids Res. 38:W96–W102. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Altermann E and Klaenhammer TR:
PathwayVoyager: Pathway mapping using the Kyoto encyclopedia of
genes and genomes (KEGG) database. BMC Genomics. 6:602005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stamoulis C and Betensky RA: A novel
signal processing approach for the detection of copy number
variations in the human genome. Bioinformatics. 27:2338–2345. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller CA, Settle SH, Sulman EP, Aldape KD
and Milosavljevic A: Discovering functional modules by identifying
recurrent and mutually exclusive mutational patterns in tumors. BMC
Med Genomics. 4:342011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ciriello G, Cerami E, Sander C and Schultz
N: Mutual exclusivity analysis identifies oncogenic network
modules. Genome Res. 22:398–406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vandin F, Upfal E and Raphael BJ: De novo
discovery of mutated driver pathways in cancer. Genome Res.
22:375–385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsaniras Champeris S, Kanellakis N,
Symeonidou IE, Nikolopoulou P, Lygerou Z and Taraviras S: Licensing
of DNA replication, cancer, pluripotency and differentiation: An
interlinked world? Semin Cell Dev Biol. 30:174–180. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pruitt SC, Bailey KJ and Freeland A:
Reduced Mcm2 expression results in severe stem/progenitor cell
deficiency and cancer. Stem Cells. 25:3121–3132. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Simon NE and Schwacha A: The Mcm2-7
replicative helicase: A promising chemotherapeutic target. Biomed
Res Int. 2014:5497192014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pittayapruek P, Meephansan J, Prapapan O,
Komine M and Ohtsuki M: Role of matrix metalloproteinases in
photoaging and photocarcinogenesis. Int J Mol Sci. 17:pii:
E8682016. View Article : Google Scholar
|
|
35
|
Chan TF, Poon A, Basu A, Addleman NR, Chen
J, Phong A, Byers PH, Klein TE and Kwok PY: Natural variation in
four human collagen genes across an ethnically diverse population.
Genomics. 91:307–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bosman FT and Stamenkovic I: Functional
structure and composition of the extracellular matrix. J Pathol.
200:423–428. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Uitto VJ and Larjava H: Extracellular
matrix molecules and their receptors: An overview with special
emphasis on periodontal tissues. Crit Rev Oral Biol Med. 2:323–354.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Deyl Z, Miksik I and Eckhardt A:
Preparative procedures and purity assessment of collagen proteins.
J Chromatogr B Analyt Technol Biomed Life Sci. 790:245–275. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dai J, Wang T, Wang W, Zhang S, Liao Y and
Chen J: Role of MAPK7 in cell proliferation and metastasis in
ovarian cancer. Int J Clin Exp Pathol. 8:10444–10451.
2015.PubMed/NCBI
|
|
40
|
Lee YS and Dutta A: The tumor suppressor
microRNA let-7 represses the HMGA2 oncogene. Genes Dev.
21:1025–1030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly
MB, Wang Y, Qian Z, Jin J, Zhang Y, et al: MicroRNA expression
signatures accurately discriminate acute lymphoblastic leukemia
from acute myeloid leukemia. Proc Natl Acad Sci USA.
104:19971–19976. 2007. View Article : Google Scholar : PubMed/NCBI
|