Introduction
Helicobacter pylori (H. pylori), which
was first isolated and discovered on the surface of the gastric
epithelium by Warren and Marshall in 1983 (1), is a gram-negative pathogenic
bacterium. H. pylori induces chronic inflammation and
epithelial damage in the stomach and duodenum. Infection induced by
H. pylori usually occurs early in life and persists if left
untreated (2). The prevalence of
H. pylori infection has increased worldwide in the past two
decades, affecting ~50% of the global population (2). H. pylori infection has been
associated with a number of upper gastrointestinal disorders,
including peptic ulcers in the stomach and duodenum, gastric
carcinoma and gastric mucosa-associated lymphoid tissue lymphoma
(3).
The precise pathophysiological process of H.
pylori-induced chronic inflammation and mucosal damage, and the
consequences of this chronic inflammation, remain elusive.
Virulence factors of H. pylori, including the
cytotoxin-associated gene A (CagA), Cag pathogenicity
island (Cag PAI) and vacuolating cytotoxin A (VacA),
have been widely investigated in past decades. The Cag PAI,
composed of a series of genes encoding members of a type IV
secretion system, is essential in the transfer of the bacterial
products of H. pylori, including Cag, into host cells
to mediate host cell damage (4).
The portion of CagA that is translocated into host cells is
phosphorylated. Phosphorylated CagA induces alterations in
cell motility and proliferation (5–8),
whereas secreted unphosphorylated CagA results in the
impairment of epithelial barrier function (9). VagA is essential for the binding of
H. pylori toxins to host cells, which disrupt the autophagy
of epithelial cells, impair tight junctions between epithelial
cells and enhance apoptosis (10–13).
The responses of host cells to H. pylori infection,
particularly the molecular mechanisms of infected cells, remain
unclear.
Members of a newly discovered class of non-coding
RNAs, microRNAs (miRNAs/miRs), have been demonstrated to exhibit
numerous functions in health and disease (14,15).
Mature miRNAs, which are composed of 19–25 nucleotides, are cleaved
from precursors of 60–110 nucleotide hairpin miRNAs by the RNase
III enzyme Dicer in the cytoplasm (16). Single-stranded miRNAs bind target
mRNAs at the 3′-untranslated region with imperfect complementarity,
which results in mRNA degradation and subsequent inhibition of
translation. miRNAs have been demonstrated to exhibit numerous
roles in physiological and pathological processes, including the
pathogenesis of cancer.
It is well established that miR-100 is involved the
pathophysiological process of various types of cancer, including
ovarian (17), cervical (18) and prostate (19) cancer. However, the effects of
miR-100 in the infection of H. pylori in gastric epithelial
cells remain unclear. The results of the present study demonstrated
that miR-100 was upregulated during H. pylori infection, and
upregulation of miR-100 may mediate gastric epithelial barrier
impairment via inhibition of mechanistic target of rapamycin kinase
(mTOR) signaling.
Materials and methods
Ethics consideration
The Ethics Committee of The Affiliated Hospital of
Medical School of Ningbo University (Ningbo, China) reviewed,
approved and supervised the proposal for the present study
(approval number: 2016-07-01). All participants involved in the
present study gave written informed consent.
Patients and volunteers
Individuals with epigastric complaints visiting the
outpatient clinic of The Affiliated Hospital of Medical School of
Ningbo University were recruited in the present study. Between July
2016 and January 2017, a total of 100 age- and sex-matched
volunteers and 98 patients with H. pylori infection were
enrolled. The clinical features of individuals in the present study
are summarized in Table I. The
diagnosis of H. pylori infection was confirmed by the
typical endoscopic appearance of the stomach and a positive urea
breath test the day prior to gastroscopy (20). Healthy volunteers were defined as
those with a normal endoscopic appearance of the stomach and a
negative urea breath test. Two double-biopsies were obtained during
gastroscopy from each individual.
 | Table I.Characteristics of study
participants. |
Table I.
Characteristics of study
participants.
|
| Volunteers
(n=100) | H. pylori
infection (n=98) | P-value |
|---|
| Age, years | 64.55±5.70 | 63.17±63.2 | 0.378 |
| Gender |
|
| 1 |
|
Male | 58 | 57 |
|
|
Female | 42 | 41 |
|
Cell culture
The GES-1 gastric epithelial cell line was purchased
from the Cell Resource Center, Shanghai Institute of Biochemistry
and Cell Biology, Chinese Academy of Sciences (Shanghai, China).
GES-1 cells were cultured in RPMI 1640 (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Thermo Fisher Scientific, Inc.; without any antibiotics) in
an incubator at 37°C with 5% CO2. Measurements for
fluorescein isothiocyanate (FITC)-dextran permeability and
resistance were performed in Transwell plates according to previous
publications (21,22). Briefly, GES-1 cells were seeded on
the upper compartment of a Transwell chamber. At 80–90% confluency,
FITC-dextran (0.5 mg/ml; purchased from Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was added into the upper apartment. The
concentration of FITC in the bottom compartment was read by an
infrared fluorescence laser reader (LI-COR Biosciences, Lincoln,
NE, USA). The electrical resistance between the upper and lower
compartments of the Transwell chambers was measured in ohms.
miR-100 mimic and inhibitor
Inhibitors for miR-100 (single-stranded chemically
modified oligonucleotide) and miR-100 mimic (double-stranded
oligonucleotides) were purchased from Thermo Fisher Scientific,
Inc. GES-1 cells (5×106) were seeded in 75
mm2 flasks. The next day, transfection with miR-100
mimic (5′-CAAGCUUGUAUCUAUAGGUAUG-3′) or miR-100 inhibitor
(5′-CAAGCUUGUAUCUAUAGGUAUG-3′) was performed using DharmaFECT 1
reagents (50 nM, GE Healthcare Dharmacon, Inc., Lafayette, CO, USA)
according to the manufacturer's protocol. Control cells were
treated by a mock transfection using a non-targeting miRNA sequence
(5′-UCACAACCUCCUAGAAAGAGUAGA-3′). The levels of miR-100 in
transfected cells were confirmed by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) 24 h
after transfection.
Infection of H. pylori
The H. pylori strain 7.13 (generously
provided by Dr. D. Scott Mereel from Uniformed Services University
of Health Services, Bethesda, MD, USA) was cultured in Brucella
broth (purchased from Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) with 10% FBS for 16 h at 37°C. Subsequently, H.
pylori was collected by centrifugation (400 × g for 10 min at
4°C) and used to infect GES-1 cells at a bacterium-to-cell ratio of
100:11 for 24 h, as described previously (23). Control cells (referred to as Ctrl)
were treated with PBS.
RT-qPCR for mature miR-100
Quantification of miRs was performed to measure the
levels of mature miR-100, as described previously (24). Total RNA was extracted using a
commercial RNeasy Micro kit (Qiagen, Inc., Valencia, CA, USA). RNA
was reverse-transcribed to cDNA with an miR-100-specific
stem-loop-like reverse transcription primer
(5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCACAAG-3′) using a
commercial kit (High Capacity cDNA Reverse Transcription kit
purchased from Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. qPCR was subsequently performed with SYBR
Green Master Mix (Thermo Fisher Scientific, Inc.) using the
following primers: Forward primer, 5′-GAGCCAACCCGTAGATCCGA-3′; and
reverse primer, 5′-GTGCAGGGTCCGAGGT-3′. The accuracy of the PCR
amplification of the mature miR-100 sequences was verified by
sequencing using Applied Biosystems 3730×l 96 capillary DNA
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Small nuclear RNA U6 was used as a housekeeping gene. Relative
expression of miR-100 was normalized with the 2−ΔΔCq
method (25).
For qPCR analyses of mRNAs of interest, total RNA (2
µg) was reverse-transcribed to cDNA using an RT-PCR kit (Verso
1-step RT-PCR kit ReddyMix; Thermo Fisher Scientific, Inc.) at 55°C
according to the manufacturer's protocol. The expression of the
genes of interest and the housekeeping gene hypoxanthine
phosphoribosyltransferase was measured using a SYBR Green Master
Mix (94°C for 2 min, 40 cycles of 94°C for 15 sec and 60°C for 1
min), according to the manufacturer's protocol. The sequences of
primers are listed in Table II.
Quantification of each mRNA was performed using the
2−ΔΔCq method.
| Table II.Primer sequences for reverse
transcription-quantitative polymerase chain reaction of mRNAs. |
Table II.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction of mRNAs.
|
| Sequence
(5′-3′) |
|---|
|
|
|
|---|
| Target gene | Forward | Reverse |
|---|
| E-cadherin |
CTGAGAACGAGGCTAACG |
TTCACATCCAGCACATCC |
| ZO-1 |
GTGTTGTGGATACCTTGT |
GATGATGCCTCGTTCTAC |
| HPRT |
GCAGACTTTGCTTTCCTTGG |
AAGCAGATGGCCACAGAACT |
| U6 |
GCCATGCTAATCTTCTCTGTATC |
CGGCAGCACATATACTAAAATTGG |
Western blotting
Whole-cell lysates from biopsies or cultured cells
were lysed in 30 µl lysis buffer [1% Triton X-100, 0.5% Nonidet
P-40, 10 mM Tris-HCl, 150 mM NaCl, (pH 7.4), 1 mM EDTA, 1 mM EGTA,
0.2 mM phenylmethylsulfonyl fluoride]. A total of 50 µg protein
measured by Lowry assays was resolved by SDS-PAGE and subsequently
transferred to nitrocellulose membranes. Membranes were blocked in
5% fat-free milk for 1 h at room temperature. Membranes were then
incubated with primary antibodies against epithelial (E)-cadherin
(1:10,000; cat. no. ab40772; Abcam, Cambridge, MA, USA), zona
occludens-1 (ZO-1; 1:1,000; cat. no. ab59720; Abcam), mTOR
(1:1,000; cat. no. 2983; Cell Signaling Technology, Inc., Danvers,
MA, USA), phosphorylated-eukaryotic translation initiation factor
4E-binding protein 1 (P-4EBP-1; 1:1,000; cat. no. 9459, Cell
Signaling Technology, Inc.), 4EBP-1 (1:1,000; cat. no. 9452, Cell
Signaling Technology, Inc.), p-P70S6K (1:1,000; cat. no. 9204; Cell
Signaling Technology, Inc.) and P70S6K (1:1,000; cat. no. 9202;
Cell Signaling Technology, Inc.) at the optimized titrations at 4°C
overnight. Following incubation with corresponding horseradish
peroxidase-conjugated secondary antibodies (anti-rabbit secondary
antibody, 1:10,000; cat. no. ab6721); anti-goat secondary antibody,
1:5,000; cat. no. ab6885; Both were purchased from Abcam) at room
temperature for 30 min, the bands were visualized using an
enhancing chemiluminescence system (Amershan ECL Western Blotting
Detection kit, GE Healthcare, Chicago, IL, USA). Densitometric
analysis was performed with ImageJ software (Windows Edition,
1.35j, National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are presented as the mean ± the standard error
of the mean. The difference between two groups was analyzed using a
two-tailed student's t-test. The statistical significance between
>2 groups was measured by one-way analysis of variance followed
by Bonferroni's post-hoc tests. Statistical analysis was performed
by GraphPad Prism 5 Windows Edition (GraphPad Software, Inc., La
Jolla, CA, USA). All experiments were performed with a minimum of
three times. P<0.05 was considered to indicate a statistically
significant difference.
Results
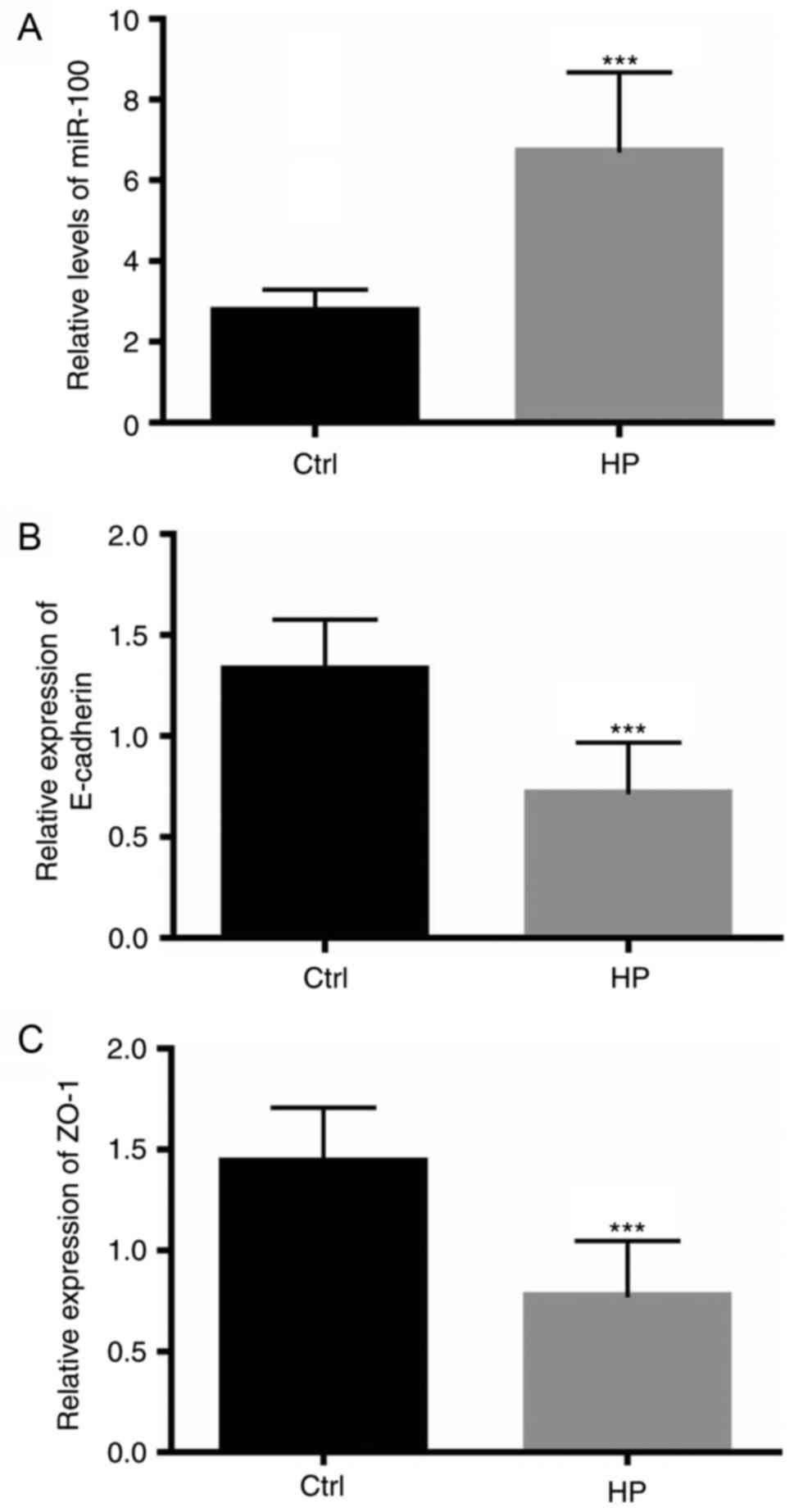
Patients with H. pylori infection
exhibit increased miR-100 levels and reduced expression of junction
proteins
The present study initially investigated whether
miR-100 may be involved in the pathophysiological process of H.
pylori-mediated gastric epithelium infection. Individuals with
epigastric complaints visiting the outpatient clinic of The
Affiliated Hospital of Medical School of Ningbo University, who
underwent upper gastrointestinal endoscopy, were recruited to the
present study. H. pylori infection was confirmed or excluded
by a urea breath test and the endoscopic appearance of the gastric
mucosa. A total of 100 healthy controls without H. pylori
infection and 98 patients infected with H. pylori were
analyzed. Two double-biopsies under endoscopy were obtained from
each individual. The level of miR-100 was assessed by RT-qPCR, as
described above. The expression of miR-100 in the gastric mucosa of
patients with H. pylori infection was significantly
upregulated compared with infection-free individuals (P<0.0001;
Fig. 1A). As the adherens
junctions and tight junctions have an important role as a barrier
to prevent the influx of luminal contents into the lamina propria
(26), the mRNA levels of
E-cadherin and ZO-1, important components of the adherens junctions
and tight junctions, respectively, were also measured. As
illustrated in Fig. 1B and C, in
patients with H. pylori infections, the mRNA levels of
E-cadherin and ZO-1 significantly decreased (P<0.0001),
indicating an impaired barrier function of the gastric epithelium
compared with infection-free controls. The protein expression
levels of E-cadherin and ZO-1 in patients with H. pylori
infection were also measured using western blotting, which
demonstrated that patients with H. pylori infection
exhibited decreased levels of the proteins of the adherens and
tight junctions (Fig. 1D and
E).
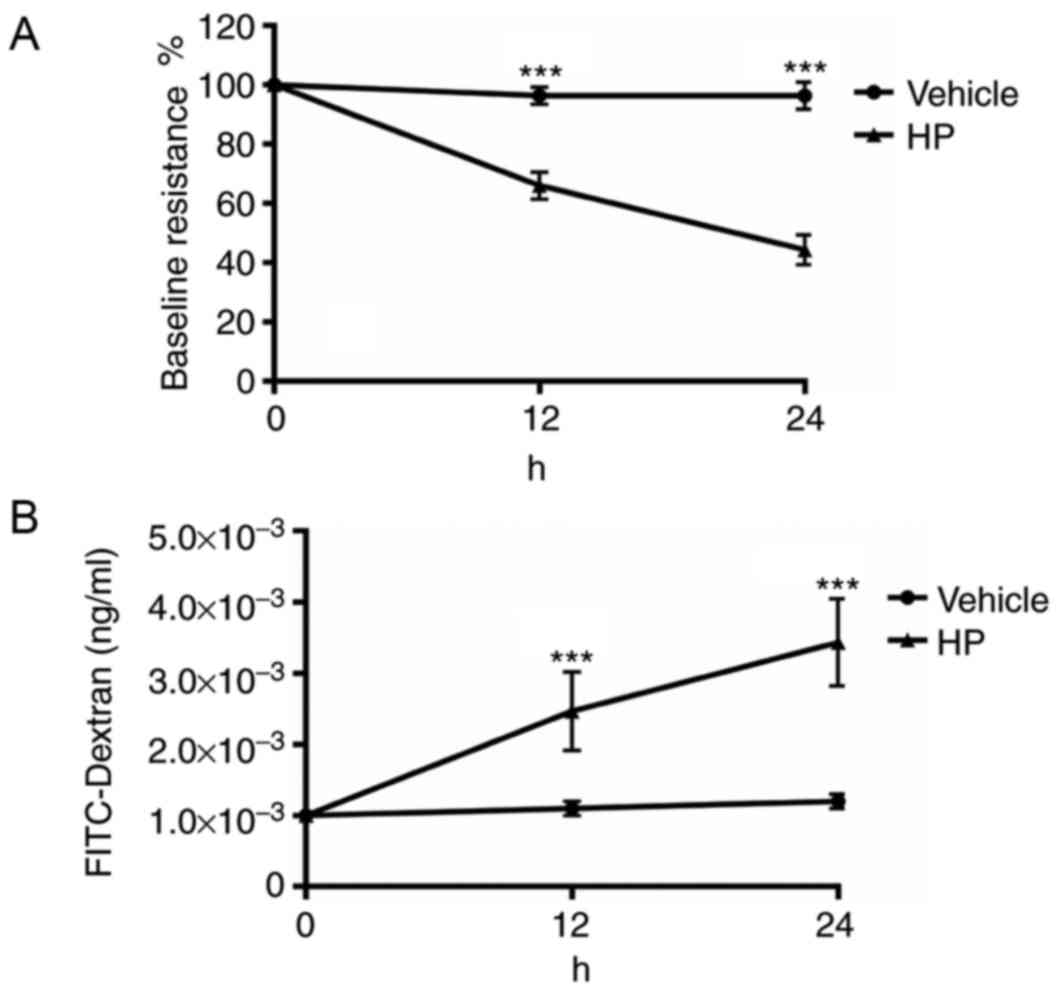
H. pylori infection increases gastric
epithelial monolayer permeability in vitro
As H. pylori infection decreased the
expression of E-cadherin and ZO-1 in the gastric epithelium, which
are vital for maintaining normal gastric epithelial barrier
function, the permeability of the cultured gastric epithelial
monolayer was assessed. The resistance of cultured GES-1 cells
treated with a PBS vehicle or H. pylori for 1 h was checked.
After 12 and 24 h of infection, the resistance of the cultured cell
monolayer was measured. The resistance prior to infection was used
as the baseline value. As demonstrated in Fig. 2A, cultured GES-1 gastric epithelial
cells had a significantly decreased level of resistance in the
H. pylori group compared with the vehicle (P<0.001).
FITC-dextran in the lower chamber of Transwells used for the
culture of GES-1 cells was also measured. As illustrated in
Fig. 2B, the lower chamber of the
Transwell in which gastric epithelial cells were infected with
H. pylori had significantly increased levels of FITC-dextran
after 12 and 24 h of infection (P<0.001). Results from both the
resistance and FITC-dextran assays indicated that gastric
epithelial cells infected with H. pylori had increased
permeability compared with controls.
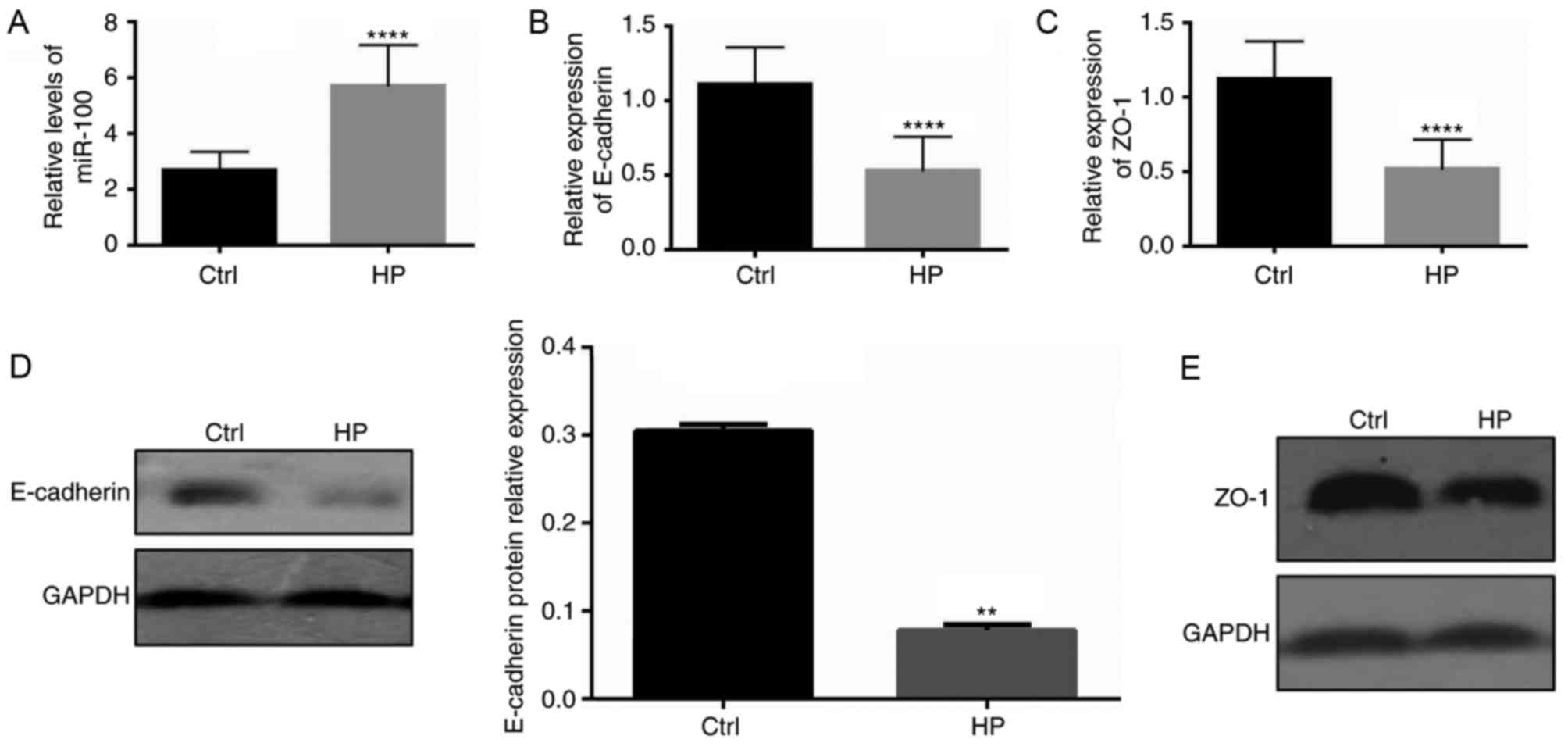
H. pylori infection increases the
levels of miR-100 and decreases the expression of E-cadherin and
ZO-1 in vitro
The present study further investigated whether
impaired barrier function of infected gastric epithelial cells
in vitro may be a result of miR-100-mediated reduction of
E-cadherin and ZO-1 expression. The levels of miR-100, as well as
the mRNA and protein expression of E-cadherin and ZO-1 junction
proteins, was assessed in cultured GES-1 gastric epithelial cells
infected with H. pylori. As demonstrated in Fig. 3A, the levels of miR-100 in cells
infected with H. pylori were significantly elevated compared
with control cells (P<0.001). However, the mRNA levels of
E-cadherin and ZO-1 were significantly downregulated in infected
cells (P<0.001; Fig. 3B and C).
Notably, the protein levels of these two junction proteins were
also decreased in infected cells (data not shown). These data
indicate the possibility that increased miR-100 levels induced by
H. pylori infection may mediate the downregulation of
E-cadherin and ZO-1 expression.
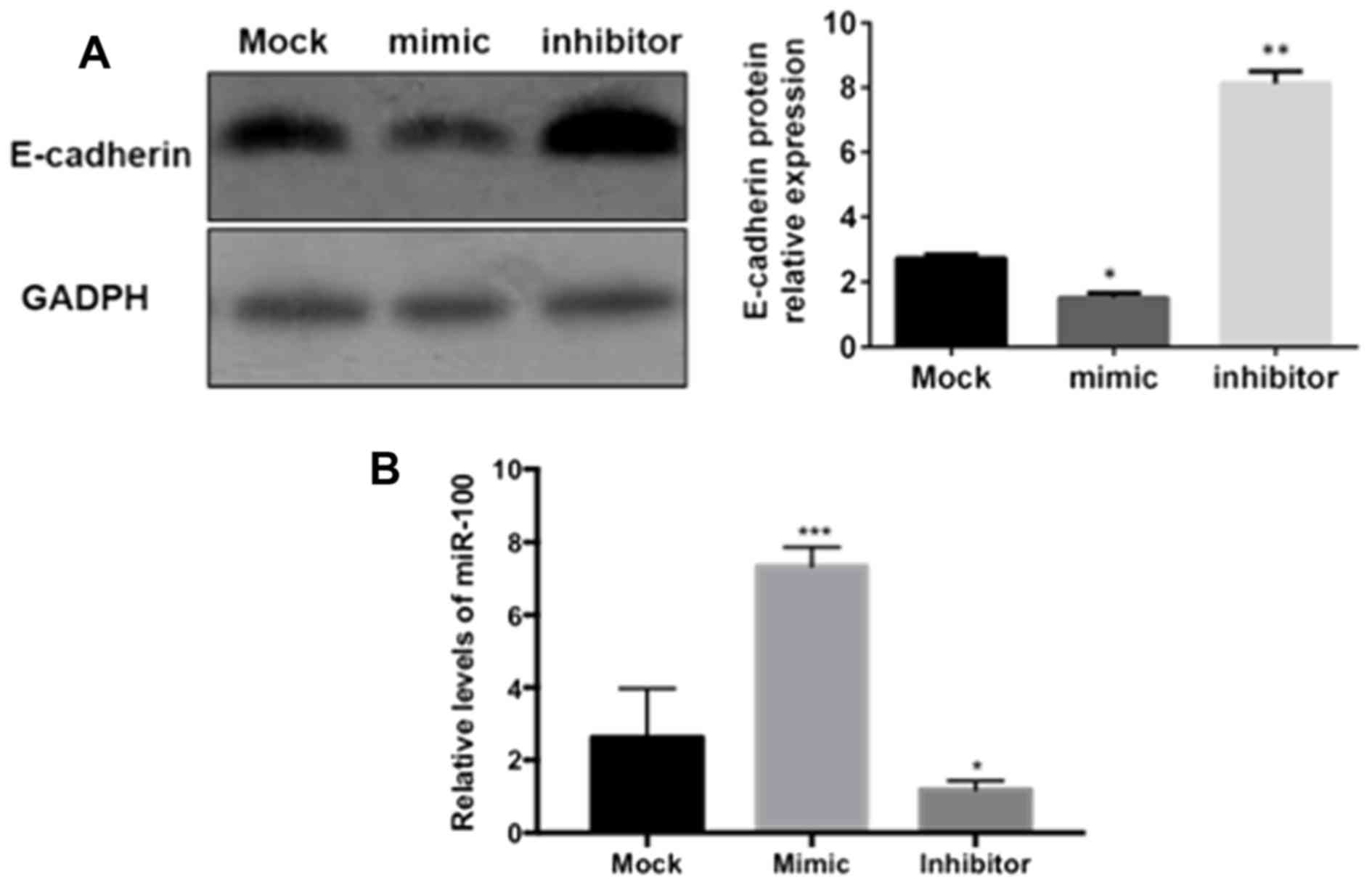
miR-100 mediates the inhibition of
E-cadherin and ZO-1 expression
In order to investigate the effects of miR-100 on
the expression of E-cadherin and ZO-1 in vitro,
‘loss-of-function’ and ‘gain-of-function’ protocols were used to
inhibit or overexpress miR-100 in GES-1 gastric epithelial cells.
As demonstrated in Fig. 4,
E-cadherin protein was significantly downregulated in gastric
epithelial cells transfected with miR-100 mimic (P<0.05), while
the protein expression of E-cadherin was significantly increased in
cells transfected with the miR-100 inhibitor (P<0.01), compared
with the mock group, as measured by western blotting. A similar
effect of miR-100 on the expression of ZO-1 in gastric epithelial
cells was also observed in the present study (data not shown).
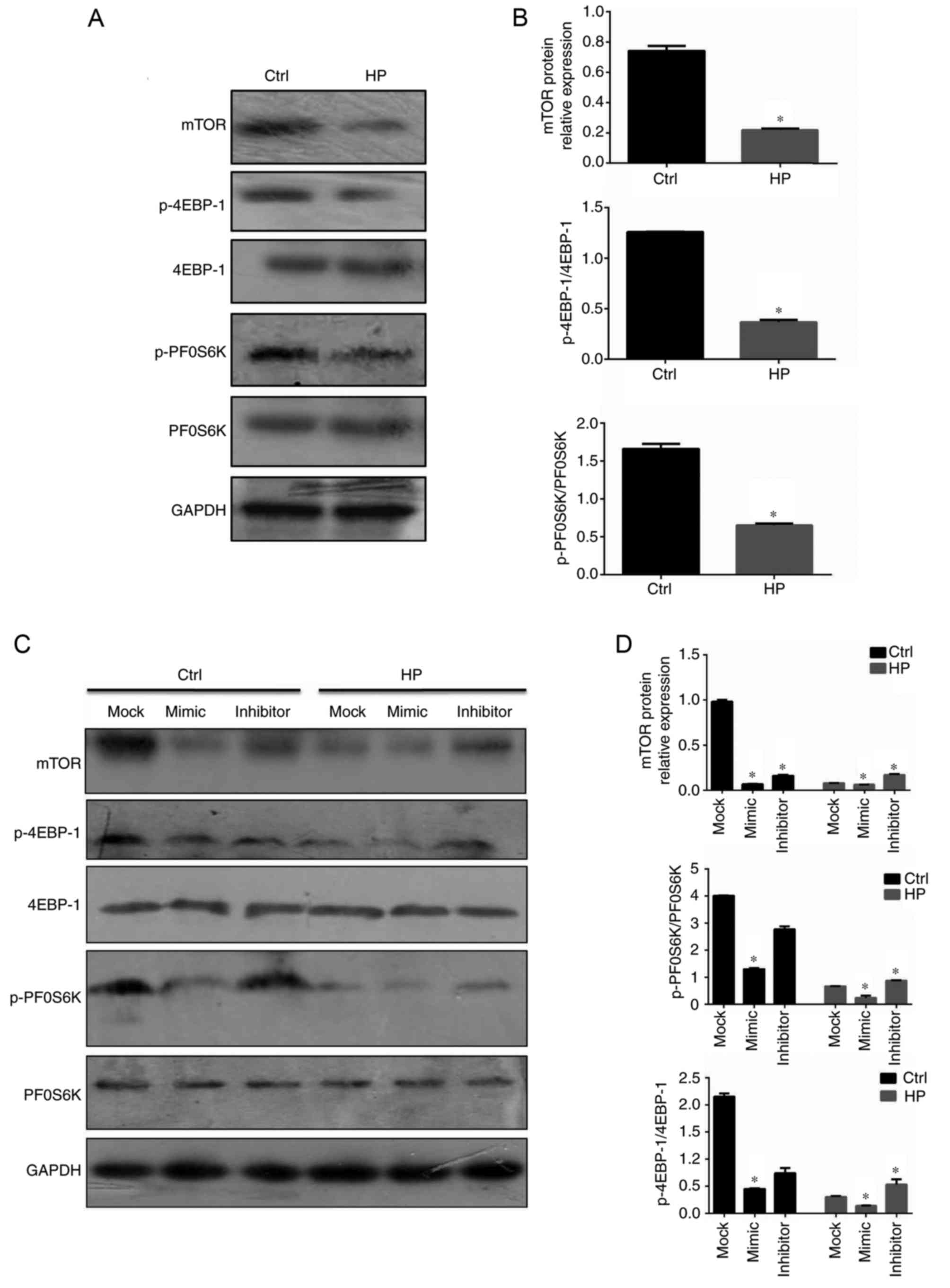
mTOR is the downstream signaling
molecule involved in miR-100-mediated regulation of E-cadherin and
ZO-1
It is well established that the mTOR signaling
pathway regulates the epithelial barrier function, including the
expression of the adherens junctions and tight junctions (27,28).
The activation status of mTOR signaling in gastric epithelial cells
infected with H. pylori was therefore assessed in the
current study. As demonstrated in Fig.
5A, in contrast to the levels of miR-100 in infected gastric
epithelial cells, mTOR and the downstream active forms of 4EBP-1
and PF0S6K were downregulated (Fig. 5A
and B). In order to assess the effects of miR-100 on the
activation of mTOR signaling, the same protocols of overexpression
and suppression of miR-100 in GES-1 cells were employed. The mimic
of miR-100 significantly inhibited the mTOR signaling pathway in
infected gastric epithelial cells; however, suppression of miR-100
significantly increased the activation of this pathway, compared
with mock-treated cells (P<0.05; Fig. 5C and D). These data indicated that
miR-100 may be a negative regulator of mTOR signaling in tuning the
barrier functions.
Discussion
In the present study, the role of miR-100 in the
impaired functioning of gastric epithelial cells infected with
H. pylori was described. The results of the present study
indicated that patients, as well as cultured cells in vitro,
infected with H. pylori exhibited increased levels of
miR-100 and decreased expression of junction-proteins. Furthermore,
cultured gastric epithelial cells infected with H. pylori
exhibited impaired barrier functions and mTOR signaling was
demonstrated to be controlled by an upregulation of miR-100.
Infections induced by H. pylori are a huge
public health burden. H. pylori infection has been reported
to be implicated in gastritis, precancerous lesions and gastric
carcinoma (29). The eventual
consequence of H. pylori infection, gastric carcinoma, is
the third leading cause of cancer-associated mortality worldwide.
As the association between H. pylori infection and gastric
carcinoma has become clear, H. pylori has been demonstrated
to be the most common pathogen that is associated with malignancy
(30). The process from chronic
H. pylori infection to gastric carcinoma is complex and
involves a number of molecular and cellular events, some of which
remains inclusive (31). Although
the pathogenesis of H. pylori has been investigated
thoroughly, the responses of host epithelial cells and
intracellular molecular events require further attention.
Based on the results of the present study, part of
the host gastric epithelial cell response to H. pylori
infection may be described as follows. Following infection, levels
of miR-100 are upregulated, this miR-100 upregulation may suppress
the activation of the mTOR signaling pathway, which subsequently
downregulates the expression of the adherens and tight junction
proteins E-cadherin and ZO-1, respectively. Decreased expression of
junction proteins leads to an impaired barrier function, which was
indicated in the present study as decreased resistance and an
increased permeability level were observed in monolayer gastric
epithelial cells infected with H. pylori. It may be
hypothesized that an impaired gastric epithelial barrier function
will result in the influx of gastric contents to the lamina propria
of the stomach, subsequently inducing chronic and persistent
inflammation. Chronic inflammation has been positively associated
with the onset of cancer (32),
therefore, miR-100 may be involved in gastric oncogenesis. However,
the mechanism by which miR-100 is upregulated in gastric epithelial
cells infected with H. pylori remains unknown at present,
and the regulation of miR-100 in H. pylori infection
requires investigation in the near future.
miRNAs are single-stranded RNA molecules that are
20–23 nucleotides in length. miRNAs control gene expression in
numerous cellular processes, including inflammation, cell cycle
regulation, stress differentiation, apoptosis, proliferation and
tumorigenesis (33). As a member
of the miRNA family, miR-100 has gained interest from biologists
and oncologists. miR-100 has been demonstrated to be associated
with ovarian (17), cervical
(18) and prostate (19) cancer. To the best of our knowledge,
the present study is the first to demonstrate that upregulation of
miR-100, as part of the host epithelial cell response to H.
pylori infection, may mediate impaired gastric barrier
functions by inhibiting mTOR signaling. These data expand on the
current understanding of host responses to H. pylori
infection.
However, there are a number of limitations
associated with the present study. As mentioned above, the
mechanism by which miR-100 is upregulated during H. pylori
infection was not investigated. In addition, the mechanism by which
miR-100 regulates E-cadherin and ZO-1 expression is yet to be
established. When the ‘loss-of-function’ and ‘gain-of function’
assays were performed for miR-100, the mRNA levels of E-cadherin
and ZO-1 were also determined in addition to the protein levels,
which indicated lower levels of mRNA in ‘gain-of-function’ assays
and increased levels of mRNA in ‘loss-of-function’ assays (data not
shown). Although these data indicate that miR-100 may affect the
transcription of E-cadherin and ZO-1, the precise underlying
mechanisms of this process require investigation in the future.
In conclusion, the results of the current study
indicated that miR-100 may impair the gastric epithelial barrier
function by affecting mTOR signaling in H. pylori
infection.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Ningbo City (grant no. 2016A610122). The
funding organization had no roles in the study design, data
collection and analysis, manuscript preparation or publication
decision.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GH designed this study, performed most experiments,
and prepared the first draft; LG performed some of repeated
experiments, and performed data analysis. GY designed this study,
supervised this study, and reviewed the draft.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of The Affiliated Hospital of Medical School of Ningbo
University, China (approval number: 2016-07-01). All participants
involved in the present study gave written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Warren JR and Marshall B: Unidentified
curved bacilli on gastric epithelium in active chronic gastritis.
Lancet. 1:1273–1275. 1983.PubMed/NCBI
|
|
2
|
Everhart JE: Recent developments in the
epidemiology of Helicobacter pylori. Gastroenterol Clin North Am.
29:559–578. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McColl KE: Clinical practice. Helicobacter
pylori infection. N Engl J Med. 362:1597–1604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Censini S, Lange C, Xiang Z, Crabtree JE,
Ghiara P, Borodovsky M, Rappuoli R and Covacci A: cag, a
pathogenicity island of Helicobacter pylori, encodes type
I-specific and disease-associated virulence factors. Proc Natl Acad
Sci USA. 93:14648–14653. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Selbach M, Moese S, Hauck CR, Meyer TF and
Backert S: Src is the kinase of the Helicobacter pylori CagA
protein in vitro and in vivo. J Biol Chem. 277:6775–6778. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tammer I, Brandt S, Hartig R, König W and
Backert S: Activation of Abl by Helicobacter pylori: A novel kinase
for CagA and crucial mediator of host cell scattering.
Gastroenterology. 132:1309–1319. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Backert S, Moese S, Selbach M, Brinkmann V
and Meyer TF: Phosphorylation of tyrosine 972 of the Helicobacter
pylori CagA protein is essential for induction of a scattering
phenotype in gastric epithelial cells. Mol Microbiol. 42:631–644.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mueller D, Tegtmeyer N, Brandt S, Yamaoka
Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A and Backert
S: c-Src and c-Abl kinases control hierarchic phosphorylation and
function of the CagA effector protein in Western and East Asian
Helicobacter pylori strains. J Clin Invest. 122:1553–1566. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saadat I, Higashi H, Obuse C, Umeda M,
Murata-Kamiya N, Saito Y, Lu H, Ohnishi N, Azuma T, Suzuki A, et
al: Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt
epithelial cell polarity. Nature. 447:330–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Palframan SL, Kwok T and Gabriel K:
Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori
pathogenesis. Front Cell Infect Microbiol. 2:922012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jain P, Luo ZQ and Blanke SR: Helicobacter
pylori vacuolating cytotoxin A (VacA) engages the mitochondrial
fission machinery to induce host cell death. Proc Natl Acad Sci
USA. 108:16032–16037. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rassow J and Meinecke M: Helicobacter
pylori VacA: A new perspective on an invasive chloride channel.
Microbes Infect. 14:1026–1033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boquet P and Ricci V: Intoxication
strategy of Helicobacter pylori VacA toxin. Trends Microbiol.
20:165–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lagos-Quintana M, Rauhut R, Lendeckel W
and Tuschl T: Identification of novel genes coding for small
expressed RNAs. Science. 294:853–858. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nagaraja AK, Creighton CJ, Yu Z, Zhu H,
Gunaratne PH, Reid JG, Olokpa E, Itamochi H, Ueno NT, Hawkins SM,
et al: A link between mir-100 and FRAP1/mTOR in clear cell ovarian
cancer. Mol Endocrinol. 24:447–463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li BH, Zhou JS, Ye F, Cheng XD, Zhou CY,
Lu WG and Xie X: Reduced miR-100 expression in cervical cancer and
precursors and its carcinogenic effect through targeting PLK1
protein. Eur J Cancer. 47:2166–2174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leite KR, Sousa-Canavez JM, Reis ST,
Tomiyama AH, Camara-Lopes LH, Sañudo A, Antunes AA and Srougi M:
Change in expression of miR-let7c, miR-100 and miR-218 from high
grade localized prostate cancer to metastasis. Urol Oncol.
29:265–269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei Y, Wang T, Song H, Tian L, Lyu G, Zhao
L and Xue Y: C-C motif chemokine 22 ligand (CCL22) concentrations
in sera of gastric cancer patients are related to peritoneal
metastasis and predict recurrence within one year after radical
gastrectomy. J Surg Res. 211:266–278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Temmesfeld-Wollbrück B, Brell B, zu Dohna
C, Dorenberg M, Hocke AC, Martens H, Klar J, Suttorp N and
Hippenstiel S: Adrenomedullin reduces intestinal epithelial
permeability in vivo and in vitro. Am J Physiol Gastrointest Liver
Physiol. 297:G43–G51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oshima T, Miwa H and Joh T: Aspirin
induces gastric epithelial barrier dysfunction by activating p38
MAPK via claudin-7. Am J Physiol Cell Physiol. 295:C800–C806. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Franco AT, Israel DA, Washington MK,
Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L,
Perez-Perez GI, Hatakeyama M, et al: Activation of beta-catenin by
carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA.
102:10646–10651. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hou J, Wang P, Lin L, Liu X, Ma F, An H,
Wang Z and Cao X: MicroRNA-146a feedback inhibits RIG-I-dependent
Type I IFN production in macrophages by targeting TRAF6, IRAK1, and
IRAK2. J Immunol. 183:2150–2158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hartsock A and Nelson WJ: Adherens and
tight junctions: Structure, function and connections to the actin
cytoskeleton. Biochim Biophys Acta. 1778:660–669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sampson LL, Davis AK, Grogg MW and Zheng
Y: mTOR disruption causes intestinal epithelial cell defects and
intestinal atrophy postinjury in mice. FASEB J. 30:1263–1275. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kaser A and Blumberg RS: Autophagy,
microbial sensing, endoplasmic reticulum stress, and epithelial
function in inflammatory bowel disease. Gastroenterology.
140:1738–1747. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ajani JA, Lee J, Sano T, Janjigian YY, Fan
D and Song S: Gastric adenocarcinoma. Nat Rev Dis Primers.
3:170362017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
de Martel C, Ferlay J, Franceschi S,
Vignat J, Bray F, Forman D and Plummer M: Global burden of cancers
attributable to infections in 2008: A review and synthetic
analysis. Lancet Oncol. 13:607–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Polk DB and Peek RM Jr: Helicobacter
pylori: Gastric cancer and beyond. Nat Rev Cancer. 10:403–414.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|