Introduction
Numerous factors, including trauma, alcohol,
steroids, coagulation disorders, sickle cell disease and fat
embolism, have been reported to be implicated in the destruction of
the femoral head (1). Avascular
necrosis of the femoral head (ANFH), as a consequence of impaired
blood supply, is a common type II collagenopathy that is associated
with collagen type II α1 chain (COL2A1) mutations (2,3). The
primary clinical manifestations of ANFH are limping gait,
discrepancy in leg length and pain on exertion, which have a
substantial effect on the quality of life of individuals (4,5).
Although the exact pathogenesis of ANFH remains to be elucidated,
evidence obtained from idiopathic osteonecrosis in twin studies
indicates that genetic factors may have a vital role in the
development of ANFH (6,7). Additionally, genetic studies have
demonstrated that various single nucleotide mutations in COL2A1 are
associated with an increased risk of ANFH. Based on the data
obtained from familial ANFH patients in three Chinese families, a
study conducted by Liu et al (2) revealed that a G to A transition in
exon 50 (c.3665G>A) or exon 30 (c.2306G>A) in COL2A1 led to
the replacement of glycine (Gly) with serine at codon 1,170 in the
Gly-X-Y domain among patients with ANFH, which did not occur in
healthy individuals. Furthermore, a study conducted by Li et
al (8) revealed that a novel
p.Gly630Ser mutation in the COL2A1 gene may have led to ANFH and
Legg-Calve-Perthes disease (LCPD) in a Chinese family.
The COL2A1 gene, which is localized at 12q13,
encodes the necessary element for type II collagen, characterized
by three homologous α1-peptide chains (9). Type II collagen is predominantly
expressed in the hyaline cartilage, as well as the vitreous body
and nucleus pulposus (10). Type
II collagen is also an essential component of collagen in the
connective tissue; the biomechanical strength of this tissue is
primarily dependent on the amount and extent of collagen fibril
crosslinking (11). Type II
collagen is composed of three homologous α1 peptide chains
(homotrimers) that twist together to form a triple-stranded helix.
The triple helical domain contains ~300 amino acids, and Gly
followed by two other amino acids (AAs) constitute the repeated
(Gly-X-Y)n sequence. If Gly in the specific Gly-X-Y
repeat is replaced by other AAs, the structure of the type II
collagen is destroyed (8,12). Over 200 mutations have been
identified in the COL2A1 gene, and the majority of these mutations
have been associated with the replacement of Gly residues in the
triple helix domain (13). In
recent decades, an increasing number of studies have indicated that
mutations in the COL2A1 gene are associated with familial or
inherited ANFH (8,14–20).
However, at present, no mutation hotspot has been confirmed. The
present study was performed to identify potential mutations in the
COL2A1 gene that may be implicated in ANFH among a unique
three-generation Chinese family, and to conduct a review of the
literature.
Case report
Ethics statement
All experimental protocols described in the present
study were approved by the Institutional Review Board of the Second
Hospital of Yueyang (Yueyang, China). Written informed consent
according to the Declaration of Helsinki was obtained from all
subjects described in the present study.
Case presentation and pedigree
analysis
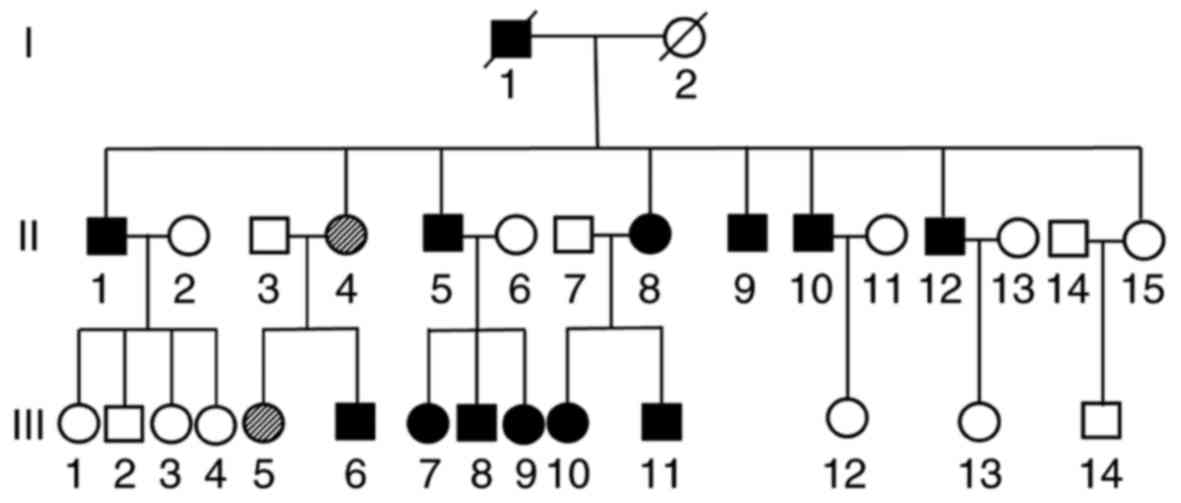
A femoral head osteonecrosis pedigree with a total
of 31 subjects (13 males and 18 females) of three generations
(Yueyang, China) was recruited in the present study in July 2016.
The proband (II-5), a male diagnosed with ANFH, visited the Second
Hospital of Yueyang (Yueyang, China) in July 2014 due to groin pain
and restricted motion of the left hip joint, which started when the
patient was 41 years old. A comprehensive survey was conducted to
obtain detailed information of the patient's medical history,
physical examination and laboratory examination. Total hip
replacement surgery was performed on his right hip joint in July
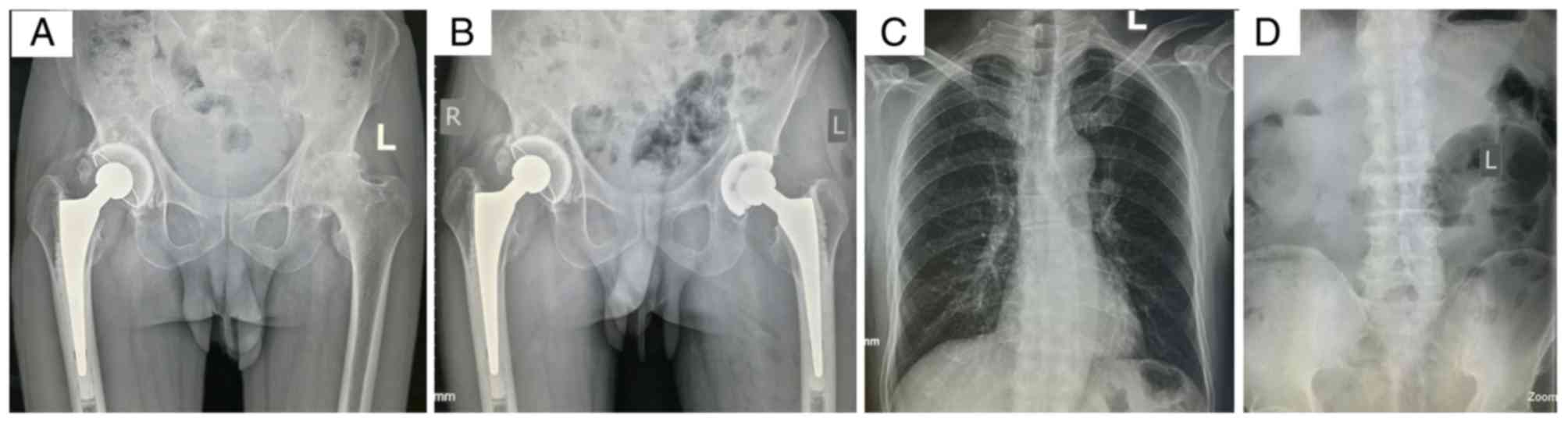
2014 in the Second Hospital of Yueyang. The radiograph revealed
that the primary pathological change in his left hip was a flat
femoral head with cystic degeneration, secondary osteoarthritis
(OA) and light subluxation, indicating a Ficat stage IV lesion
(21) (Fig. 1A). The patient was treated with
total hip arthroplasty (Fig. 1B)
and a post-operation biopsy confirmed the diagnosis.
The X-ray of the patient's spine was normal
(Fig. 1C and D) and facial
features were unremarkable. There were no obvious abnormalities in
the patient's neurological system or limbs. A total of 11 members
of the proband's family (II-1, II-8, II-9, II-10, II-12 and III-6
to III-11) had similar clinical manifestations observed in X-rays
(data not shown). Due to the aggregation of similar hip disorders
in this family, a pedigree analysis was conducted to determine the
potential cause (Fig. 2). Subjects
of the first generation had passed away.
A total of 12 family members (54.5% of the
offspring) were clinically diagnosed with ANFH (Fig. 2). Radiographic data revealed the
typical features of the affected hips, including cysts, collapse,
joint space narrowing, secondary OA and subluxation, indicating
lesion stages ranging from Ficat II to IV. No family members had a
history of trauma, alcohol, steroid use or any other high-risk
factors.
Nucleic acid isolation
Blood samples of 22 family members from the same
lineage as the proband and 20 sporadic ANFH volunteers were
collected in July 2016 at the Second Hospital of Yueyang. Diagnois
was conducted by an independent pathologist and imaging doctors.
Characteristics of affected family members and sporadic ANFH
volunteers are presented in Tables
I and II, respectively.
Genomic DNA was extracted from peripheral blood samples (200 µl)
using the TIANamp Blood DNA kit (Tiangen Biotech Co., Ltd.,
Beijing, China), according to the manufacturer's protocol, and
stored at −20°C prior to experimentation. The concentration and
purity of the genomic DNA was measured using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) at 260/280 nm. The process of blood sample collection and
mutation detection was double-blinded. No authors had access to
information that could identify individual participants during or
following data collection.
 | Table I.Summary of findings of the affected
members. |
Table I.
Summary of findings of the affected
members.
| Family members | Age | Sex | Age at onset | Major symptoms | Radigraphic
findings | Genotype | Affected hip |
|---|
| II 1 | 79 | M | 76 | Groin
pain/limping | Cystic/collapse/joint
space narrowing/secondary OA | Nomal | Bilateral |
| II 4 | 75 | F |
| Asymptomatic | Normal | Mutation |
|
| II 5 | 71 | M | 60 | Groin
pain/limping/restricted motion | Cystic/joint space
narrowing/secondary OA | Mutation | Bilateral |
| II 8 | 68 | F | 20 | Groin
pain/limping | Cystic/joint space
narrowing/secondary OA | Mutation | Bilateral |
| II 9 | 66 | M | 30 | Groin
pain/limping/restricted motion | Cystic/joint space
narrowing/secondary OA | Mutation | Bilateral |
| II 10 | 64 | M |
| Groin pain | Cystic | Mutation | Right |
| II 12 | 62 | M | 30 | Groin
pain/limping |
Cystic/collapse/joint space narrowing | Mutation | Right |
| III 5 | 58 | F |
| Asymptomatic | Normal | Mutation |
|
| III 6 | 50 | M | 49 | Groin pain | Cystic | Mutation | Right |
| III 7 | 46 | F | 32 | Groin
pain/limping |
Cystic/collapse/joint space narrowing | Mutation | Bilateral |
| III 8 | 45 | M | 30 | Groin
pain/limping | Cystic/joint space
narrowing | Mutation | Bilateral |
| III 9 | 39 | F | 23 | Groin
pain/limping | Cystic/joint space
narrowing | Mutation | Bilateral |
| III 10 | 48 | F | 38 | Groin
pain/limping | Cystic/joint space
narrowing | Mutation | Bilateral |
| III 11 | 41 | M | 40 | Groin pain | Normal | Mutation | Left |
| Table II.Clinical data of 20 volunteer
patients with sporadic avascular necrosis of the femoral head. |
Table II.
Clinical data of 20 volunteer
patients with sporadic avascular necrosis of the femoral head.
| Sporadic
members | Age | Sex | Age of onset | Major symptoms | Radiographic
findings | Affected hip |
|---|
| 1 | 57 | F | 42 | Groin pain,
limping | Cysts, collapse,
joint space narrowing, secondary OA | Right |
| 2 | 64 | F | 63 | Groin pain,
limping | Cysts, joint space
narrowing, secondary OA | Left |
| 3 | 67 | M | 66 | Groin pain,
limping, restricted motion | Cysts, joint space
narrowing, secondary OA | Right |
| 4 | 51 | M | 47 | Groin pain,
limping | Cysts, joint space
narrowing, secondary OA | Right |
| 5 | 66 | M | 64 | Groin pain,
limping | Cysts, joint space
narrowing, collapse | Bilateral |
| 6 | 85 | M | 79 | Groin pain,
limping | Cysts, joint space
narrowing, secondary OA | Left |
| 7 | 77 | M | 67 | Groin pain,
limping | Cysts, collapse,
joint space narrowing | Left |
| 8 | 59 | M | 55 | Groin pain,
limping | Cysts, collapse,
joint space narrowing | Left |
| 9 | 62 | M | 52 | Groin pain,
limping, restricted motion | Cysts, collapse,
joint space narrowing, secondary OA | Left |
| 10 | 66 | F | 63 | Groin pain,
limping | Cysts, subluxation,
joint space narrowing | Bilateral |
| 11 | 63 | F | 60 | Groin pain,
limping | Cysts, joint space
narrowing, secondary OA | Left |
| 12 | 64 | M | 62 | Groin pain,
limping | Cysts, joint space
narrowing | Left |
| 13 | 75 | F | 67 | Groin pain,
limping | Cysts, joint space
narrowing | Right |
| 14 | 47 | M | 46 | Groin pain,
limping | Cysts, joint space
narrowing, secondary OA | Left |
| 15 | 64 | M | 62 | Groin pain,
limping | Cysts, collapse,
joint space narrowing, secondary OA | Left |
| 16 | 67 | F | 62 | Groin pain,
limping | Cysts, collapse,
joint space narrowing, secondary OA | Left |
| 17 | 67 | F | 61 | Groin pain,
limping, restricted motion | Cysts, collapse,
joint space narrowing, secondary OA | Left |
| 18 | 71 | M | 67 | Groin pain,
limping | Cysts, collapse,
joint space narrowing, secondary OA | Left |
| 19 | 64 | M | 63 | Groin pain,
limping, restricted motion | Cysts, collapse,
joint space narrowing, secondary OA | Bilateral |
| 20 | 68 | M | 62 | Groin pain,
limping, restricted motion | Cysts, collapse,
joint space narrowing, secondary OA | Bilateral |
COL2A1 gene mutation analysis
Primers were designed based on the sequences of exon
50 in the COL2A1 gene, as previously described (9). The primer sequences were as follows:
COL2A1-exon 50, TTTCCCAGCACTGATCATGG (forward) and
GCCTCTCGCTGTCAGACAGA (reverse). Polymerase chain reaction (PCR) was
performed using Pfu DNA Polymerase (Tiangen Biotech Co., Ltd.) in
the T100 Thermal Cycler (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) with 100 ng genomic DNA as the template. The PCR was conducted
with the following thermocycling conditions: 95°C for 5 min,
followed by 35 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C
for 60 sec, and final extension at 72°C for 5 min. The PCR products
were subsequently sequenced by Sangon Biotech Co., Ltd (Shanghai,
China). The sequencing results were compared to the National Center
for Biotechnology Information reference sequence (NG_008072.1)
(22) using DNAMAN 6.0 software
(Lynnon LLC, San Ramon, CA, USA).
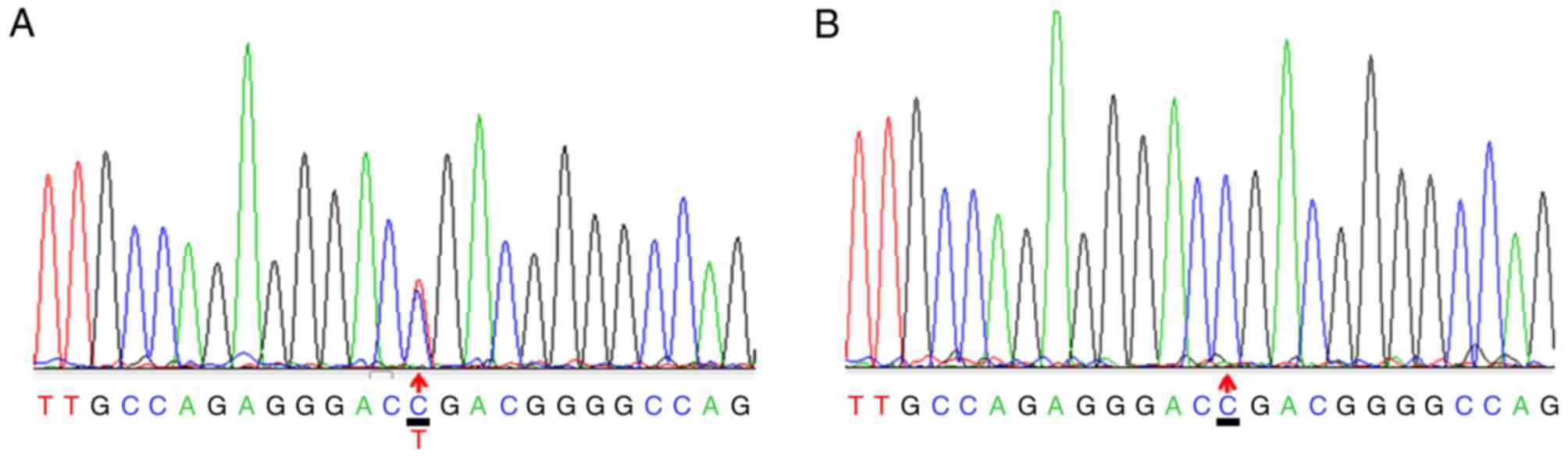
A heterozygous c.3508G>A mutation within exon 50
of the COL2A1 gene was identified in the proband (Fig. 3A) and other affected members in
this family, excluding subject II-1. The repeated analysis obtained
the same results. Genetic analysis revealed that this mutation was
inherited in an autosomal dominant manner. No mutation was found at
c.3508G>A in subject II-1, in which onset began at 76 years old.
However, the patient received sequential bilateral total hip
arthroplasty. Subjects II-2 and III-5 were also confirmed to be
asymptomatic carriers and require future follow-up. The mutation
described above was not detected in the other family subjects or
the 20 sporadic ANFH patients (Fig.
3B).
Histology
To observe the pathological alterations, cartilage
with the most severe necrosis area in the margin of the femoral
head from mutant (II-5 and II-9) and control (sporadic members)
groups was collected. The cartilage specimens were fixed in 10%
neutral-buffered formalin overnight at 4°C, dehydrated in an
alcohol gradient, and embedded in low-melting point paraffin.
Continuous 5-µm thick tissue sections were cut and fixed onto
silicified slides using neutral resin size at room temperature. The
sections were deparaffinised and rehydrated with deionized
Millipore water (EMD Millipore, Billerica, MA, USA). Sections were
stained with 1% toluidine blue with borate for 20 min at 20–25°C,
rinsed with tap water, dehydrated, immersed in xylene for 10 min,
and then mounted with neutral resins (17).
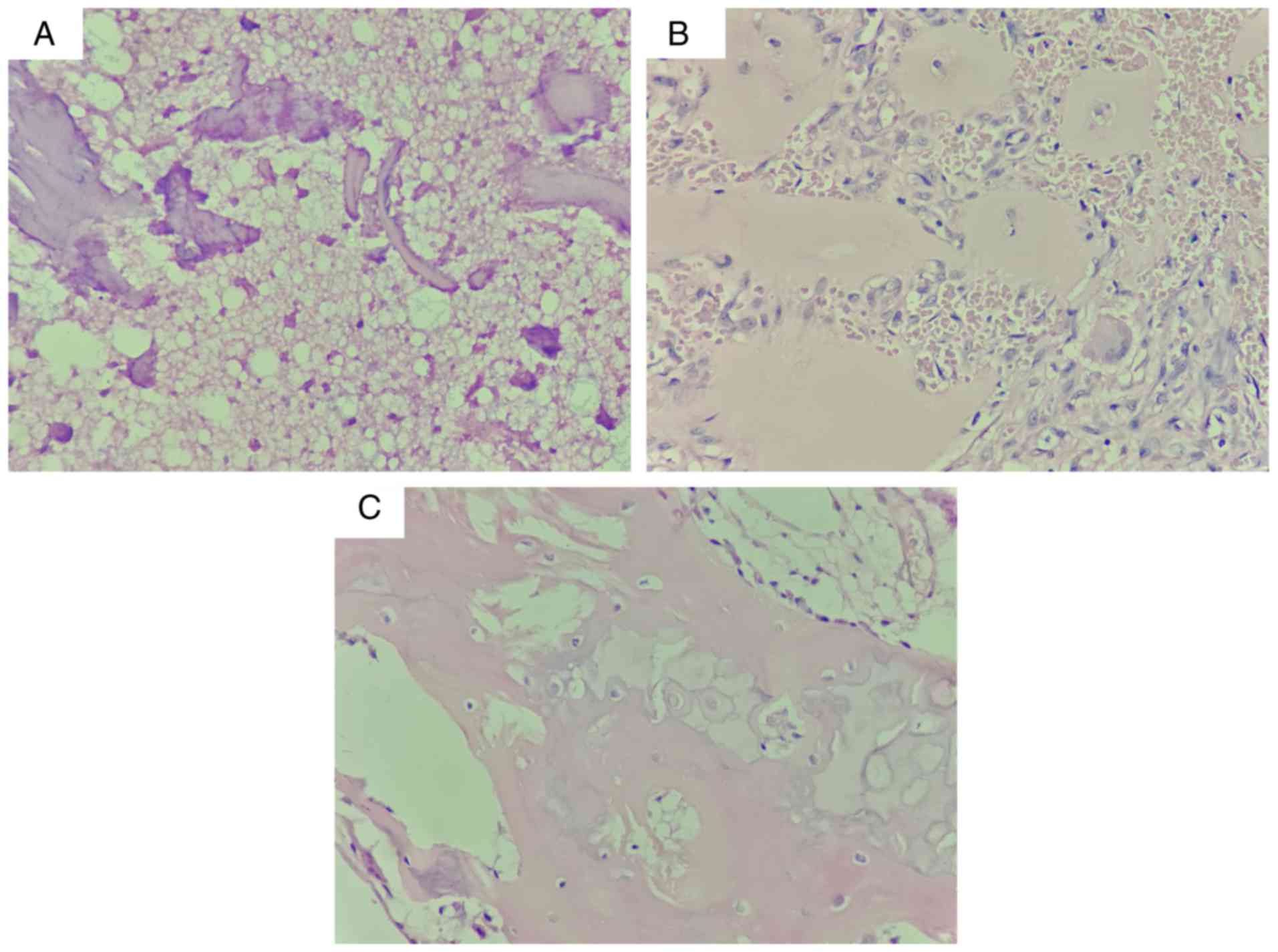
The presence of necrotic trabeculae or newly formed
trabeculae, necrotic bone marrow and fibrous tissue was observed in
inherited ANFH (Fig. 4A), subject
II-1 without the COL2A1 c.3508G>A mutation (Fig. 4B) and sporadic ANFH (Fig. 4C). Furthermore, empty lacunae
without osteocytes were observed in both Fig. 4B and C. No marked differences were
observed between inherited and sporadic ANFH.
Discussion
The present study identified a heterozygous
c.3508G>A mutation in the COL2A1 gene, which resulted in
inherited ANFH in a large Chinese family. Unexpectedly, one family
member with ANFH (II-1) had a normal genotype compared with the
other affected family members; further investigation into the
identification of the possible mutated genes is required. In recent
decades, >200 mutations have been identified in the COL2A1 gene,
including single substitution, splice-site mutations and partial
deletions (2,3,8).
Type II collagen is a major component of the articular cartilage,
which reduces articular friction and absorbs load pressure during
movement. COL2A1 mutations have been associated with various human
disorders, which are collectively termed type II collagenopathies
and include spondyloepiphyseal dysplasia congenita, Kniest
dysplasia, Stickler dysplasia, otospondylomegaepiphyseal dysplasia
and spondyloepiphyseal dysplasia with premature onset arthrosis
(23–26).
The c.3508G>A transition identified in the
present study resulted in the replacement of Gly by serine at codon
1,170 in the affected family members, leading to a hip disorder
that manifested as inherited ANFH. This did not occur in control
family members. Liu et al (2) revealed that a c.3665G>A transition
(p.G1170S) or c.2306G>A transition (p.G717S) in the COL2A1 gene
led to ANFH in three Chinese families. However, the differing
histopathology between inherited and sporadic ANFH and the
histopathological characteristics of the mutant subjects has yet to
be further discussed (2). All
subjects with ANFH from the three families were confirmed to have
different COL2A1 mutations. However, in the present study, no
similar mutations were detected in one of the family members
(subject II-1), which strongly indicates that acquired factors may
have an irreversible role in the pathogenesis of ANFH.
Additionally, it was reported that a c.4148G>A (18) or c.1774G>A mutations (19) in the COL2A1 gene may also lead to
familial ANFH. Notably, evidence from clinical studies has
demonstrated that a single base substitution in the COL2A1 gene is
also implicated in the development of LCPD (8,14,25)
and premature hip OA, which are two other types of type II
collagenopathies (16,27). However, it remains to be determined
whether these diseases occur separately or simultaneously in a
single family with COL2A1 mutations. Although a c.3508G>A
transition was not reported to be associated with idiopathic
osteonecrosis of the femoral head (iONFH) in sporadic Japanese
patients, studies have indicated that a c.3508G>A mutation may
be the root cause of familial Ionfh (3). However, it remains unclear whether
the G>A transition (p.G1170S) is the hotspot mutation in COL2A1
that leads to inherited hip disorders in Chinese individuals
(3). Hence, this requires further
investigation.
Type II collagen is predominantly expressed in the
matrix and chondrocytes. The morphology and density of chondrocytes
vary according to the different depths of normal articular
cartilage surface, which is divided into three zones: Superficial
zone (10–20%), middle zone (40–60%), and deep zone (30%) (11). One histological study revealed that
the deep zone in inherited ANFH dramatically increased to 70%, from
30% in normal samples, and the superficial zone dramatically
decreased to 10% (17). As mature
chondrocytes are primarily distributed in the superficial zone,
these results indicated that the number of mature chondrocytes is
significantly decreased in inherited ANFH. Furthermore, although
mutated type II collagen was reported to be secreted into the
matrix, the non-uniform arrangement and distribution of the
pathological collagen indicated that the biomechanical
characteristics of the pathological cartilage was destroyed. As
endochondral ossification occurs in infant development of the
femoral head, the biomechanical properties of the subchondral bone
may also be markedly weakened (27). These features are notably different
from sporadic ANFH, which begins with subchondral bone necrosis and
does not affect the articular cartilage (28–30).
No marked differences between inherited and sporadic ANFH were
demonstrated in the present study. This may due to the different
biopsy locations or the small sample size.
The process of type II collagen synthesis and
assembly has been reported to be impaired in chondrocytes with a
COL2A1 mutation (31,32). The pro-α1 peptide chain encoded by
COL2A1 forms a triple-helix structure within the endoplasmic
reticulum and Golgi bodies of chondrocytes, which integrates into
numerous fibrin networks following secretion from the extracellular
matrix (10,11). The Gly-X-Y triple-helix motif is
crucial for the proper crosslinking of the pro-α1 peptide chain to
form functional type II collagen. The deletion of sequences for
maintaining the Gly-X-Y triple-helix domain lead to type II
collagenopathies (33,34). Mortier et al reported that
there are numerous excessive post-translational modifications in
type II collagen in individuals carrying a Gly-substituted mutation
(35). These excessively modified
type II collagens prevent the protein from folding into its
fundamental triple-helical domain structure (35). In addition, an increase in
hydroxymethyl may cause local structural damage and a loosened
super helix structure, resulting in pathological alterations
(8,11). However, why this mutation only
appears to affect the hip joint remains unknown and suggests that
other biomechanical factors may be involved, which require further
investigation.
In conclusion, the present study identified a
heterozygous c.3508G>A mutation in COL2A1 that was implicated in
familial ANFH, and was compatible with autosomal dominant
inheritance. The present study may provide a novel molecular basis
for genetic counseling and the molecular diagnosis of ANFH. Further
studies are required to investigate the potential mechanism
underling COL2A1 mutation-induced inherited ANFH.
Acknowledgements
The authors thank Professor Xie Hui and Dr Chen
Tuanhui from the Movement System Injury and Repair Research Center
of Xiangya Hospital, Central South University (Changsha, China) for
their methodological support with gene mutation analysis.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FL and ZX conceived and designed the study. QL, JH
and WL performed the experiments. QL, JH and NZ provided the
mutants. FL and ZX wrote the paper. ZX reviewed and edited the
manuscript. All authors read and approved the manuscript.
Ethics approval and consent to
participate
All experimental protocols described in the present
study were approved by the Institutional Review Board of the Second
Hospital of Yueyang (Yueyang, China). Written informed consent was
obtained from all patients, or parent, guardian or next of kin
described in the present study.
Consent for publication
All the experimental protocols described in this
study were approved by The Ethics Committee of Hunan Normal
University (Yueyang, China). Written informed consent was obtained
from all patients, or parent, guardian or next of kin for the
publication of any associated data and accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Malizos KN, Karantanas AH, Varitimidis SE,
Dailiana ZH, Bargiotas K and Maris T: Osteonecrosis of the femoral
head: Etiology, imaging and treatment. Eur J Radiol. 63:16–28.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu YF, Chen WM, Lin YF, Yang RC, Lin MW,
Li LH, Chang YH, Jou YS, Lin PY, Su JS, et al: Type II collagen
gene variants and inherited osteonecrosis of the femoral head. N
Engl J Med. 352:2294–2301. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sakamoto Y, Yamamoto T, Miyake N,
Matsumoto N, Iida A and Nakashima Y: Research Committee on
Idiopathic Osteonecrosis of the Femoral Head of the Ministry of
Health, Labour and Welfare of Japan, Iwamoto Y and Ikegawa S:
Screening of the COL2A1 mutation in idiopathic osteonecrosis of the
femoral head. J Orthop Res. 35:768–774. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zalavras CG and Lieberman JR:
Osteonecrosis of the femoral head: Evaluation and treatment. J Am
Acad Orthop Surg. 22:455–464. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moya-Angeler J, Gianakos AL, Villa JC, Ni
A and Lane JM: Current concepts on osteonecrosis of the femoral
head. World J Orthop. 6:590–601. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Glueck CJ, Glueck HI, Welch M, Freiberg R,
Tracy T, Hamer T and Stroop D: Familial idiopathic osteonecrosis
mediated by familial hypofibrinolysis with high levels of
plasminogen activator inhibitor. Thromb Haemost. 71:195–198.
1994.PubMed/NCBI
|
|
7
|
Nobillot R, Le Parc JM, Benoit J and
Paolaggi JB: Idiopathic osteonecrosis of the hip in twins. Ann
Rheum Dis. 53:7021994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li N, Yu J, Cao X, Wu QY, Li WW, Li TF,
Zhang C, Cui YX, Li XJ, Yin ZM and Xia XY: A Novel p. Gly630Ser
mutation of COL2A1 in a Chinese family with presentations of
Legg-Calvé-Perthes disease or avascular necrosis of the femoral
head. PloS One. 9:e1005052014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen WM, Liu YF, Lin MW, Chen IC, Lin PY,
Lin GL, Jou YS, Lin YT, Fann CS, Wu JY, et al: Autosomal dominant
avascular necrosis of femoral head in two Taiwanese pedigrees and
linkage to chromosome 12q13. Am J Hum Genet. 75:310–317. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mow VC GW and Chen FH: Structure and
function of articular cartilage and meniscusLippincott Williams and
Wilkins. Philadelphia, PA: pp. 181–258. 2005
|
|
11
|
Eyre D: Collagen of articular cartilage.
Arthritis Res. 4:30–35. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arnold WV and Fertala A: Skeletal diseases
caused by mutations that affect collagen structure and function.
Int J Biochem Cell Biol. 45:1556–1567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gelse K, Pöschl E and Aigner T:
Collagens-structure, function, and biosynthesis. Adv Drug Deliv
Rev. 55:1531–1546. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Al-Omran AK and Sadat-Ali M:
Legg-Calvé-Perthes disease in two generations of male family
members: A case report. J Orthop Surg (Hong Kong). 21:258–261.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kannu P, Irving M, Aftimos S and
Savarirayan R: Two novel COL2A1 mutations associated with a
Legg-Calvé-Perthes disease-like presentation. Clin Orthop Relat
Res. 469:1785–1790. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Su P, Li R, Liu S, Zhou Y, Wang X, Patil
N, Mow CS, Mason JC, Huang D and Wang Y: Age at onset-dependent
presentations of premature hip osteoarthritis, avascular necrosis
of the femoral head, or Legg-Calvé-Perthes disease in a single
family, consequent upon a p. Gly1170Ser mutation of COL2A1.
Arthritis Rheum. 58:1701–1706. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su P, Zhang L, Peng Y, Liang A, Du K and
Huang D: A histological and ultrastructural study of femoral head
cartilage in a new type II collagenopathy. Int Orthop.
34:1333–1339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kannu P O, Rielly DD, Hyland JC and Kokko
LA: Avascular necrosis of the femoral head due to a novel C
propeptide mutation in COL2A1. Am J Med Genet A. 155A:1759–1762.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kishiya M, Nakamura Y, Ohishi H, Furukawa
K and Ishibashi Y: Identification of a novel COL2A1 mutation
(c.1744G>A) in a Japanese family: A case report. J Med Case Rep.
8:2762014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L, Pan H and Zhu ZA: A genetic
pedigree analysis to identify gene mutations involved in femoral
head necrosis. Mol Med Rep. 10:1835–1838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ficat RP: Idiopathic bone necrosis of the
femoral head. Early diagnosis and treatment. J Bone Joint Surg Br.
67:3–9. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Robin NH, Moran RT and Ala-Kokko L: Homo
sapiens collagen type II alpha 1 chain (COL2A1), RefSeqGene on
chromosome 12GeneReviews((R)). Adam MP, Ardinger HH, Pagon RA, et
al: University of Washington; Seattle, WA: pp. 249–252. 1993
|
|
23
|
Higuchi Y, Hasegawa K, Yamashita M, Tanaka
H and Tsukahara H: A novel mutation in the COL2A1 gene in a patient
with Stickler syndrome type 1: A case report and review of the
literature. J Med Case Rep. 11:2372017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang X, Deng X, Xu H, Wu S, Yuan L, Yang
Z, Yang Y and Deng H: Identification of a novel mutation in the
COL2A1 gene in a Chinese family with spondyloepiphyseal dysplasia
congenita. PloS One. 10:e01275292015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Miyamoto Y, Matsuda T, Kitoh H, Haga N,
Ohashi H, Nishimura G and Ikegawa S: A recurrent mutation in type
II collagen gene causes Legg-Calve-Perthes disease in a Japanese
family. Hum Genet. 121:625–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spranger J, Winterpacht A and Zabel B: The
type II collagenopathies: A spectrum of chondrodysplasias. Eur J
Pediatr. 153:56–65. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kannu P, Bateman JF, Randle S, Cowie S, du
Sart D, McGrath S, Edwards M and Savarirayan R: Premature arthritis
is a distinct type II collagen phenotype. Arthritis Rheum.
62:1421–1430. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yoshida M: Experimental avascular necrosis
of the femoral head-a microangiographic and histological study.
Nihon Seikeigeka Gakkai Zasshi. 65:56–69. 1991.(In Japanese).
PubMed/NCBI
|
|
29
|
Mont MA, Zywiel MG, Marker DR, Mcgrath MS
and Delanois RE: The natural history of untreated asymptomatic
osteonecrosis of the femoral head: A systematic literature review.
J Bone Joint Surg Am. 92:2165–2170. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Catto M: A histological study of avascular
necrosis of the femoral head after transcervical fracture. J Bone
Joint Surg Br. 47:749–776. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bonaventure J, Cohen-Solal L, Ritvaniemi
P, Van Maldergem L, Kadhom N, Delezoide AL, Maroteaux P, Prockop DJ
and Ala-Kokko L: Substitution of aspartic acid for glycine at
position 310 in type II collagen produces achondrogenesis II, and
substitution of serine at position 805 produces hypochondrogenesis:
Analysis of genotype-phenotype relationships. Biochem J.
307:823–830. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ritvaniemi P, Körkkö J, Bonaventure J,
Vikkula M, Hyland J, Paassilta P, Kaitila I, Kääriäinen H, Sokolov
BP and Hakala M: Identification of COL2A1 gene mutations in
patients with chondrodysplasias and familial osteoarthritis.
Arthritis Rheum. 38:999–1004. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tiller GE, Polumbo PA, Weis MA, Bogaert R,
Lachman RS, Cohn DH, Rimoin DL and Eyre DR: Dominant mutations in
the type II collagen gene, COL2A1, produce spondyloepimetaphyseal
dysplasia, Strudwick type. Nat Genet. 11:87–89. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tiller GE, Weis MA, Polumbo PA, Gruber HE,
Rimoin DL, Cohn DH and Eyre DR: An RNA-splicing mutation (G+5IVS20)
in the type II collagen gene (COL2A1) in a family with
spondyloepiphyseal dysplasia congenita. Am J Hum Genet. 56:388–395.
1995.PubMed/NCBI
|
|
35
|
Mortier GR, Weis MA, Nuytinck L, King LM,
Wilkin DJ, De Paepe A, Lachman RS, Rimoin DL, Eyre DR and Cohn DH:
Report of five novel and one recurrent COL2A1 mutations with
analysis of genotype-phenotype correlation in patients with a
lethal type II collagen disorder. J Med Genet. 37:263–271. 2000.
View Article : Google Scholar : PubMed/NCBI
|