Introduction
Vascular dementia (VaD) is the second most prevalent
form of dementia, after Alzheimer's disease (AD), and accounts for
~20% of all dementia cases (1).
According to world prevalence data, the number of individuals
affected by dementia is expected to increase to 70 million by 2030
(2). VaD is characterized by
hypoxia, oxidative stress, and inflammation, which may lead to
symptoms of mood disorders and difficulties with problem solving,
memory, thinking, reasoning, and executive functions such as
judgment (3–5). Moreover, the white matter, basal
ganglia, and hippocampus are vulnerable to damage, and disruption
of the blood-brain barrier subsequently leads to memory impairment
(6–8). Although numerous studies on the
pathophysiology of VaD have been conducted, the pathological
mechanisms behind this disorder have yet to be fully understood
(3,8–10).
Studies into the genetic basis of dementia have been conducted
since 1993, when it was reported that apolipoprotein E
(APOE) 4 was a risk factor for AD (11).
Previous genetics studies on VaD have identified
several single-nucleotide polymorphisms (SNPs), such as
methylenetetrahydrofolate 677C/T (rs1801133), paraoxonase 1 L55M
(rs854560), transforming growth factor-β1 +29C/T (rs1800470), and
tumor necrosis factor-α −850C/T (rs1799724) (12). Although SNPs may be crucial to
verifying the pathogenesis of the disease, VaD is more affected by
lifestyle-associated risk factors, such as age, diabetes,
hypertension, and the metabolic syndrome, compared with AD
(8). Therefore,
epigenetic-associated changes in the expression of a gene (that is,
without alterations in the DNA sequence) may present a more
suitable approach for studying VaD (13).
Epigenetic changes include DNA methylation, histone
modification, chromatin remodeling, and microRNA regulation
(14). DNA methylation, one of the
most widely studied epigenetic changes that regulate many cellular
processes (15), is mediated by
DNA methyltransferases during early development and throughout the
lifespan of an organism (16).
Methyl group additions to the cytosine residue that precedes a
guanine, the so-called cytosine-guanine (CpG) dinucleotides, have
been reported to significantly modify gene expression (17,18).
Previous studies have concentrated mainly on how the position of
the methylation in the transcriptional unit affects its association
to gene expression. The majority of these studies focused on CpG
islands (CGIs), which are dense repetitions of CpG nucleotides
located mainly in the gene promoter region and are known to repress
long-term gene expression (13,19,20).
The methyl-CpG-binding domain proteins function by binding to the
promoter region of the transcription site and blocking gene
expression (21).
To the best of the authors' knowledge, there are
currently no studies that used the epigenetic approach of VaD. The
present study may be meaningful as an initial study on the
genome-wide DNA methylation differences in VaD. The aim of the
present study was to investigate the differences in DNA methylation
in the hippocampus, which serves an important role in VaD
pathogenesis.
Materials and methods
Animal preparation
Male Wistar rats (n=4; weight, 80±10 g; age 4 weeks;
Orient Bio, Seongnam, Korea) were used in this experiment. Animals
were housed in a controlled environment at 25±2°C, 50–55% humidity,
and 12-h light/dark cycle, with ad libitum access to food
and water. The rats were randomly divided into two groups: The
sham-operated animals (Sham group; n=2) and BCCAO-operated animals
(VaD group; n=2). The BCCAO or sham surgery was performed when the
rats were 12 weeks old, and the animals were sacrificed 6 weeks
post-surgery. The experimental procedures were performed in
accordance with the animal care guidelines of The National
Institutes of Health (Bethesda, MD, USA), and all animal
experiments were approved by The Committee for the Care and Use of
Laboratory Animals at Kyung Hee University [Seoul, Korea; approval
no. KHUASP(SE)-15-082].
BCCAO model establishment
The BCCAO model is an animal model of permanent VaD;
it is established through the permanent ligation of the common
carotid artery to induce chronic cerebral hypoperfusion, which
leads to damage to the white matter, disruption of the blood-brain
barrier, nerve injury of the cerebral cortex and hippocampus,
oxidative stress, and inflammation (22). Rats were anesthetized with 50 mg/kg
body weight Zoletil 50 (Virbac Laboratories, Carros, France); body
temperature was maintained at 36.5–37.5°C using a heating pad.
Following a midline incision to expose the common carotid arteries,
the vessel ligations were carefully performed to avoid damage to
the vagus nerve. Each carotid artery was double ligated with 3-0
silk sutures (Ailee, Seoul, Korea). Similar surgical procedures
were performed on the Sham-operated animals, except for the vessel
ligation step. To prevent postoperative pain and infection, the
animals were maintained in a warm and clean environment during a
three day recovery period.
Radial arm maze test
A behavioral study was conducted to determine
whether the VaD model had been induced by the BCCAO surgery. The
radial arm maze test was conceived by Olton and Samuelson in 1976
to measure spatial working memory in rats (23). The radial arm maze apparatus
comprises eight arms (length, 50 cm; width, 10 cm), spaced
equidistantly. The apparatus was placed 1 m above the floor, and a
small water bowl was located at the end of each arm. The rats were
deprived of water for 48 h and subsequently moved to the room where
the radial arm maze was located and allowed acclimate for 1 h to
prevent bias caused by anxiety. Rats were placed into the apparatus
and were allowed to move freely for 8 min and to drink water. Three
observers that were independent of the present study were recruited
to count the number of errors in revisitation of the same arm each
time and to calculate the data. If their spatial working memory
were perfect, the rats would explore a novel arm each time to find
water.
DNA extraction and methylation
profiling
To explore the DMGs, genomic DNA was extracted from
10 mg hippocampal tissue using the DNeasy Blood & Tissue kit
(Qiagen, Inc., Valencia, CA, USA), according to the manufacturer's
protocol.
In the present study, the MBD-seq method was used,
owing to its low cost, high specificity, and efficiency (24–26).
Genomic DNA (1.5 µg) was sent to the Theragen Etex Bio Institute
(Suwon, Korea) for the generation of recombinant methyl-binding
domain protein to enrich 5-methylcytosine-modified regions of the
genome for subsequent massively parallel sequencing analysis.
Fragmentation of the genomic DNA was achieved with a Covaris S220
ultrasonicator (duty cycle, 10%; intensity, 5; 200 cycles per burst
for 180 sec; Covaris, Woburn, MA, USA) to obtain sequences with an
average length of 200 bp. Methylated fragments were captured using
the MethylCap kit (Diagenode Inc., Denville, NJ, USA), according to
the manufacturer's protocol. The captured DNA fragments were
purified using QIAquick PCR purification columns (Qiagen, Inc.),
and the purified DNA was used for library preparation.
Quantification of the captured DNA was performed with the Quant-iT
PicoGreen dsDNA Assay kit (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), and 20 ng of chromatin immunoprecipitation
(ChIP)-enriched, fragmented input DNA was used in the TruSeq ChIP
Library Preparation kit (Illumina, Inc., San Diego, CA, USA),
according to the manufacturer's protocol. The library was examined
with a bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA,
USA) and quantified. The denatured and amplified libraries were
loaded onto Illumina NextSeq 500 using the High Output v2 kit (1×72
cycles, 2 index reads; Illumina, Inc.).
Bisulfite genomic sequencing
The aforementioned genomic DNA (500 ng) from each
hippocampal sample was treated by bisulfite conversion with the
EZ-96 DNA Methylation kit (Zymo Research Corp., Irvine, CA, USA),
according to the manufacturer's protocol. To determine the
methylation patterns in the genes, the following primers were used:
Vascular endothelial growth factor A (VEGFA)-1, forward
5′-AGGTGAGGTTTGAGTTTTTTATTTA-3′, reverse
5′-CTAAACCATCAAACACCCAAAAA-3′; VEGFA-2, forward
5′-TTGTAGGGTTTTATTTTGTTATTAGG-3, reverse
5′-AAATACAAATATCCACTACACCCTC-3′; kinase insert domain receptor
(KDR)-1, forward 5′-GAAGTTTTTTTAAGTGGTTTATTTTGTT-3′, reverse
5′-AAAAACTTTTCAAAATCCAAATTCA-3′; and KDR-2,
5′-GGATTTTGAAAAGTTTTTTGGGTT-3′, reverse
5′-ATATAACTTTCTTTCCATTCCTTCCA-3. Both genes were analyzed by using
two primers divided into 300–400 bp for accurate analysis. PCR
amplification of these regions was achieved by using a PCR KOD FX
kit (Toyobo Life Science, Osaka, Japan). Amplification was carried
out with the following thermocycling conditions: 95°C for 4 min; 35
cycles of 95°C for 30 sec, 55°C for 30 sec, 72°C for 30 sec and
final extension at 72°C for 7 min. PCR products were purified using
a QIAquick PCR purification kit (Qiagen, Inc.) and quantified using
Quant-iT™ PicoGreen™ dsDNA Assay kit (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. PCR
product sizes were confirmed through agarose gel electrophoresis.
Polymerase chain reaction (PCR) products were subsequently
processed to produce a DNA sequencing library using the TruSeq Nano
DNA LT kit (Illumina Inc.), according to the manufacturer's
protocol. For each library, an amplified PCR product size of
approximately 450 bp was determined with a bioanalyzer using the
Agilent DNA 1000 ChIP kit, and library quantification was performed
on a CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories
Inc., Hercules, CA, USA). Serial sequencing of each library was
conducted using Illumina MiSeq, and the generation of clusters of
DNA libraries in flow cells and of 250 bp paired-end reads (2×250)
were performed using the MiSeq 500 Cycle v2 kit (Illumina, Inc.).
The raw image data were transformed into sequence data by base
calling and stored in the FASTQ format.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was performed using the T100 thermal cycler
and CFX-96 real-time PCR detection systems (Bio-Rad Laboratories,
Inc.). RNA was extracted from hippocampal whole tissue (10 mg)
using the RNeasy mini kit (Qiagen, Inc.). cDNA synthesis using the
ReverTra Ace-α kit (Toyobo Life Sciences) according to the
manufacturer's protocol. PCR was performed with the Thunderbird
SYBR qPCR Mix (Toyobo Life Science); all cDNA samples were diluted
1:10. The PCR mixture (20 µl) contained cDNA (2 ng/µl), 1X SYBR
Green qPCR mix, and 250 µM of each primer set. The following
primers were used: VEGFA forward, 5′-CCTCCTCCTCCTGGGAAC-3′ and
reverse, 5′-AGGGTAAGCCACTCACACA-3′; KDR forward,
5′-GCAGCCAAGTCCGAATCC-3′ and reverse, 5′-TCCCGCATCTCTTTCACTCA-3′;
GAPDH forward, 5′-CTCCCATTCTTCCACCTTTGAT-3′ and reverse,
5′-CACCACCCTGTTGCTGTAG-3. Thermocycling conditions were as follows:
Initial denaturation at 95°C for 1 min, followed by a single cycle
of denaturation at 95°C for 10 sec, annealing at 58°C for 10 sec,
and extension at 72°C for 20 sec. Data were analyzed with CFX
Manager v1.5 software (Bio-Rad Laboratories, Inc.) and the relative
expression of genes were quantified using the 2−ΔΔCq
method (27).
Data analysis
MBD-seq reads were mapped with Bowtie v1.0.0
(www.bowtie-bio.sourceforge.net). Genome-wide and
meta-gene profile analyses were performed using in-house PERL and R
scripts. Highly methylated regions (HMRs) were called with MACS
v1.4.2 (www.liulab.dfci.harvard.edu/MACS; P<0.01), and
differentially methylated regions (DMRs) were analyzed using an
in-house PERL script. DMGs were selected by in-house scripts, and
were defined with at least 1 DMR between a region located 2 kbp
upstream and 500 bp downstream from the transcription start site.
Gene Ontology (GO) and functional annotation analysis for
significant genes were performed on the DAVID Bioinformatics
Resource v6.8 server (david.abcc.ncifcrf.gov) (28). To identify potential functional
pathways, protein-protein interaction (PPI) network analysis was
conducted using the STRING v10.0 database (www.string-db.org). Putative binding sites at the
promoter regions of VEGFA and KDR were also analyzed.
Transcription factor binding sites were predicted by AliBaba2.1
using the TRANSFAC 4.0 transcription factor database (www.gene-regulation.com/pub/programs/alibaba2/index.html).
Statistical analysis
Statistical analyses were performed using SPSS v22.0
(IBM Corp., Armonk, NY, USA). All data are presented as the mean ±
standard error of the mean. All experiments were repeated at least
three times. Statistical comparisons between groups were processed
with the unpaired Student t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Confirmation of cognitive impairment
induced by BCCAO
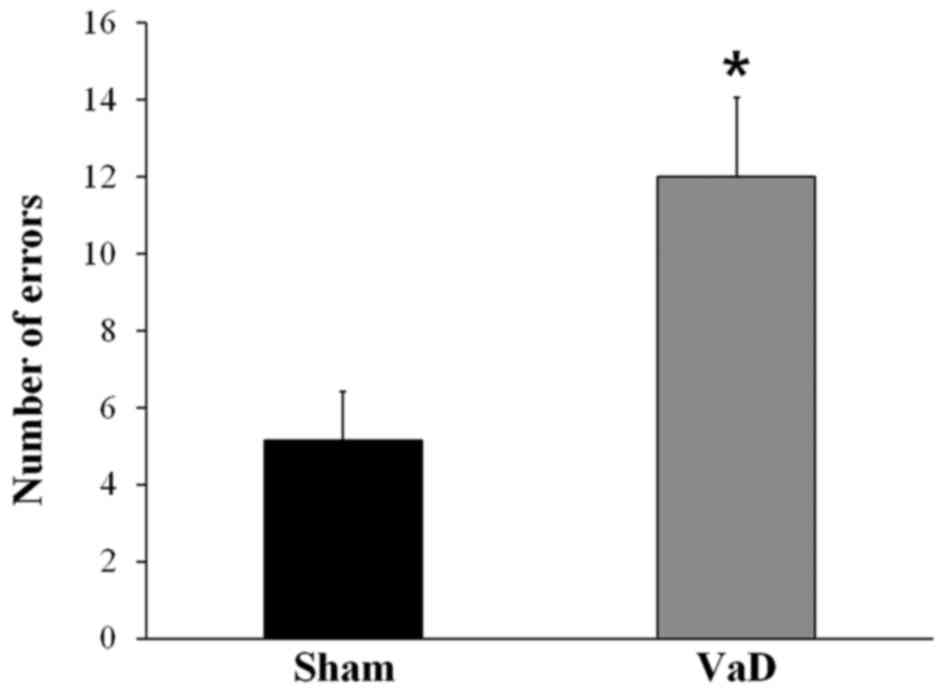
Alternation behavior in the radial arm apparatus was
recorded to assess the spatial working memory performance. Rats in
the VaD group had a significantly higher number of errors
(12.00±2.07) in the radial arm maze test compare with rats in the
Sham group (5.14±1.28; P<0.05; Fig.
1). These results confirmed that cognitive impairment had been
induced by BCCAO.
Identification of DMRs
The aim of the present study was to uncover the DNA
methylation changes associated with cognitive impairment by
comparing the gene methylated regions in an animal model of VaD. To
investigate the difference in DNA methylation between the Sham and
VaD groups, DMRs were analyzed in the promoter regions that may
affect the regulation of mRNA expression. Promoter regions were
defined as being between 2 kbp upstream and 500 bp downstream from
the transcription start site. DMRs were identified by HMR peaks
with a fold enrichment ratio of >2.0 between Sham and VaD
samples, or if a HMR peak occurred in only one sample.
Of the 112,665 reliable HMRs detected, a total of
95,238 DMRs were identified, of which 1,250 (1.3%) DMRs were
specifically located in the promoter region. Of the 27,030
hypermethylated DMRs detected, 397 (1.5%) were associated with the
promoter, and of the 68,208 hypomethylated DMRs detected, 853
(1.3%) were associated with the promoter.
Functional annotation of DMGs
A total of 1,180 DMGs were identified based on the
detected promoter region DMRs, of which 384 (32.5%) were
hypermethylated and 796 (67.5%) were hypomethylated. There were
fewer hypermethylated DMGs identified in the VaD group compared
with the Sham group, with fold enrichment ratios ranging from 20 to
2,388 (log2 ratios of 1.00–11.22). GO analysis of the DMG sets was
performed using DAVID to verify the functions associated with the
genes.
The top 10 GO biological processes for the 1,180
genes are listed in Table I. The
10 functional categories of GO biological processes: Regulation of
nucleotide biosynthetic process; regulation of cyclic (c)AMP
biosynthetic process; multicellular organism reproduction;
regulation of lyase activity; cAMP-mediated signaling; regulation
of adenylate cyclase activity; sexual reproduction; G-protein
signaling, coupled to cyclic nucleotide second messenger; antigen
receptor-mediated signaling pathway; and spermatogenesis. STRING
identified interactions among proteins coded by 72 genes. PPI
network analysis demonstrated that seven DMGs were closely
connected, including cluster of differentiation 247,
ζ-chain-associated protein kinase 70, protein tyrosine phosphatase
receptor type C, lymphocyte-specific protein tyrosine kinase, KDR,
epidermal growth factor receptor, and VEGFA (Fig. 2). Among these, VEGFA and
KDR were selected as candidate genes for VaD for further
validation studies, due to their high connectivity in the PPI
network and association with VaD.
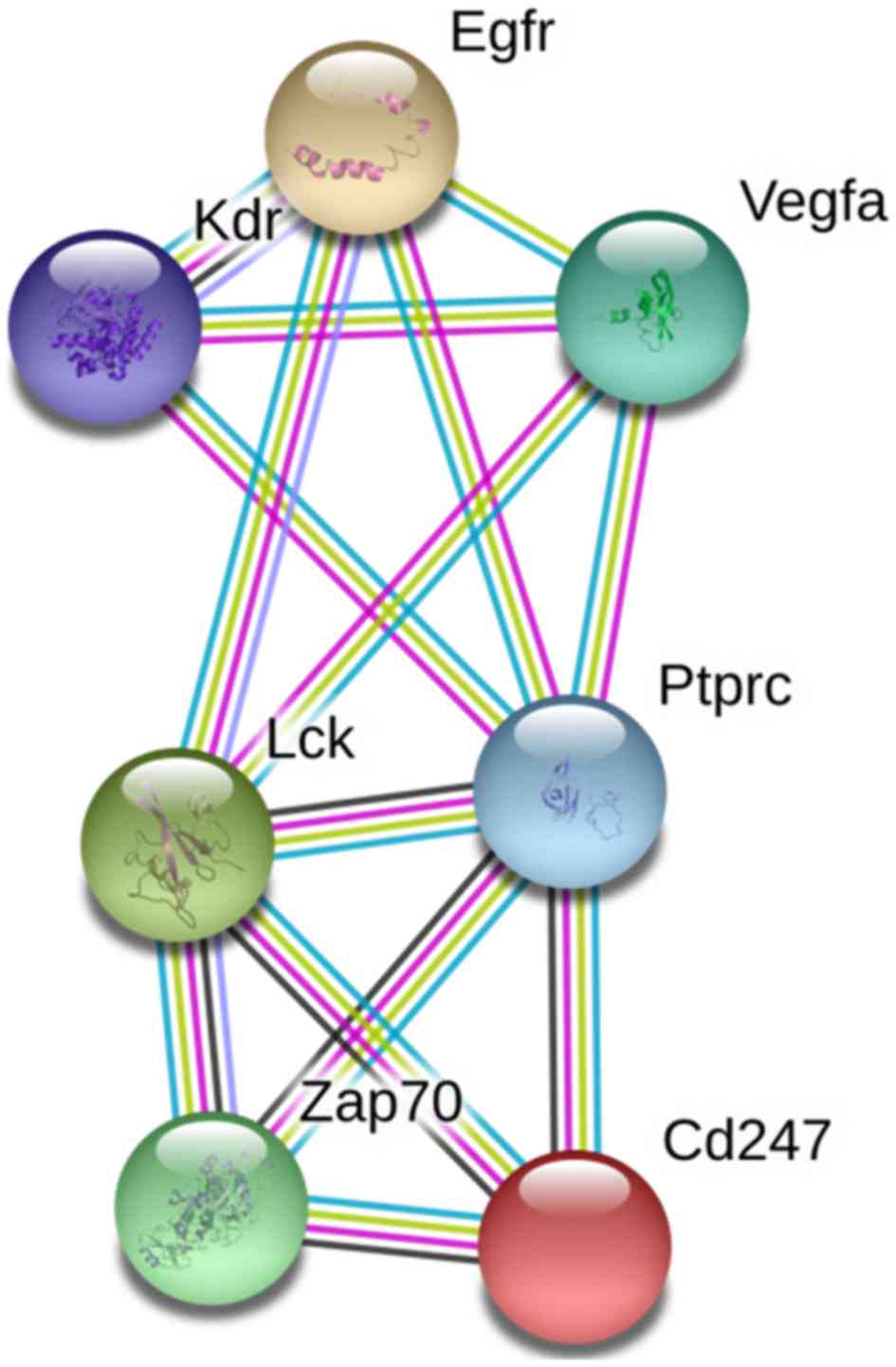 | Figure 2.PPI network analysis. PPI network
analysis was conducted using STRING, and seven genes were
demonstrated to be well-connected in the VaD group. PPI,
protein-protein interaction; cAMP, cyclic adenosine monophosphate;
Egfr, epidermal growth factor receptor; Kdr, kinase insert domain
receptor; Lck, lymphocyte-specific protein tyrosine kinase; Ptprc,
protein tyrosine phosphatase receptor type C; VaD, vascular
dementia; VEGFA, vascular endothelial growth factor A; Zap,
ζ-chain-associated protein kinase. |
 | Table I.Top 10 GO biological processes for
1,180 differentially methylated genes. |
Table I.
Top 10 GO biological processes for
1,180 differentially methylated genes.
| GO term | Count | Fold
enrichment | P-value | FDR | Genes |
|---|
| Regulation of
nucleotide biosynthetic process | 18 | 3.37 | <0.001 | <0.01 | ACR, ADCY3,
GNAZ, AVP, LHCGR, ABCA1, SSTR5, S1PR3, ADRB2, HRH3, HTR7, GIPR,
GNAS, NOS3, ADM2, GUCA2B, GLP1R, GNG7 |
| Regulation of cAMP
biosynthetic process | 16 | 3.34 | <0.001 | <0.01 | ACR, ADCY3,
GNAZ, AVP, LHCGR, ABCA1, SSTR5, S1PR3, ADRB2, HRH3, HTR7, GIPR,
GNAS, ADM2, GLP1R, GNG7 |
| Multicellular
organism reproduction | 45 | 1.84 | <0.001 | <0.01 | ACR, NANOS3,
RAD51C, PCDHA8, H1FNT, SPATA20, FKBP6, LHCGR, PCDHGA9, ADAD1, MAEL,
HP, BCL2L1, SOHLH1, SOHLH2, KRT9, VDR, WNT4, POU5F2, NOS3, DAZL,
TGM4, FAS, PIWIL4, DND1, SPATA3, B4GALNT1, PCDHGA12, EGFR, B4GALT1,
AVP, PCDHGA11, MAK, TNP1, CELSR2, THEG, KDR, APRT, LEP, REC8, ESR2,
TXNDC2, P2RX1, TXNDC8, VEGFA |
| Regulation of lyase
activity | 15 | 3.28 | <0.001 | <0.01 | ACR, ADCY3,
GNAZ, LHCGR, SSTR5, S1PR3, ADRB2, HRH3, HTR7, GIPR, GNAS, NOS3,
ADM2, GLP1R, GNG7 |
| cAMP-mediated
signaling | 15 | 3.13 | <0.001 | <0.01 | ADCY3, GNAZ,
LHCGR, RIMS2, LEP, SSTR5, S1PR3, ADRB2, HTR7, GIPR, GNAS, ADM2,
GLP2R, MC3R, GLP1R |
| Regulation of
adenylate cyclase activity | 14 | 3.26 | <0.001 | <0.01 | ACR, ADCY3,
GNAZ, LHCGR, SSTR5, S1PR3, ADRB2, HRH3, HTR7, GIPR, GNAS, ADM2,
GLP1R, GNG7 |
| Sexual
reproduction | 38 | 1.85 | <0.001 | <0.01 | ACR, NANOS3,
RAD51C, PCDHA8, H1FNT, SPATA20, FKBP6, PCDHGA9, ADAD1, MAEL, HP,
BCL2L1, SYCP2, SOHLH1, SOHLH2, KRT9, WNT4, POU5F2, CD4, NOS3, DAZL,
FAS, PIWIL4, DND1, SPATA3, B4GALNT1, PCDHGA12, B4GALT1, PCDHGA11,
MAK, TNP1, CELSR2, THEG, LEP, REC8, TXNDC2, ADAM1A, TXNDC8 |
| G-protein
signaling, coupled to cyclic nucleotide second messenger | 14 | 3.18 | <0.001 | <0.01 | ADCY3, GNAZ,
LHCGR, SSTR5, S1PR3, ADRB2, HRH1, HTR7, GIPR, GNAS, ADM2, GLP2R,
MC3R, GLP1R |
| Antigen
receptor-mediated signaling pathway | 9 | 4.67 | <0.001 | <0.01 | PTPRC, LAT2,
CD19, LAX1, CD247, LCK, ZAP70, CACNB4, SKAP1 |
|
Spermatogenesis | 28 | 2.05 | <0.001 | <0.01 | NANOS3, RAD51C,
H1FNT, SPATA20, PCDHA8, FKBP6, ADAD1, PCDHGA9, MAEL, HP, BCL2L1,
SOHLH1, SOHLH2, KRT9, POU5F2, FAS, SPATA3, PIWIL4, B4GALNT1,
PCDHGA12, PCDHGA11, MAK, TNP1, CELSR2, THEG, REC8, TXNDC2,
TXNDC8 |
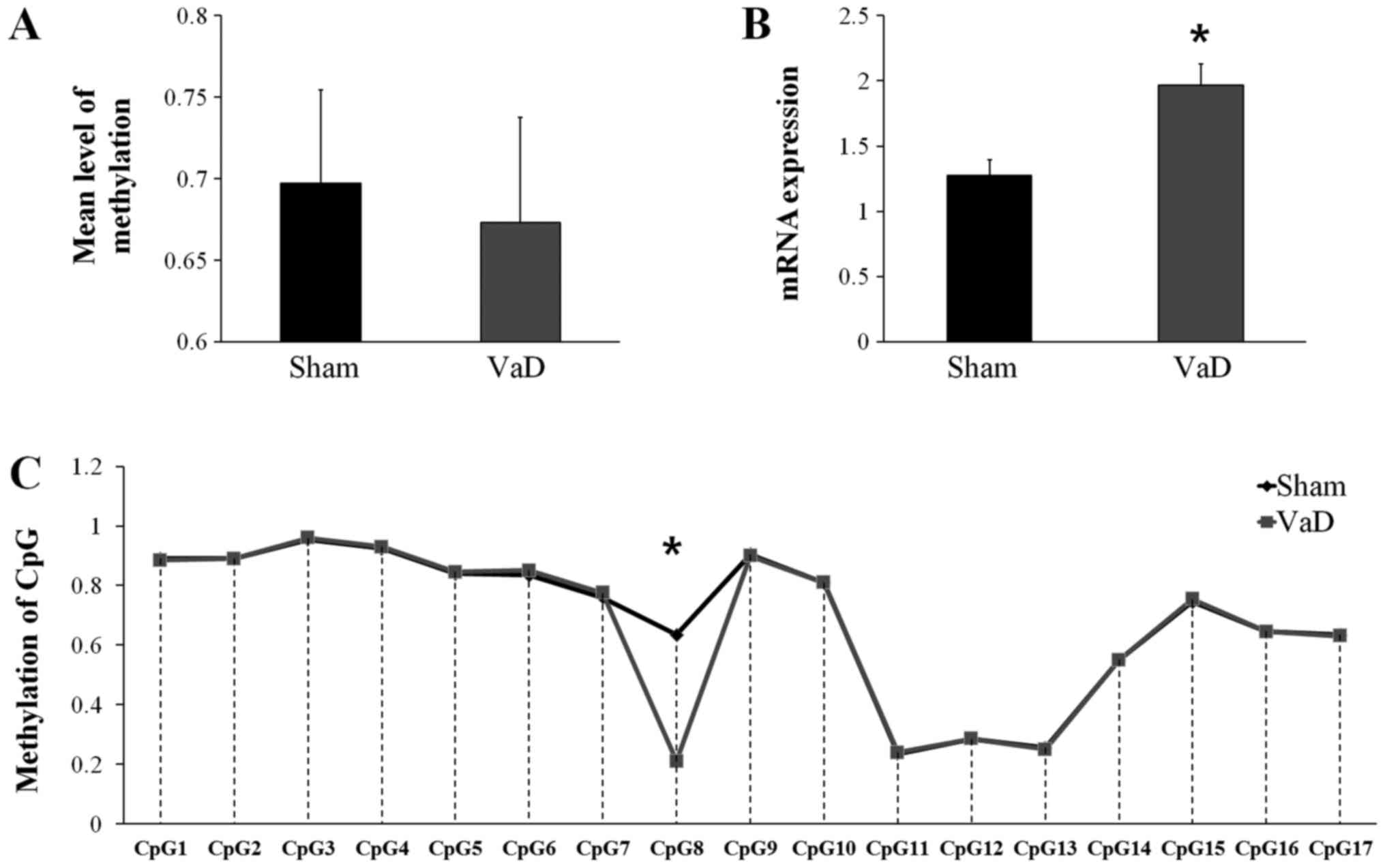
Validation of VEGFA methylation and
relative mRNA expression levels
Bisulfite sequencing was used to confirm the
differences in promoter region methylation of VEGFA.
Although the mean CGI methylation status of VEGFA was
slightly higher in the Sham group (0.6971±0.0575) compare with the
VaD group (0.6729±0.0647; Fig.
3A), the difference was not statistically significant. However,
VEGFA mRNA expression was significantly higher in the VaD
group (1.9647±0.2804) compared with expression in the Sham group
(1.2759±0.2111; P>0.05; Fig.
3B), and the methylation status in CpG8 was significantly lower
in the VaD group compared with the Sham group (Fig. 3C). CpG8 is the eighth CGI in the
promoter region of VEGFA, 544 bp away from the transcription start
site, in the forward strand.
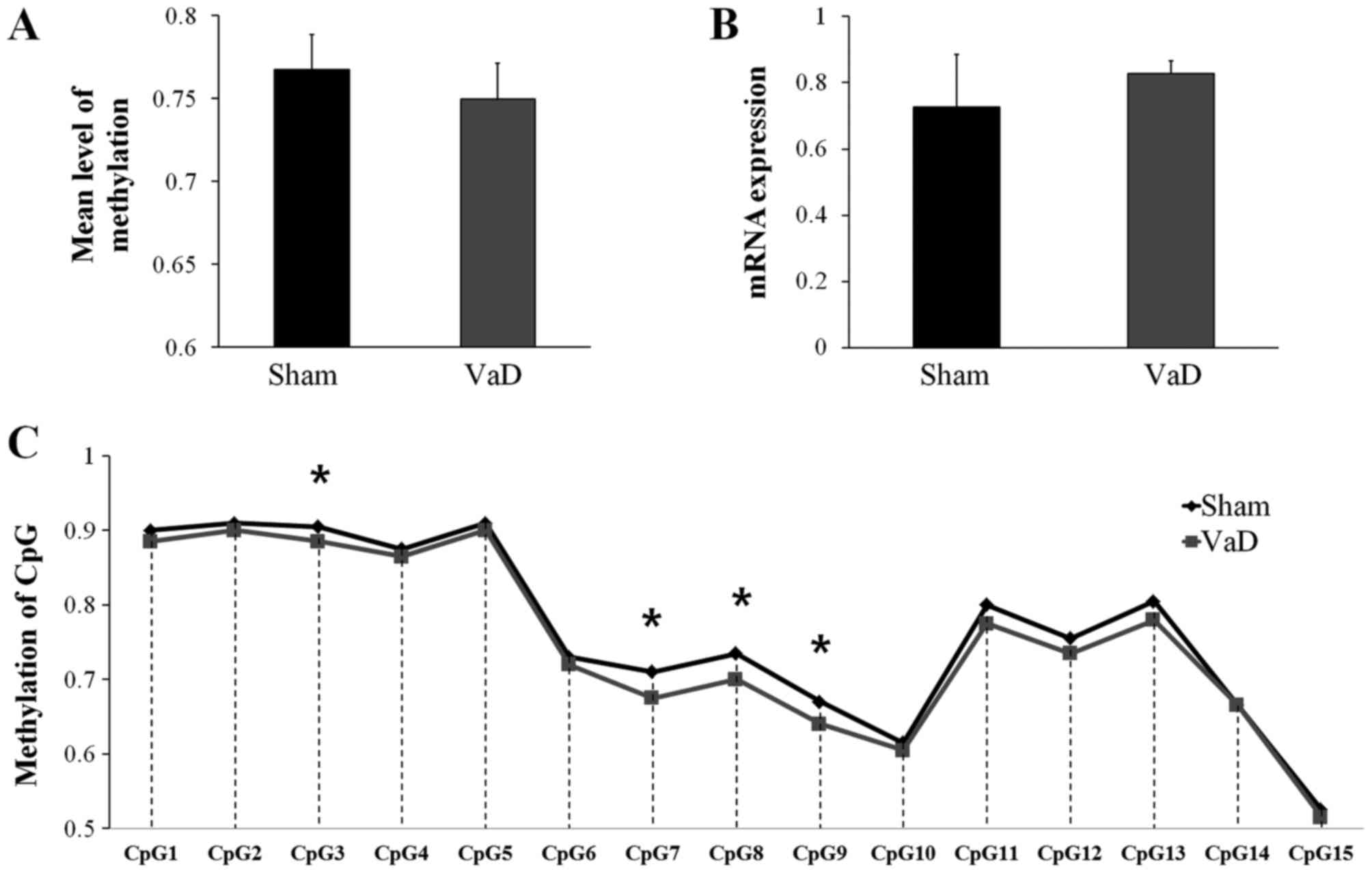
Validation of KDR methylation and
relative mRNA expression
Although the mean methylation level of the CGIs in
KDR promoter region was higher in the Sham group
(0.7673±0.1177) than in the VaD group (0.7497±0.1185), the
difference was not statistically significant (Fig. 4A). KDR mRNA expression
levels in the Sham group (0.7275±0.1568) were lower compared with
expression in the VaD group (0.8276±0.0383), but the difference was
not statistically significant (Fig.
4B). However, significant differences were identified in the
methylation status of four CGIs (CpG3, CpG7, CpG8, and CpG9) in the
promoter region on the forward strand between the two groups
(Fig. 4C).
Predicted transcription factor binding
analysis
To further examine the regulatory roles of
site-specific DNA methylation in the promoter region of
VEGFA and KDR, predicted transcription factor
analysis was performed by AliBaba2.1 programs using the TRANSFAC
4.0 transcription factor database. CpG8 in VEGFA was
predicted to be a binding site for serum response factor (SRF).
Putative transcription factor binding sites for CpGs of KDR
were as follows: SRF in CpG3, activator protein (AP)-4 in CpG7 and
CpG8, and immunoglobulin transcription factor (ITF) 2 in CpG9 (data
not shown).
Discussion
The present study investigated DMGs in the
hippocampus between Sham and VaD model animals, as well as their
functional categories, to identify candidate genes that may be
associated with cognitive function. To verify the successful
establishment of the BCCAO-induced VaD model rat model, cognitive
impairment was confirmed by the commonly used radial arm maze test
(29,30).
Alterations of DNA methylation in the promoter
region are a way to regulate gene expression (31,32).
It has been suggested that specific diseases, including
neurodegenerative disease, may occur by characterized
hypermethylated and hypomethylated CpGs (33). Gene expression may be regulated by
promoter-site CGI methylation, where hypermethylation of the CpGs
suppresses gene transcription, and hypomethylation of the promoter
region allows transcription factor binding and activation of gene
transcription (34,35). A previous methylation analysis
using 122 cortical tissues from patients with AD revealed that the
Ankyrin 1 gene was hypermethylated in the superior temporal gyrus
and prefrontal cortex (36). In
another study using 708 autopsied brains from individuals with AD,
11 DMRs were identified, which were associated with several genes,
including ankyrin-1, cadherin-23, disco-interacting protein homolog
A, inactive rhomboid protein 2, 60S ribosomal protein L13 and E3
ubiquitin-protein ligase RNF34 (37). In the present study, 72 DMGs were
identified and annotated to 10 categories of GO biological
processes and may be associated with VaD. Through PPI network
analysis, seven genes were classified as closely interacting and
may be related to the VEGF signaling pathway.
VEGFA and KDR are involved in the VEGF
signaling pathway (38–40). In the present study, rats in the
VaD group exhibited relative hypomethylation, although this was not
significant. However, VEGFA mRNA expression increased in the
hippocampus, compared with rats in the Sham group. A number of
putative transcription factors that bind to the hypomethylated
sites were analyzed in the present study, and these may contribute
to increased VEGFA expression. VEGFA is a crucial regulator
of angiogenesis, skeletal growth, and ovarian angiogenesis
(41). In the present study, CpG8
in the VEGFA promoter was confirmed to be hypomethylated in
the VaD group and was predicted to be a binding site for the
transcription factor SRF. SRF is a downstream mediator of VEGF
signaling pathway and serves a crucial role for VEGF-induced
angiogenesis (42,43). KDR is a VEGF receptor and is
crucial for endothelial cell proliferation, survival, and
migration, as well as neuroprotection or improved neurovascular
coupling (41,44,45).
In the present study, putative transcription factors, such as SRF,
Ap-4, and ITF-2, were investigated if they were predicted bind to
hypomethylated CpG in KDR, but there were few studies on the
association between these transcription factors and KDR.
Consistent with previous studies, upregulated VEGFA mRNA
expression in the VaD model rat was confirmed; this may serve an
important role in compensating for cellular damage through
neurobehavioral recovery and neurovascular remodeling after hypoxic
brain injury (46,47). It has also been demonstrated that
increased expression of VEGFA under hypoxic conditions and
increased VEGF-promoted angiogenesis and recovery of neurological
damage in animal models of stroke (48,49).
In the present study, hippocampal damage caused by VaD may have
increased the expression of VEGFA to compensate for the
impaired cognitive function through neurovascular remodeling via
angiogenesis.
In summary, using methylation analysis, the present
study identified DMGs in the hippocampal tissues of Sham and VaD
model rats. These preliminary data may contribute to our
understanding of the pathophysiology of VaD, which may lead to the
development of methods for the recovery of cognitive function;
however, further investigations are required.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from The
National Research Foundation of Korea funded by The Ministry of
Education, Science and Technology (grant no.
NRF-2017R1A2B4012775).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors designed the experiments. YooJK, MKS and
JML were responsible for the animals, establishment of the animal
model and the behavioural test. JMP and YJK performed the
methylation profiling analysis, polymerase chain reaction and wrote
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by The
Committee for the Care and Use of Laboratory Animals at Kyung Hee
University [Seoul, Korea; approval no. KHUASP(SE)-15-082].
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kalaria RN, Maestre GE, Arizaga R,
Friedland RP, Galasko D, Hall K, Luchsinger JA, Ogunniyi A, Perry
EK, Potocnik F, et al: Alzheimer's disease and vascular dementia in
developing countries: Prevalence, management, and risk factors.
Lancet Neurol. 7:812–826. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prince M, Bryce R, Albanese E, Wimo A,
Ribeiro W and Ferri CP: The global prevalence of dementia: A
systematic review and metaanalysis. Alzheimers Dement. 9(63): 75.
e22013.
|
|
3
|
Iadecola C: The pathobiology of vascular
dementia. Neuron. 80:844–866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gill R, Tsung A and Billiar T: Linking
oxidative stress to inflammation: Toll-like receptors. Free Radic
Biol Med. 48:1121–1132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu H and Zhang J: Cerebral hypoperfusion
and cognitive impairment: The pathogenic role of vascular oxidative
stress. Int J Neurosci. 122:494–499. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park HR, Park M, Choi J, Park K, Chung HY
and Lee J: A high-fat diet impairs neurogenesis: Involvement of
lipid peroxidation and brain-derived neurotrophic factor. Neurosci
Lett. 482:235–239. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Cui X, Zacharek A, Cui Y, Roberts
C and Chopp M: White matter damage and the effect of matrix
metalloproteinases in type 2 diabetic mice after stroke. Stroke.
42:445–452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Venkat P, Chopp M and Chen J: Models and
mechanisms of vascular dementia. Exp Neurol. 272:97–108. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khan A, Kalaria RN, Corbett A and Ballard
C: Update on vascular dementia. J Geriatr Psychiatry Neurol.
29:281–301. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Murray ME, Meschia JF, Dickson DW and Ross
OA: Genetics of vascular dementia. Minerva Psichiatr. 51:9–25.
2010.PubMed/NCBI
|
|
11
|
Travis J: New piece in alzheimer's puzzle.
Science. 261:828–830. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun J, Tan L, Wang H, Tan M, Tan L, Li J,
Xu W, Zhu X, Jiang T and Yu J: Genetics of vascular dementia:
Systematic review and meta-analysis. J Alzheimers Dis. 46:611–629.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jaenisch R and Bird A: Epigenetic
regulation of gene expression: How the genome integrates intrinsic
and environmental signals. Nat Genet. 33 Suppl:S245–S254. 2003.
View Article : Google Scholar
|
|
14
|
Fessele KL and Wright F: Primer in
genetics and genomics, article 6: Basics of epigenetic control.
Biol Res Nurs. 20:103–110. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maloney B and Lahiri DK: Epigenetics of
dementia: Understanding the disease as a transformation rather than
a state. Lancet Neurol. 15:760–774. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Niculescu MD and Zeisel SH: Diet, methyl
donors and DNA methylation: Interactions between dietary folate,
methionine and choline. J Nutr. 132 8 Suppl:2333S–2335S. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cong L, Jia J, Qin W, Ren Y and Sun Y:
Genome-wide analysis of DNA methylation in an APP/PS1 mouse model
of alzheimer's disease. Acta Neurol Belg. 114:195–206. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sanchez-Mut JV and Gräff J: Epigenetic
alterations in alzheimer's disease. Front Behav Neurosci.
9:3472015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Borgel J, Guibert S, Li Y, Chiba H,
Schübeler D, Sasaki H, Forné T and Weber M: Targets and dynamics of
promoter DNA methylation during early mouse development. Nat Genet.
42:1093–1100. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Valinluck V, Tsai H, Rogstad DK, Burdzy A,
Bird A and Sowers LC: Oxidative damage to methyl-CpG sequences
inhibits the binding of the methyl-CpG binding domain (MBD) of
methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res.
32:4100–4108. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Farkas E, Luiten PG and Bari F: Permanent,
bilateral common carotid artery occlusion in the rat: A model for
chronic cerebral hypoperfusion-related neurodegenerative diseases.
Brain Res Rev. 54:162–180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Olton DS, Collison C and Werz MA: Spatial
memory and radial arm maze performance of rats. Learn Motiv.
8:289–314. 1977. View Article : Google Scholar
|
|
24
|
Harris RA, Wang T, Coarfa C, Nagarajan RP,
Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al:
Comparison of sequencing-based methods to profile DNA methylation
and identification of monoallelic epigenetic modifications. Nat
Biotechnol. 28:1097–1105. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lan X, Adams C, Landers M, Dudas M,
Krissinger D, Marnellos G, Bonneville R, Xu M, Wang J, Huang TH, et
al: High resolution detection and analysis of CpG dinucleotides
methylation using MBD-seq technology. PLoS One. 6:e222262011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Serre D, Lee BH and Ting AH: MBD-isolated
genome sequencing provides a high-throughput and comprehensive
survey of DNA methylation in the human genome. Nucleic Acids Res.
38:391–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (-Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sim YJ: Treadmill exercise alleviates
impairment of spatial learning ability through enhancing cell
proliferation in the streptozotocin-induced alzheimer's disease
rats. J Exerc Rehabil. 10:81–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hussein AM, Aher YD, Kalaba P, Aher NY,
Dragačević V, Radoman B, Ilic M, Leban J, Beryozkina T, Ahmed ABMA,
et al: A novel heterocyclic compound improves working memory in the
radial arm maze and modulates the dopamine receptor D1R in frontal
cortex of the sprague-dawley rat. Behav Brain Res. 332:308–315.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huidobro C, Fernandez AF and Fraga MF: The
role of genetics in the establishment and maintenance of the
epigenome. Cell Mol Life Sci. 70:1543–1573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Goldberg AD, Allis CD and Bernstein E:
Epigenetics: A landscape takes shape. Cell. 128:635–638. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Klein H and De Jager PL: Uncovering the
role of the methylome in dementia and neurodegeneration. Trends Mol
Med. 22:687–700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ling C and Groop L: Epigenetics: A
molecular link between environmental factors and type 2 diabetes.
Diabetes. 58:2718–2725. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu B, Russanova VR, Gravina S, Hartley S,
Mullikin JC, Ignezweski A, Graham J, Segars JH, DeCherney AH and
Howard BH: DNA methylome and transcriptome sequencing in human
ovarian granulosa cells links age-related changes in gene
expression to gene body methylation and 3′-end GC density.
Oncotarget. 6:3627–3643. 2015.PubMed/NCBI
|
|
36
|
Lunnon K, Smith R, Hannon E, De Jager PL,
Srivastava G, Volta M, Troakes C, Al-Sarraj S, Burrage J, Macdonald
R, et al: Methylomic profiling implicates cortical deregulation of
ANK1 in alzheimer's disease. Nat Neurosci. 17:1164–1170. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Jager PL, Srivastava G, Lunnon K,
Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe
C, et al: Alzheimer's disease: Early alterations in brain DNA
methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci.
17:1156–1163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma J, Sawai H, Ochi N, Matsuo Y, Xu D,
Yasuda A, Takahashi H, Wakasugi T and Takeyama H: PTEN regulate
angiogenesis through PI3K/akt/VEGF signaling pathway in human
pancreatic cancer cells. Mol Cell Biochem. 331:161–171. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Waldner MJ and Neurath MF: Targeting the
VEGF signaling pathway in cancer therapy. Expert Opin Ther Targets.
16:5–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee S, Chen TT, Barber CL, Jordan MC,
Murdock J, Desai S, Ferrara N, Nagy A, Roos KP and Iruela-Arispe
ML: Autocrine VEGF signaling is required for vascular homeostasis.
Cell. 130:691–703. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ferrara N, Gerber H and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Franco CA and Li Z: SRF in angiogenesis:
Branching the vascular system. Cell Adh Migr. 3:264–267. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chai J, Jones MK and Tarnawski AS: Serum
response factor is a critical requirement for VEGF signaling in
endothelial cells and VEGF-induced angiogenesis. FASEB J.
18:1264–1266. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ahmad S, Hewett PW, Wang P, Al-Ani B,
Cudmore M, Fujisawa T, Haigh JJ, le Noble F, Wang L, Mukhopadhyay D
and Ahmed A: Direct evidence for endothelial vascular endothelial
growth factor receptor-1 function in nitric oxide-mediated
angiogenesis. Circ Res. 99:715–722. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Morin-Brureau M, Lebrun A, Rousset MC,
Fagni L, Bockaert J, de Bock F and Lerner-Natoli M: Epileptiform
activity induces vascular remodeling and zonula occludens 1
downregulation in organotypic hippocampal cultures: Role of VEGF
signaling pathways. J Neurosci. 31:10677–10688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dzietko M, Derugin N, Wendland M, Vexler Z
and Ferriero D: Delayed VEGF treatment enhances angiogenesis and
recovery after neonatal focal rodent stroke. Transl Stroke Res.
4:189–200. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shimotake J, Derugin N, Wendland M, Vexler
ZS and Ferriero DM: Vascular endothelial growth factor receptor-2
inhibition promotes cell death and limits endothelial cell
proliferation in a neonatal rodent model of stroke. Stroke.
41:343–349. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Waltenberger J, Mayr U, Pentz S and
Hombach V: Functional upregulation of the vascular endothelial
growth factor receptor KDR by hypoxia. Circulation. 94:1647–1654.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yang J, Liu H and Liu X: VEGF promotes
angiogenesis and functional recovery in stroke rats. J Invest Surg.
23:149–155. 2010. View Article : Google Scholar : PubMed/NCBI
|