Introduction
Spinal nociceptive pain continues to be a serious
clinical challenge. Despite having been evaluated in numerous
studies, the underlying cellular and molecular mechanisms that
control the induction and maintenance of spinal nociceptive pain
remain to be fully elucidated (1,2).
These mechanisms are reported to involve a combination of neural
excitability and spinal synaptic plasticity, with unique
characteristics (3,4). In addition, nerve injury elicits
neuronal alterations, including the promotion of synapse formation
during the development of pain (5). The spinal cord is the main center of
pain information transmission and integration. The central
sensitization of spinal dorsal horn neurons is involved in the
primary mechanism of spinal nociceptive pain, which is mainly
manifested in the long-term potentiation (LTP) effect of synaptic
transmission. LTP is one of two important forms of synaptic
plasticity, and its role in pain is mainly achieved by synaptic
morphological plasticity and functional plasticity. A previous
study found that the synaptic membrane area, the post synaptic
density, the number of synaptic vesicles, the synaptic gap, and the
density of total synapses were increased significantly in rats
suffering from neuropathic pain (6). In addition, γ-aminobutyric acid and
glutamate receptors are involved in the regulation of spinal
nociceptive pain as essential inhibitory and excitatory receptors,
respectively, in synapses (7).
Several studies have shown that signal transduction
have important in the development and progression of spinal
nociceptive pain. Currently, the majority of investigations have
focused on the role of signal transduction via receptor tyrosine
kinases (RTKs). EphrinB and EphB receptors are the most important
subfamily of RTKs in humans. Their activation depends on the close
proximity (or adhesion) of cells, allowing the ligand to bind to
the receptor and activate signal transduction. They are involved in
numerous vital developmental processes and in the maintenance of
pain by regulating revascularization, cell migration, axon guidance
and synaptic plasticity (8,9). In
addition, ephrinB/EphB are important in physical changes in the
brain and peripheral tissues. Eph family members are numerous, and
according to their sequence homology, distribution, and mutual
affinity with the ligand, they are divided into two partially
overlapping families, A and B. There are 10 EphA receptors
(EphA1-EphA10), six EphB receptors (EphB1-EphB6) and three ephrinB
ligands (ephrinB1-ephrinB3). Ephrin is a transmembrane protein with
a transmembrane domain and a cytoplasmic domain. The cytoplasmic
domain is highly conserved and contains five potential tyrosine
phosphorylation sites, whereas the carboxy terminus forms a PDZ
(discoid homology region) domain-binding motif. Eph receptors are
also transmembrane proteins with extracellular (containing
ligand-binding domains), transmembrane and intracellular domains, a
cysteine-rich structure and two fibronectin repeats. The
intracellular region has two membrane-proximal tyrosine residues,
comprising the classical protein tyrosine kinase binding domain,
the sterile α-motif domain, and the PDZ binding domain. Ephrin-A
ligands typically bind specifically to the EphA receptor, and
Ephrin-B ligands bind specifically to the EphB receptor; however,
the EphA4 receptor has a ligand-binding domain with a broader
specificity, which binds to the majority of ephrin-A, ephrin-B2 and
ephrin-B3 ligands. EphrinB/EphB can regulate the activity of
post-synaptic N-methyl-D-aspartate (NMDA) receptors by regulating
the release of L/D-serine from astrocytes and regulating synaptic
plasticity via glutamine-glutamate cycling. In addition,
ephrinB/EphB can induce central sensitization and is involved in
nociceptive information modulation by activating the downstream
mitogen-activated protein kinase pathway and the Src family kinase
(10).
In addition to tryosine protein kinase
(TPK)-dependent signal transduction, calpains are activated during
the sensitization of nociceptive neurons through synaptic
plasticity (11). Calpain-1, which
belongs to the cysteine protease enzyme family, is a
calcium-activated neutral protease. Central nervous system injury
or disease can increase intracellular Ca2+
concentrations, leading to excessive activation of calpains and
scaffold proteins, membrane protein degradation (12), mitochondrial permeability changes,
or phosphorylation of calpastatin and calpain and other unknown
pathways mediating central cell death or apoptosis (13,14).
Differences have been observed in the time and location of
calpain-1 following its activation; a cleaved caspase-3 protein
fragment activated by calpain-1 and caspase-3 increased the
activity of calpains in turn (15,16).
A preliminary experiment showed that numerous caspases and calpains
are present in pre- and post-synaptic compartments of neurons, and
are important in modulating synaptic plasticity (17). From the information mentioned
above, it is clear that the ephrinB/EphB signaling pathway is vital
in the development of nociceptive pain by regulating synaptic
plasticity, and that the pathway formed by calpain-1 and caspase-3
also regulates synaptic plasticity and neural circuitry. However,
whether the pathway formed by calpain-1 and caspase-3 is involved
in processing ephrinB/EphB signaling pathway-induced nociceptive
pain remains to be fully elucidated. Therefore, it was hypothesized
in the present study that ephrinB/EphB signaling is involved in the
processing of nociceptive pain via the actions of calpain-1 and
caspase-3.
Materials and methods
Animals
All experiments involved adult male Kunming mice
(22–25 g) and were approved by the Animal Experimental Center,
Second Military Medical University (Shanghai, China; production
license no. SCXK (hu)2012-0003). The mice were supplied by the
Institutional Animal Care and Use Committees and handled in
accordance with the regulations of the Ethics Committee of the
International Association for the Study of Pain (https://www.iasp-pain.org) and experimental methods.
The mice were housed in individual cages in a
temperature-controlled (18–28°C) room with alternating 12-h
light/dark cycles. Food was withheld for 8 h prior the start of the
experiments, and all animals had free access to water. Monitoring
of health problems was performed three times each day and no deaths
occurred during the experiments.
Surgical procedures
Drug injection model
A total of 164 mice were used in the experiments; 64
mice were injected with either ephrinB1-Fc or ephrinB2-Fc (0.1 and
0.5 µg). The saline injection group was used as a comparison group
with the ephrinB injection groups to indicate the effect of ephrinB
injection; the blank control group (control group) was used to
verify that intrathecal injection of saline alone had no effect on
the mice. A total of 72 mice were injected with MDL28170 (an
inhibitor of calpain-1), and with the respective solvent as a
control, with saline and ephrinB as a control, and dimethyl
sulfoxide (DMSO) and MDL28170 as a control The method of injection
was as described by Hylden and Wilcox (18). Briefly, 10-µl trace syringes were
used and inserted vertically between the L5 and L6 vertebrae of the
experimental mice. If the tail of the mice exhibited a sudden
slight swing, entry into the subarachnoid space had occurred. The
volume of drug or contrast solvent was 5 µl, which were injected
into the subarachnoid space over a 20 sec period, and the trace
syringes were held in place for 10 sec, the total duration being 30
sec.
Chronic constrictive injury (CCI)
model
Of the 164 mice as described above, 28 mice were
used to establish the CCI model. The main protocol was described by
Bennett and Xie (19). Briefly,
the mice were deeply anesthetized using sodium pentobarbital (45
mg/kg, intraperitoneally). The left sciatic nerve was exposed at
the mid-thigh and a 4-0 silk thread was used to ligate it loosely
in four regions at ~1-mm intervals. For the mice in the sham group,
the nerve was exposed but no ligation or damage was induced.
Drugs
The ephrinB1-Fc (cat. no. 80106-R02H) chimera (EphB
receptor activator), and EphB2-Fc (cat. no. 51367-M02H) chimera
(EphB receptor-blocking reagent), were purchased from Sino
Biological, Inc. (Beijing, China). The ephrinB2-Fc chimera was
purchased from R&D Systems, Inc. (Minneapolis, MN, USA).
Anti-calpain-1 antibody (cat. no. ab28258) was purchased from Abcam
(Cambridge, UK). The caspase-3 rabbit monoclonal antibody was
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
The calpain- and cathepsin B-selective inhibitor, MDL28170, was
purchased from Cene Operation (Ann Arbor, MI, USA). Each drug was
dissolved in normal saline (NS); the dose and the time of treatment
were based on the results of preliminary experiments.
Behavioral assessment
The measurement of mechanical allodynia was
represented by the mechanical withdrawal threshold (MWT). The MWT
was assessed using von Frey hairs according to the up-and-down
method described by Chaplan et al (20). Briefly, the mice were placed in
individual plastic boxes (20×20×15 cm) on a metal mesh floor and
allowed to acclimate for ~1 h prior to assessment. Beginning with
0.16 g and ending with 4.0 g, the von Frey filament was used to
touch the plantar surface of the hind paw, which was held for 6–8
sec. In the event of the absence of paw withdrawal, the next higher
stimulus was selected, whereas a weaker stimulus was applied in the
opposite situation. This was repeated 10 times and the stimulation
strength was determined as the average corresponding to a 50%
response rate.
The measurement of thermal hyperalgesia was
determined by the thermal withdrawal latency (TWL). The TWL was
assessed as described by Hargreaves et al (21). Briefly, the mice were placed in
individual plastic boxes (7×9×11 cm) on a temperature-controlled
glass floor and allowed to acclimate for 1 h prior to assessment.
The radiant heat source was focused on a region of the hind paw and
was moved away immediately when the mice lifted up or licked their
hind paw. The stimulus was shut off automatically at 20 sec to
prevent tissue damage. This was repeated four times for each hind
paw at 5-min intervals, and the average was calculated as the paw
TWL.
Immunohistochemistry
The mice were deeply anesthetized with sodium
pentobarbital (65 mg/kg intraperitoneal injection) and perfused
transcardially with 40 ml ice-cold phosphate-buffered saline (PBS)
and 60 ml 4% paraformaldehyde (PFA). The spinal cord of each mouse
was then exposed and the L4-5 vertebrae were removed; these samples
were post-fixed in 4% PFA for 6 h and transferred into 30% sucrose
in phosphate buffer overnight at 4°C. Transverse series sections
(15-µm) were cut on a cryostat and stored in phosphate buffer.
Following washing in PBS, the tissue sections were incubated in PBS
containing 5% normal goat serum (R&D Systems, Inc.,
Minneapolis, MN, USA) and 0.3% Triton X-100 at 37°C for ~30 min.
For the caspase-3 assay, the sections were incubated in primary
polyclonal Rabbit-anti-caspase-3 antibody (cat. no. 5A1E; 1:1,500;
Cell Signaling Technology, Inc.) at 4°C overnight and then
incubated in biotinylated Donkey-Anti-Rabbit antibody
(594-conjugated; cat. no. 110781; 1:400; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) at 37°C for 1 h and
avidin-biotin-peroxidase complex (cat. no. AK-5001; 1:100; Vector
Laboratories, Inc., Burlingame, CA, USA) at room temperature for 2
h. Finally, the sections were treated with 0.05% diaminobenzidine
for 10 min. The sections were rinsed in PBS, mounted on
gelatin-coated slides, air-dried, dehydrated with alcohol, cleared
with xylene, and cover-slipped for examination under an inverted
fluorescent microscope (Carl Zeiss AG; Oberkochen, Germany). To
analyze the expression of caspase-3, five L4-5 spinal cord sections
were examined per animal, selecting the sections with the highest
number of positive neurons. The average total number of positive
neurons in the spinal cord was calculated.
Following perfusion, as described above, the spinal
cord was exposed and the L4-5 vertebrae were removed and post-fixed
in 4% PFA for 6 h; these samples were transferred to 30% sucrose in
phosphate buffer for ~2 days. The spinal cord was removed from the
sucrose, blocked in optimum cutting temperature compound, and
incubated at −20°C for ~20 min. The tissues were sectioned at a
16-µm thickness on a freezing microtome. All samples were incubated
in blocking solution containing 1% bovine serum albumin, 5% donkey
serum (both from Abcam), and 0.3% Triton X-100 at room temperature
for 2 h. The sections were then incubated with the following
primary antibodies at room temperature overnight:
Rabbit-anti-caspase-3 antibody (cat. no. 5A1E; 1:100, Cell
Signaling Technology, Inc.), Rabbit anti-calpain-1 antibody (cat.
no. ab28258; 1:100, Abcam), Mouse anti-neuronal nuclei antigen
(NeuN) antibody (cat. no. MAB377; 1:500, FITC-conjugated, Chemicon;
EMD Millipore, Billerica, MA, USA), Goat anti-glial fibrillary
acidic protein (GFAP) antibody (cat. no. sc-6170; 1:500, Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), and Goat anti-ionized
calcium-binding adapter molecule 1 (IBA-1) antibody (cat. no.
Ab5076; 1:200, Abcam). On the second day, the sections were washed
four times with PBS, incubated with specific secondary antibodies
for 2 h at room temperature, and washed again as described above.
Corresponding to the above primary antibodies, the corresponding
secondary antibody was selected from Donkey Anti-Rabbit antibody
(594-conjugated; cat. no. 110781; 1:400; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA), Goat Anti-Mouse
(FITC-conjugated; cat. no. E031210-011:100; EarthOx LLC, Millbrae,
CA, USA) and Donkey Anti-Goat (488-conjugated; cat. no. 109911;
1:400, Jackson ImmunoResearch Laboratories, Inc.). Finally, the
sections were rinsed and mounted on a gelatin-coated slide. Images
of the sections were captured using a fluorescence microscope
connected to a charge-coupled device spot camera for separate or
combined viewing of red, green, and blue fluorescence.
Western blot analysis
The mice were deeply anesthetized, and the L4-5
segments were rapidly removed and stored in liquid nitrogen or
homogenized in lysis buffer at pH 7.4 containing a protease
inhibitor cocktail. The homogenates were incubated for 30 min on
ice, vortexed for 10 sec on the highest setting every 5 min, and
then centrifuged at 13,000 × g, 4°C for 15 min. This was repeated
following collection of the supernatants, the second supernatants
were collected and the protein concentration was measured using the
Bradford method (22). The samples
were heated at 99°C for 10 min to denature the proteins, then the
equivalent amounts of proteins (30 µg) were separated using 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis and
transferred onto a polyvinylidene fluoride membrane (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The membranes were
incubated with rabbit anti-calpain-1 antibody (1:400) at 4°C
overnight. The membranes were washed with Tris-buffered saline
containing Tween 20 (TBST) 4 times for 5 min each, incubated for 2
h with the secondary antibody (Donkey Anti-Rabbit antibody;
594-conjugated; cat. no. 110781; 1:4,000; Jackson ImmunoResearch
Laboratories, Inc.) at room temperature and then washed with TBST 4
times for 5 min each. The proteins were detected using enhanced
chemiluminescence western detection reagents. Western blot
densitometry analysis was performed using Image J software version
1.8.0 (NIH, Bethesda, MD, USA).
Statistical analysis
All data in the present study are shown as the mean
± standard deviation. Statistical analysis between two groups was
performed using Student's t-test, whereas more than two groups were
analyzed using one-way analysis of variance (ANOVA) followed by a
Tukey's post hoc test. Differences in mechanical allodynia and
thermal hyperalgesia over time were assessed using two-way ANOVA
with repeated measures. Statistical analyses of the data were
generated using GraphPad Prism 5 (Graph Pad Software, Inc., San
Diego, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
EphrinB/EphB signaling is involved in
nociceptive pain and regulates time- and dose-dependent mechanical
allodynia and thermal hyperalgesia
Intrathecally injected ephrinB activates their EphB
receptors and produces long-lasting MWT and TWL (23,24).
However, which subtype mainly mediating nociceptive pain remains to
be elucidated. Therefore, for optimal results, the present study
examined the possible roles of ephrinB1-Fc and ephrinB2-Fc using
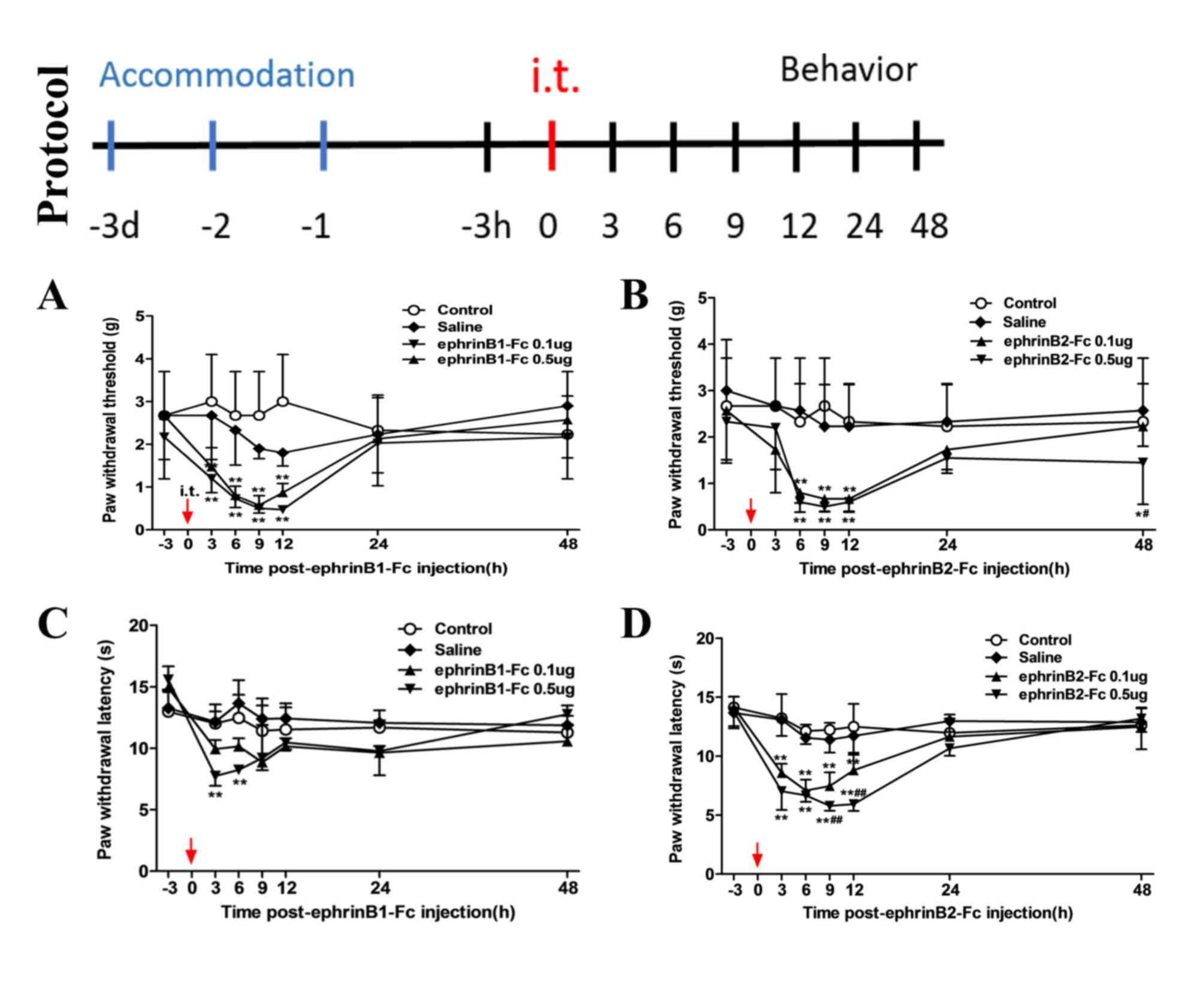
the experimental protocol shown in Fig. 1. The mice were acclimated for 3
days prior to the experiment, followed by intrathecal injection.
Behavioral assessments were performed 3 h prior to intrathecal
injection, and 3, 6, 9, 12, 24, 48 h following intrathecal
injection. From the results, it was found that the changes in MWT
induced by intrathecal injection of ephrinB1-Fc (Fig. 1A) and the changes in MWT induced by
intrathecal injection of ephrinB2-Fc (Fig. 1B) were essentially the same, with
pain observed at 3 h post-injection, which lasted ~9–12 h to reach
a peak and returning to a normal level at ~48 h. However, the
change in TWL following ephrinB1-Fc injection (Fig. 1C) was small and was sustained for
less time compared with the TWL following ephrinB2-Fc injection
(Fig. 1D). Therefore, ephrinB2-Fc
injection was used in subsequent experiments. As shown in the
behavioral assessments following ephrinB2-Fc injection, regardless
of the MWT (Fig. 1B) or TWL
(Fig. 1D), there were no
differences between the control and saline group. Compared with the
control and saline groups, the MWT and TWL were significantly
decreased in the 0.1 and 0.5 µg ephrinB2-Fc injection group
(**P<0.01), with 0.5 µg injection inducing larger decreases
(##P<0.01 vs. 0.1 µg group). The role of EphB during
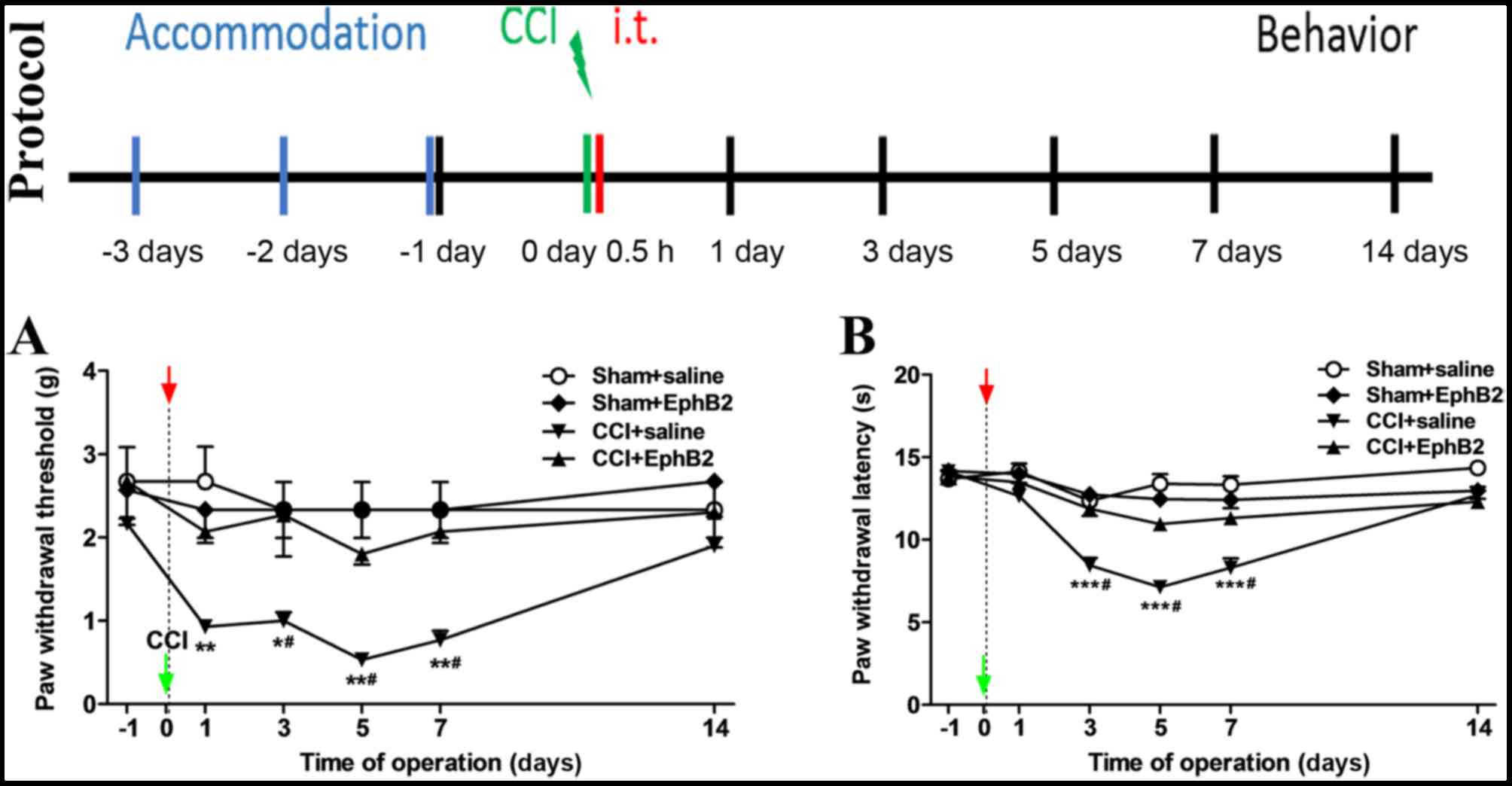
the development of spinal nociceptive pain in CCI mice was then
assessed using the experimental protocol shown in Fig. 2. The mice were acclimated for 3
days prior to the experiment, CCI models were constructed and
ephB2-Fc was intrathecally injection at 0.5 h post-surgery.
Behavioral assessments were performed at 1, 3, 5, 7 and 14 days
post-surgery. As revealed by the MWT (Fig. 2A) and TWL (Fig. 2B), no differences were observed
between the sham + saline group and sham + EphB2-Fc group; however,
compared with the sham + saline group and sham + EphB2-Fc group,
the CCI + saline group showed a distinct decrease (***P<0.001,
**P<0.01 and *P<0.05). Compared with the CCI + saline group,
the pain experienced in the CCI + EphB2-Fc group was decreased
(#P<0.05). Therefore, the intrathecal injection of
ephrinB2-Fc resulted in time-and dose-dependent mechanical
allodynia and thermal hyperalgesia in mice, and intrathecal
injection of EphB2-Fc attenuated or reversed the pain in the CCI
model mice.
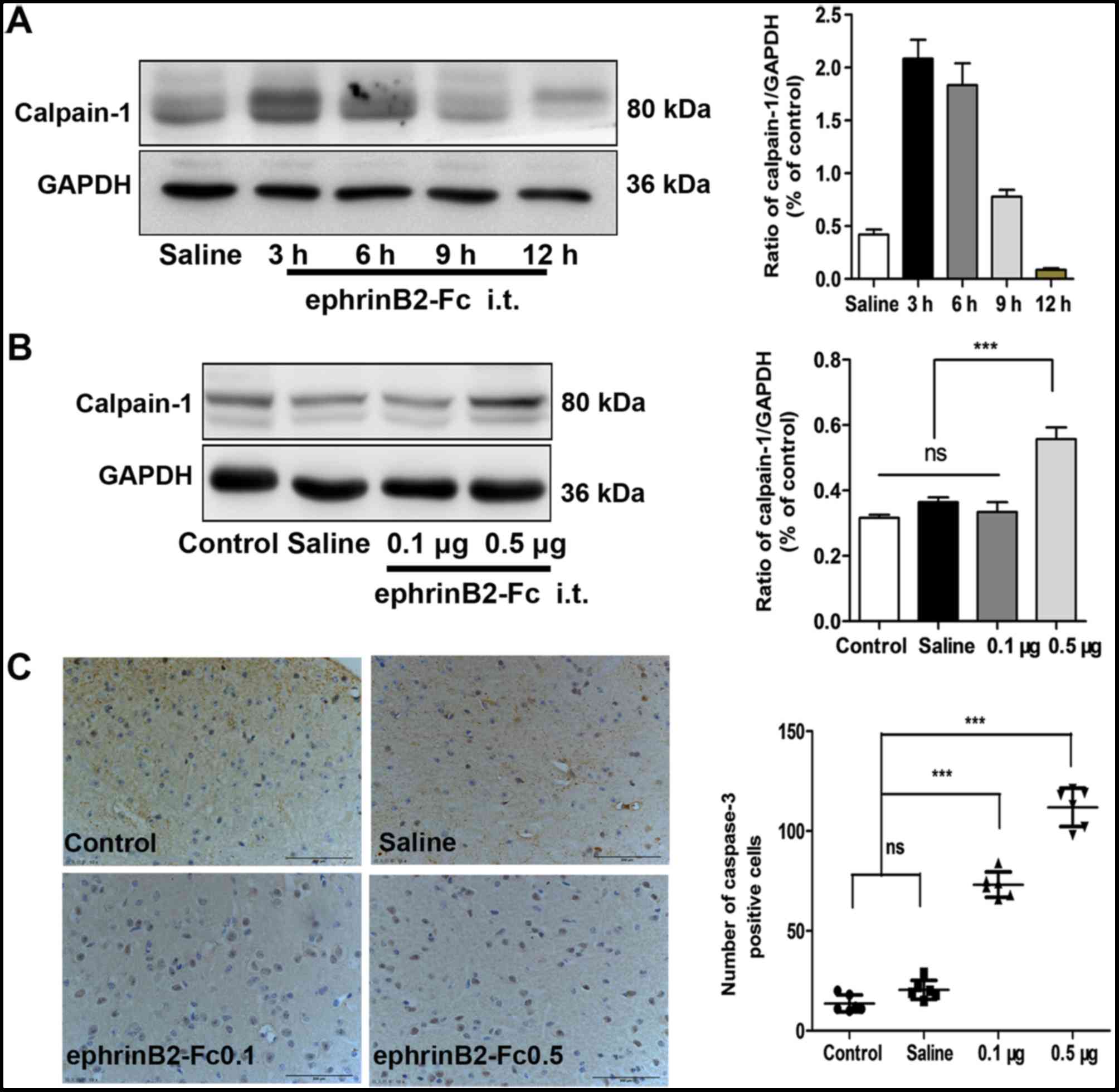
Intrathecal injection of ephrinB2-Fc
induces upregulated expression of calpain-1 and caspase-3
To investigate whether the pathways of calpain-1 and
caspase-3 were responsible for the observed results, the levels of
calpain-1 and caspase-3 were examined during the processing of
nociceptive pain by ephrinB2-Fc injection. To determine the
appropriate time point in subsequent experiments, the level of
calpain-1 following injection was detected. As demonstrated by
western blot analysis (Fig. 3A),
the highest level of calpain-1 was detected at 3 h post-injection,
which was selected as the time point for the subsequent
experiments. At 3 h post-injection (Fig. 3B) the levels of calpain-1 were
similar among the control group, saline injection group and 0.1 µg
ephrinB2-Fc injection group; however, compared with its levels in
these three groups, the level of calpain-1 in the 0.5 µg
ephrinB2-Fc injection group was significantly increased
***P<0.001). The immunohistochemistry showed that the level of
caspase-3 was similar between the control and saline injection
group (P>0.05), but was significantly increased in the
ephrinB2-Fc 0.1 and 0.5 µg injection groups ***P<0.001; Fig. 3C). Therefore, intrathecal injection
of ephrinB2-Fc not only induced pain behavior in mice but also
increased the levels of calpain-1 and caspase-3 in the spinal
cord.
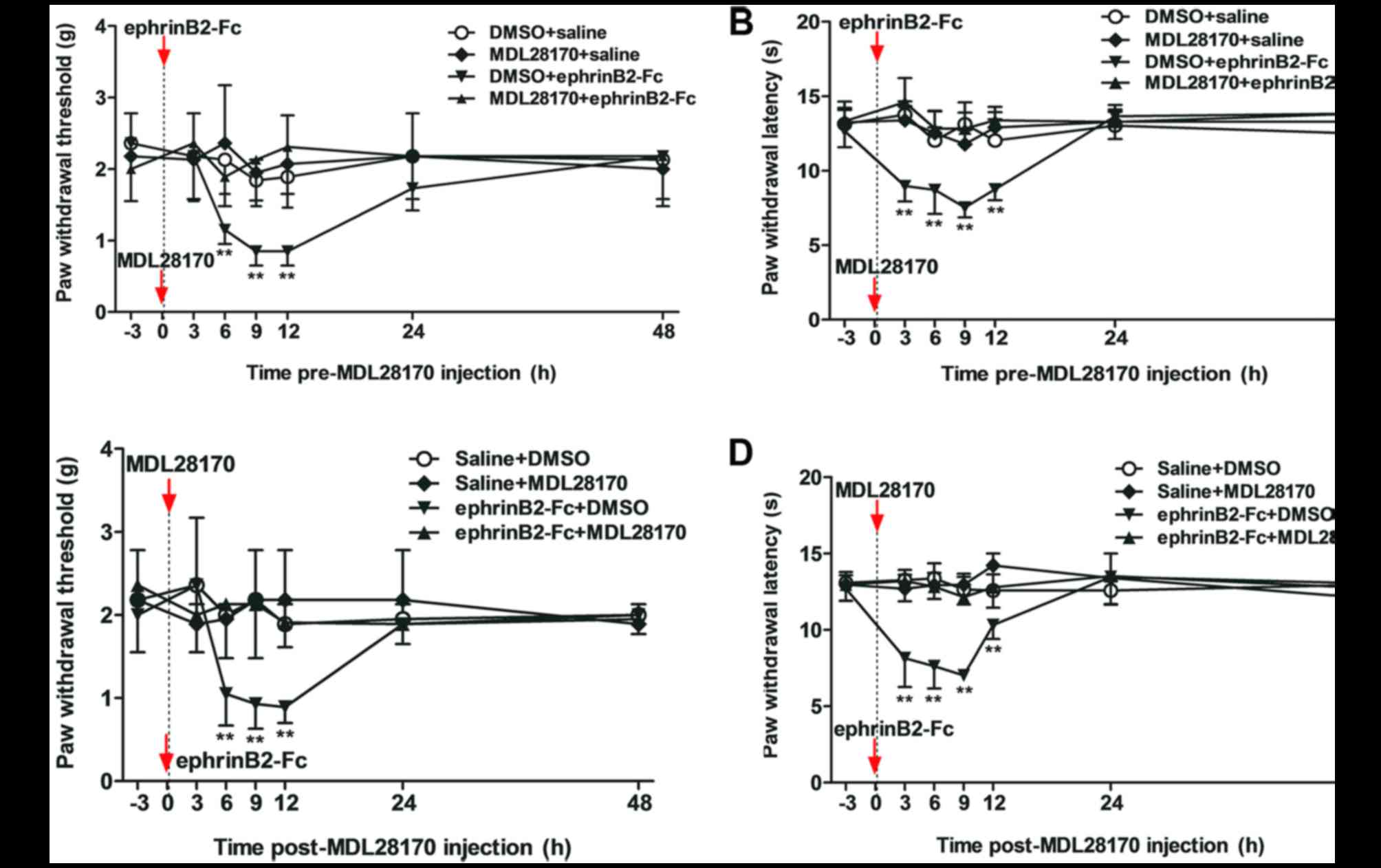
Inhibition of calpain-1 by MDL28170
reduces pain behaviors and decreases the level of caspase-3
The present study evaluated the potential connection
between calpain-1 and caspase-3, and the function of calpain-1 in
the processing of nociceptive pain induced by ephrinB2-Fc
injection. MDL28170 (1.0 µg), a selective inhibitor, was
administered at 30 min prior to (pre-) or following (post-)
ephrinB2-Fc injection. These two injection time points were
selected to detect whether calpain-1 is involved in the generation
or maintenance of spinal nociceptive pain. The behavioral
assessment (Fig. 4A and B) showed
that pre-injection with MDL28170 (1.0 µg) for 30 min prior to
ephrinB2-Fc administration inhibited the ephrinB2-Fc-induced change
in MWT and TWL (**P<0.01). Post-treatment with MDL28170 at 30
min following ephrinB2-Fc injection also inhibited the
ephrinB2-Fc-induced change in MWT and TWL (**P<0.01; Fig. 4C and D)
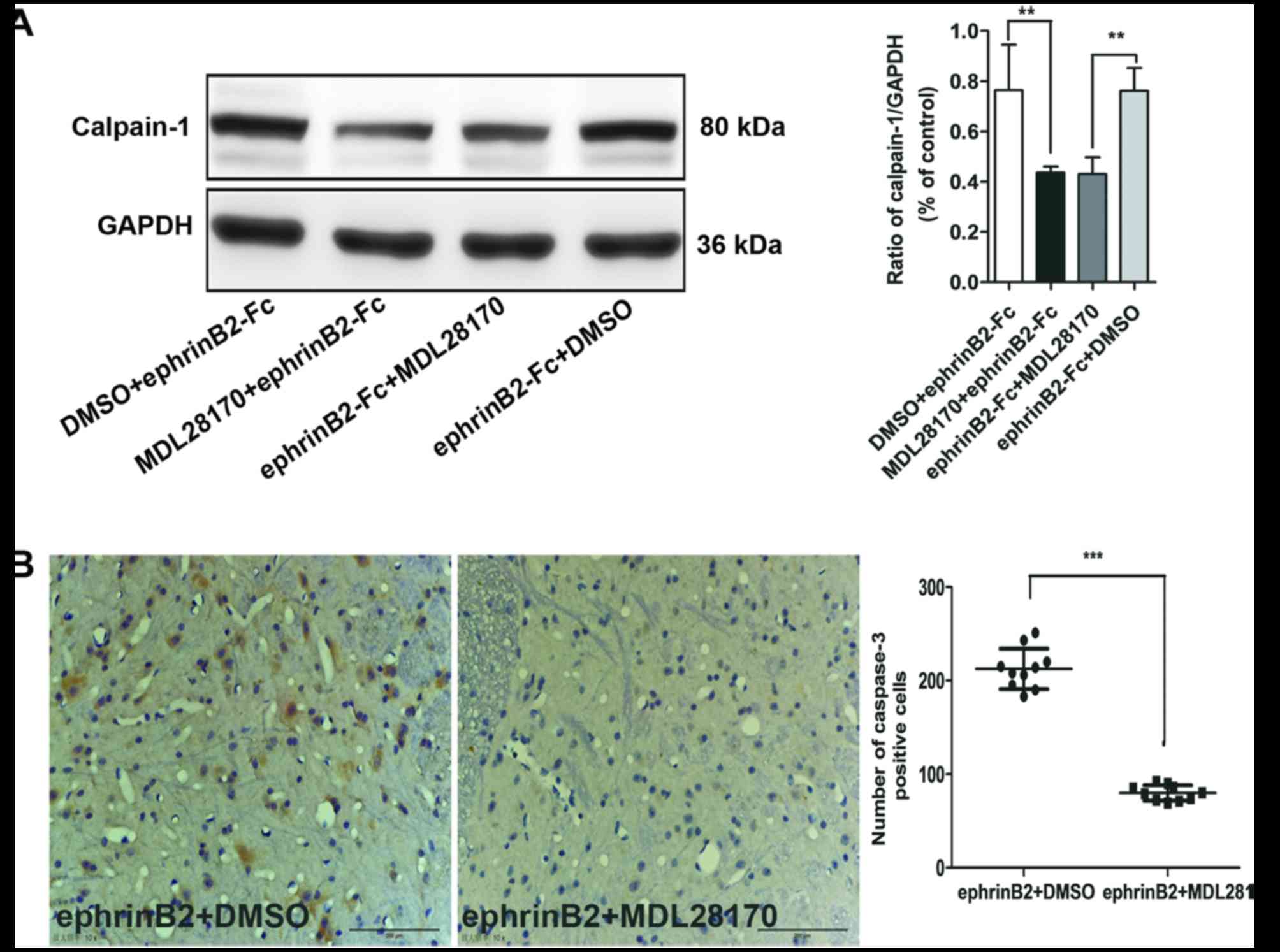
Compared with the ephrinB + DMSO group, the levels
of calpain-1 (Fig. 5A) and
caspase-3 (Fig. 5B) in the spinal
cord were decreased in the ephrinB2 + MDL28170 group, regardless of
whether MDL28170 was added prior to or following treatment
***P<0.001 and **P<0.01). Therefore, pre- or post-injection
of MDL28170, an inhibitor of calpain-1, reduced the pain behaviors
and decreased the activation of calpain-1 and caspase-3, as
evidenced by their decreased protein levels.
Calpain-1 and caspase-3 are involved
in nociceptive pain through neurons
To determine the cell phenotypes responsible for the
increased levels of calpain-1 and caspase-3 in the spinal cord
following the administration of ephrinB2-Fc, double
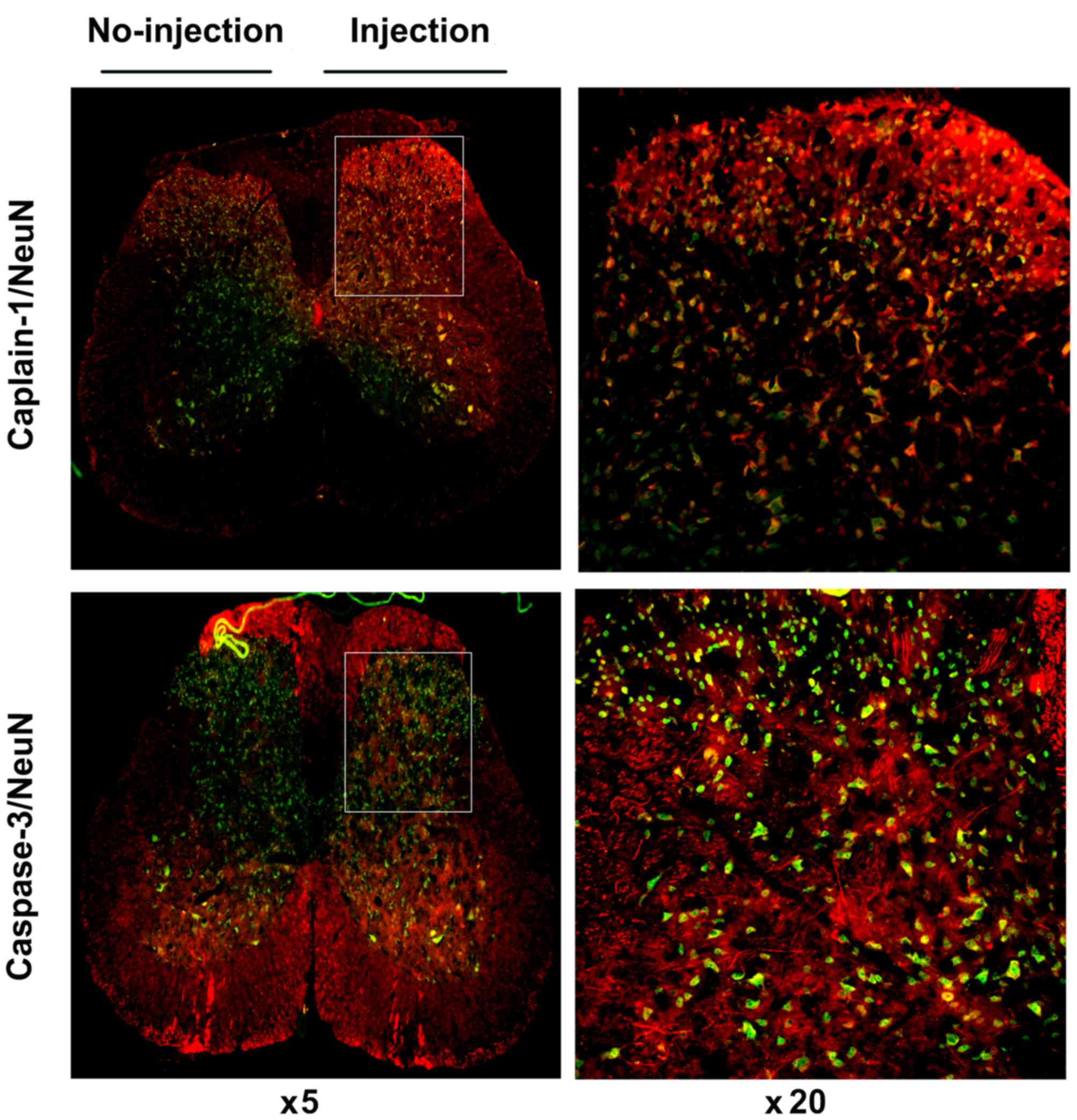
immunofluorescent labeling was performed. As shown in Fig. 6A and B, at 6 h post-ephrinB2-Fc
injection, calpain-1 or caspase-3 colocalized with NeuN (a neuron
marker), but not with GFAP (an astrocyte marker) or IBA-1 (a
microglial cell marker) in the spinal cord. Images of complete
spinal cord staining were captured, showing the contrast between
the injection side (right side) and non-injection side (left side).
As shown in Fig. 7, the expression
levels of calpain-1 and caspase-3 in the spinal cord following
injection were increased significantly, compared with those in the
non-injected side, and co-staining was observed, particularly in
the spinal dorsal horn. These results showed that the levels of
calpain-1 or caspase-3 increased in neurons following activation of
the ephrinB2/EphB2 signaling pathway and neurons may be the targets
in this process.
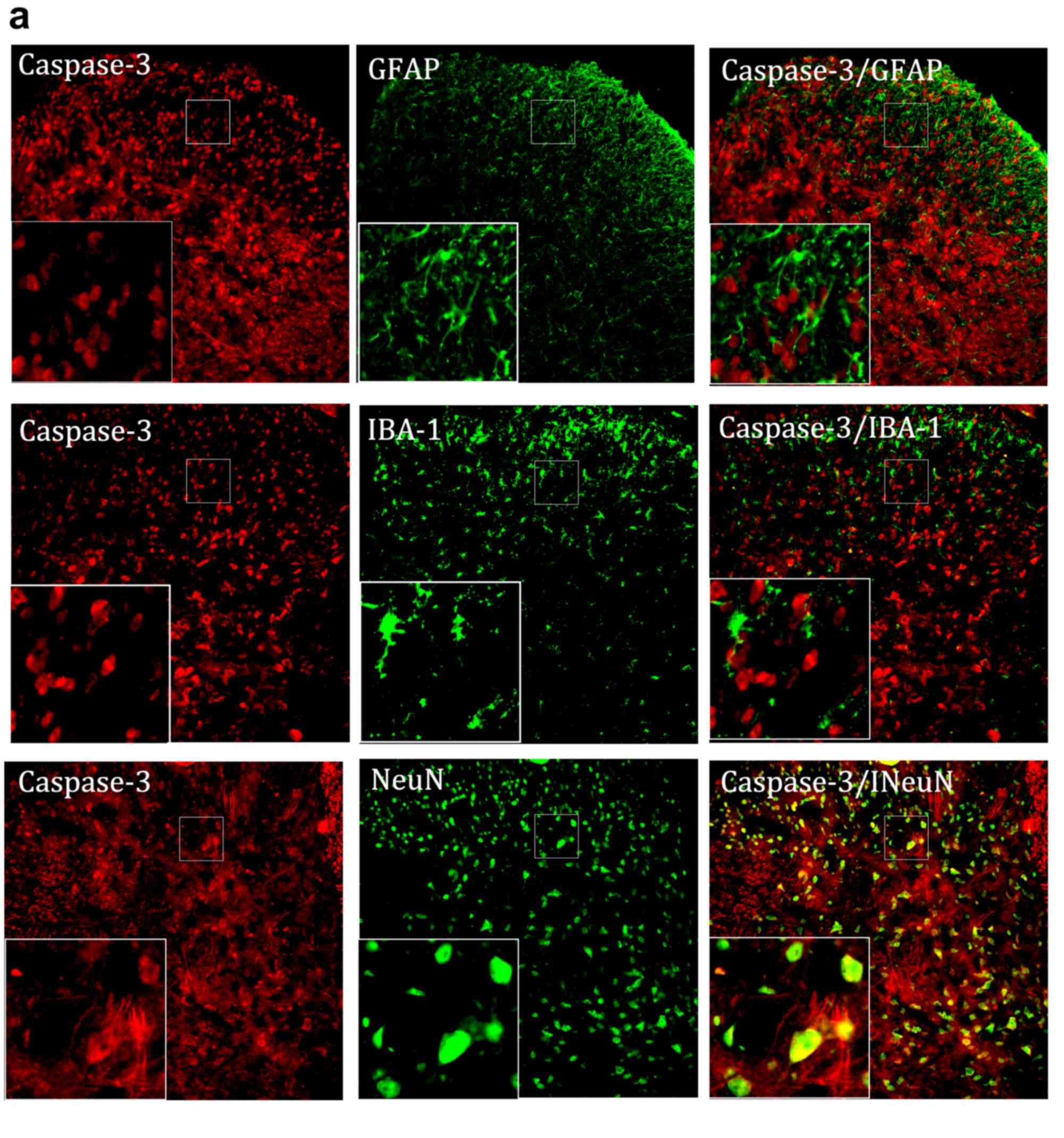 | Figure 6.Immunofluorescence staining of
calpain-1 and caspase-3. (A) Representative immunofluorescence
staining of calpain-1 (red) and its colocalization with neurons
(NeuN, green), astrocyte cells (GFAP, green) and microglial cells
(IBA-1, green) in the spinal cord. (B) Representative
immunofluorescence staining of caspase-3 (red) and its
colocalization with neurons (NeuN, green), astrocyte cells (GFAP,
green) and microglial cells (IBA-1, green) in the spinal cord.
Magnification, ×200 and ×400. NeuN, neuronal nuclei antigen; GFAP,
glial fibrillary acidic protein; IBA-1, ionized calcium-binding
adapter molecule 1. |
Discussion
EphrinB ligands and their EphB receptors comprise
the largest family of TPKs, which are vital in the regulation of
spinal nociceptive information via several mechanisms. Battaglia
et al found that ephrinB/EphB signaling contributed to
spinal nociceptive pain by injecting ephrinB into mice (25). EphrinB ligands and their EphB
receptors can bind to NMDA receptors to exert a positive regulatory
function, and nociceptive pain induced by ephrinB2-Fc injection is
antagonized by the NMDA receptor antagonist MK801 (26,27).
EphB receptors and ephrinB ligands are key regulators of the
synaptic body. Intrathecal injection of ephrinB2-Fc can activate
ephrin signaling, mainly due to the ephrinB2 ligand binding to its
receptor. The activation of
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)
receptor function can be achieved by inducing LTP; however,
inhibiting the binding of glutamate receptor-interacting protein,
an EphB and AMPA binding protein, can significantly inhibit the
production of LTP. With the exception of AMPA receptors, ephrinB2
interacts with the metabotropic glutamate receptor, mGlu1, and with
endogenous small interfering RNAs, which may be the method by which
it regulates synaptic plasticity (28). At the cellular level, ephrinB2 is
involved in pain mainly via its expression in neuronal cells. By
transducing external stimuli to the nucleus and then into cellular
effector signaling pathways, two types of densely packed cell form
a network and the confined protruding regulatory domains form a
complete intercellular scar, separating astrocytes and fibroblasts
from each other, which is detrimental to axonal regeneration
(29). In the present study,
spinal nociceptive pain and hyperalgesia were found to be
associated with ephrinB2/EphB2 signaling. Animal experiments
confirmed that, compared with the normal and saline injection
groups, the intrathecal injection of ephrinB1-Fc or ephrinB2-Fc
significantly decreased the pain threshold of mechanical allodynia
and thermal hyperalgesia in a time- and dose-dependent manner. The
levels of calpain-1 were upregulated in spinal cord neurons,
reaching the highest level 3 h following injection, but were
reduced below background levels at 12 h. This result may be due to
the pain-induced timeliness of the intrathecal injection of
ephrinB2-Fc. From the behavioral assessments, it was observed that
the pain began 3 h following intrathecal injection of ephrinB2-Fc,
which corresponded with the highest expression of calpain-1. This
may be the response of the body to a sudden onset of pain. The pain
began at 3 h and reached the lowest level at 9–12 h, following
which the pain gradually returned to the normal level. During this
period, the corresponding change in calpain-1 in the spinal cord
was gradually decreased; the levels of calpain-1 did not alter with
the development of pain, which indicated that, compared with the
stage of development, calpain-1 is more important in the initial
stage of pain. In a CCI model of sciatic nerve injury, intrathecal
injection of EphB2-Fc attenuated or reversed pain in the CCI model
mice. This may be an important target of the function of
EphB-dependent NMDA receptor in the pain process. These results
explain the potential role of ephrinB2/EphB2 in the regulation of
nociceptive pain.
In addition to receptor proteins, ephrinB/EphB
signaling can activate numerous Ca2+-dependent signals
(30) and is involved in
nociceptive neuron sensitization (10,31).
Following excitatory and ischemic insult, calpains can contribute
to cytoskeleton remodeling by changing the shape of dendritic
spines (32) and are involved in
the structural plasticity of synapses. Calpain-1 and caspase-3 have
several similar substrates, including cytoskeletal proteins,
protein kinases and transcription factors (33). Alterations in calpain levels can
trigger the activation of caspase-3 (34,35).
In ischemia and hypoxia, caspase-3 can be activated by calpain-1 to
promote apoptosis, which alters the intracellular calcium
concentration. A selective inhibitor of calpain-1, MDL28170, which
can penetrate the blood–brain barrier rapidly and exert its
activity by inhibiting brain cysteine protease activity following
systemic administration (36,37),
can also reduce neuronal apoptosis and relieve nerve damage
following ischemia. Calpains not only modulate the caspase cascade
positively and negatively during neuronal death, but also regulate
synaptic plasticity and neural circuitry (38). The results of the present study
showed a similar trend, in that the expression of calpain-1 was
increased following ephrinB2-Fc injection in a dose-dependent
manner, but was reduced to a low level at 12 h. This is consistent
with the pain behavioral assessments following the intrathecal
injection of ephrinB2-Fc: Pain began at 3 h and gradually decreased
to normal at 12 h post-injection, during which calpain-1 levels
reduced from their highest level to a low level. The level of
calpain-1 changed synchronously with the change of pain, which
showed that calpain-1 was crucial in the pain response induced by
ephrinB2-Fc injection. The administration of MDL28170 (1.0 µg),
whether as pre-treatment or post-treatment, relieved the mechanical
allodynia and thermal hyperalgesia induced by ephrinB2-Fc
injection. In addition, the data indicated a close association
between calpain-1 and caspase-3. The levels of calpain-1 and
caspase-3 increased in neurons following activation of the
ephrinB2/EphB2 signaling pathway, and increased levels of caspase-3
were followed by the upregulation of calpain-1; these increases
were inhibited by MDL28170 (1.0 µg) administration. These results
suggested that calpain-1 and caspase-3 are key in the development
of spinal nociceptive pain, and that caspases and calpains may be
involved in neuronal physiology and neurodegenerative disorders via
the ephrinB-EphB signaling pathway. These findings suggested that
the TPK-ephrinB-EphB signaling pathway is the main upstream target
of calpains and caspases during the regulation of neuronal calcium
homeostasis.
Previous studies have shown that the numbers of
neurons, astrocytes and microglia increase during neuropathic pain
(39,40). To examine which cells mediated the
effects observed in the present study, double immunofluorescent
labeling was performed, which showed that calpain-1 and caspase-3
were co-localized with NeuN, but not with GFAP or IBA-1, in the
spinal cord. This supports the finding that the interaction of
calpain-1 and caspase-3 is involved in nociceptive pain in neurons;
however, their specific roles require further investigation.
The present study found that the ephrinB/EphB
signaling pathway was involved in the processing of nociceptive
pain through the interaction of calpains and caspases. However,
only calpain-1 and caspase-3 were examined, rather than all
subtypes of the two proteins. It was hypothesized that the role of
each signaling molecule in the whole network is interrelated,
allowing the entire network to maintain a high degree of
consistency. Inhibiting one of the interactions in this network
results in a change of the entire network. Additionally, synaptic
plasticity is likely to be central to this process; however, the
function of various molecules and signal pathways were only
validated in in vivo experiments. No corresponding in
vitro experiment was performed to assess changes in axonal
function, which is a limitation of the present study. Follow-up
investigations aim to focus on in vitro experiments, to
provide further evidence.
Intrathecal injection models and CCI models were
used throughout the present study to describe spinal nociceptive
and neuropathic pain. Spinal cord nociceptive pain refers to
nociceptive pain, including inflammation, crushing, trauma and
pathological changes, in the spinal cord when subjected to various
external stimuli. Nociceptive pain is the most common type of pain
experienced. It develops when the nociceptive nerve fibers are
triggered by inflammation, chemicals or physical events, for
example stubbing a toe on a piece of furniture (41,42).
Depending on their timescale, pain can be divided into acute pain
and chronic pain. Neuropathic pain is a type of pain syndrome
caused by damage to the central or peripheral nervous system
(43), and has become one of the
most urgent problems to address. The main features of neuropathic
pain include positive symptoms, including progressive pain,
paroxysmal pain and tactile allodynia, and unhealthy phenomena,
including sensory loss at the site of painful (44). Predominantly based on chronic pain,
neuropathic pain usually continues for 2–3 weeks or longer, leading
to permanent damage to the body and psychology to a certain extent.
In the present study, normal mice and CCI mice were intrathecally
injected with ephrinB2-Fc and EphB2-Fc. The pain caused by
ephrinB2-Fc injection differs from neuropathic pain; its duration
lasts only ~12 h, and is equivalent to acute pain. The reason for
including the CCI model in experiments was mainly to achieve a
combination of acute pain and chronic neuropathic pain to better
illustrate the role of the ephrinB/EphB signaling pathway in pain,
not just neuropathic pain.
In conclusion, the results of the present study
demonstrated that calpain-1 and caspase-3 were involved in the
generation and maintenance of spinal nociceptive pain. Calpain-1 is
upstream of caspase-3 in the signaling pathway, and their
interaction in the nociceptive stimulus induced by the
ephrinB2/EphB2 signaling pathway occurs via neurons. These findings
revealed the role of ephrinB/EphB signaling in the processing of
spinal nociception and suggested that calpain-1 may be a novel
target for changes in synaptic plasticity during
ephrinB/EphB-induced spinal nociceptive pain.
Acknowledgements
The authors would like to thank the Department of
Anesthesiology, The Third Affiliated Hospital of Second Military
Medical University (Shanghai, China) for their assistance and
expertise.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81371253 and 81171054) and
the Shanghai Education Innovation Fund Project (grant no.
1477083).
Availability of data and materials
The data that support the findings of this study are
an important part of the follow-up study. To avoid affecting the
publication of subsequent articles, the relevant data of the
manuscript cannot be made publicly available.
Authors' contributions
MY contributed to the conception of the study and
manuscript preparation, WC performed the experiments, YZ
contributed significantly to analysis, RY and YRW performed the
data analyses and wrote the manuscript, and HY helped perform the
analysis and offered constructive discussions.
Ethics approval and consent to
participate
All experiments were approved by the Animal
Experimental Center, Second Military Medical University (production
license no. SCXK (hu)2012-0003).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Doth AH, Hansson PT, Jensen MP and Taylor
RS: The burden of neuropathic pain: A systematic review and
meta-analysis of health utilities. Pain. 149:338–344. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao J, Yuan G, Cendan CM, Nassar MA,
Lagerström MC, Kullander K, Gavazzi I and Wood JN:
Nociceptor-expressed ephrin-B2 regulates inflammatory and
neuropathic pain. Mol Pain. 6:772010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li XY, Ko HG, Chen T, Descalzi G, Koga K,
Wang H, Kim SS, Shang Y, Kwak C, Park SW, et al: Alleviating
neuropathic pain hypersensitivity by inhibiting PKMzeta in the
anterior cingulate cortex. Science. 330:1400–1404. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen ZY, Shen FY, Jiang L, Zhao X, Shen
XL, Zhong W, Liu S, Wang ZR and Wang YW: Attenuation of neuropathic
pain by inhibiting electrical synapses in the anterior cingulate
cortex. Anesthesiology. 124:169–183. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen ZL, Yu WM and Strickland S:
Peripheral regeneration. Ann Rev Neurosci. 30:209–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiang XY, Zhang HM, Hu NW, Zhou LJ, Zhang
T and Liu XG: A quantitative study of the synaptic alterations in
spinal dorsal horn during the induction and maintenance of
long-term potentiation. Sheng Li Xue Bao. 56:397–402. 2004.(In
Chinese). PubMed/NCBI
|
|
7
|
Kuner R: Central mechanisms of
pathological pain. Nat Med. 16:1258–1266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guan XH, Lu XF, Zhang HX, Wu JR, Yuan Y,
Bao Q, Ling DY and Cao JL: Phosphatidylinositol 3-kinase mediates
pain behaviors induced by activation of peripheral ephrinBs/EphBs
signaling in mice. Pharmacol Biochem Behav. 95:315–324. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klein R: Bidirectional modulation of
synaptic functions by Eph/ephrin signaling. Nat Neurosci. 12:15–20.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhuang Z, Huang J and Cepero ML: Eph
signaling reglates gliotransmitter release. Commum Integr Biol.
4:223–226. 2011. View Article : Google Scholar
|
|
11
|
Bali KK, Venkataramani V, Satagopam VP,
Gupta P, Schneider R and Kuner R: Transcriptional mechanisms
underlying sensitization of peripheral sensory neurons by
granulocyte-/granulocyte-macrophage colony stimulating factors. Mol
Pain. 9:482013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang XH, Jiang JT, Yang XY, Han JC and
Zheng QS: Licochalcone A induces T24 bladder cancer cell apoptosis
by increasing intracellular calcium levels. Mol Med Rep.
14:911–919. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pereira MB, Santos AM, Gonçalves DC,
Cardoso AC, Consonni SR, Gozzo FC, Oliveira PS, Pereira AH,
Figueiredo AR, Tiroli-Cepeda AO, et al: αB-crystallin interacts
with and prevents stress-activated proteolysis of focal adhesion
kinase by calpain in cardiomyocytes. Nat Comm. 5:51592014.
View Article : Google Scholar
|
|
14
|
Fang J, Liu X, Bolanos L, Barker B,
Rigolino C, Cortelezzi A, Oliva EN, Cuzzola M, Grimes HL,
Fontanillo C, et al: A calcium- and calpain-dependent pathway
determines the response to lenalidomide in myelodysplastic
syndromes. Nat Med. 22:727–734. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pörn-Ares MI, Samali A and Orrenius S:
Cleavage of the calpain inhibitor, calpastatin, during apoptosis.
Cell Death Differ. 5:1028–1033. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li M, Wang X, Yu Y, Yu Y, Xie Y, Zou Y, Ge
J, Peng T and Chen R: Coxsackievirus B3-induced calpain activation
facilitates the progeny virus replication via a likely mechanism
related with both autophagy enhancement and apoptosis inhibition in
the early phase of infection: An in vitro study in H9c2 cells.
Virus Res. 179:177–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chan SL and Mattson MP: Caspase and
calpain substrates: roles in synaptic plasticity and cell death. J
Neurosci Res. 58:167–190. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hylden JL and Wilcox GL: Intrathecal
morphine in mice: A new technique. Eur J Pharmacol. 67:313–316.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bennett GJ and Xie YK: A peripheral
mononeuropathy in rat that produces disorders of pain sensation
like those seen in man. Pain. 33:87–107. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chaplan SR, Bach FW, Pogrel JW, Chung JM
and Yaksh TL: Quantitative assessment of tactile allodynia in the
rat paw. J Neurosci Methods. 53:55–63. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hargreaves K, Dubner R, Brown F, Flores C
and Joris J: A new and sensitive method for measuring thermal
nociception in cutaneous hyperalgesia. Pain. 32:77–88. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou XL, Wang Y, Zhang CJ, Yu LN, Cao JL
and Yan M: COX-2 is required for the modulation of spinal
nociceptive information related to ephrinB/EphB signalling. Eur J
Pain. 19:1277–1287. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xia WS, Peng YN, Tang LH, Jiang LS, Yu LN,
Zhou XL, Zhang FJ and Yan M: Spinal ephrinB/EphB signalling
contributed to remifentanil-induced hyperalgesia via NMDA receptor.
Eur J Pain. 18:1231–1239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Henderson JT, Georgiou J, Jia Z, Robertson
J, Elowe S, Roder JC and Pawson T: The receptor tyrosine kinase
EphB2 regulates NMDA-dependent synaptic function. Neuron.
32:1041–1056. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grunwald IC, Korte M, Wolfer D, Wilkinson
GA, Unsicker K, Lipp HP, Bonhoeffer T and Klein R:
Kinase-independent requirement of EphB2 receptors in hippocampal
synaptic plasticity. Neuron. 32:1027–1040. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu S, Liu WT, Liu YP, Dong HL, Henkemeyer
M, Xiong LZ and Song XJ: Blocking EphB1 receptor forward signaling
in spinal cord relieves bone cancer pain and rescues analgesic
effect of morphine treatment in rodents. Cancer Res. 71:4392–4402.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hoogenraad CC, Milstein AD, Ethell IM,
Henkemeyer M and Sheng M: GRIP1 controls dendrite morphogenesis by
regulating EphB receptor trafficking. Nature Neurosci. 8:906–915.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bundesen LQ, Scheel TA, Bregman BS and
Kromer LF: Ephrin-B2 and EphB2 regulation of astrocyte-meningeal
fibroblast interactions in response to spinal cord lesions in adult
rats. J Neurosci. 23:7789–800. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nwankwo JO, Lei JX, Xu J, Rivera A, Gupta
K and Chishti AH: Genetic inactivation of calpain-1 attenuates pain
sensitivity in a humanized mouse model of sickle cell disease.
Haematologica. 101:e397–e400. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dosemeci A and Reese TS: Effect of calpain
on the composition and structure of postsynaptic densities.
Synapse. 20:91–97. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang KK: Calpain and caspase: Can you tell
the difference? Trends Neurosci. 23:20–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amini M, Ma CL, Farazifard R, Zhu G, Zhang
Y, Vanderluit J, Zoltewicz JS, Hage F, Savitt JM, Lagace DC, et al:
Conditional disruption of calpain in the CNS alters dendrite
morphology, impairs LTP, and promotes neuronal survival following
injury. J Neurosci. 33:5773–5784. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mouatt-Prigent A, Karisson JO, Agid Y and
Hirsch EC: Increased M-calpain expression in the mesencephalon of
patients with Parkinson's disease but not in other
neurodegenerative disorders involving the mesencephalon: A role in
nerve cell death? Neuroscience. 73:979–987. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Urthaler F, Wolkowicz PE, Digerness SB,
Harris KD and Walker AA: MDL-28170, a membrane-permeant calpain
inhibitor, attenuates stunning and PKC epsilon proteolysis in
reperfused ferret hearts. Cardiovasc Res. 35:60–67. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li P, Wendy H, He QP, Miyashita H,
Siddiqui M and Shuaib A: Postischemic treatment with calpain
inhibitor MDL 28170 ameliorates brain damage in a gerbil model of
global ischemia. Neurosci Lett. 247:17–20. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Peng J, Gu N, Zhou L, Eyo B U, Murugan M,
Gan WB and Wu LJ: Microglia and monocytes synergistically promote
the transition from acute to chronic pain after nerve injury. Nat
Commun. 7:120292015. View Article : Google Scholar
|
|
38
|
Xu JY, Jiang Y, Liu W and Huang YG:
Calpain inhibitor reduces cancer-induced bone pain possibly through
inhibition of osteoclastogenesis in rat cancer-induced bone pain
model. Chin Med J (Engl). 128:1102–1107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gu N, Peng J, Murugan M, Wang X, Eyo UB,
Sun D, Ren Y, DiCicco-Bloom E, Young W, Dong H and Wu LJ: Spinal
microgliosis due to resident microglial proliferation is required
for pain hypersensitivity after peripheral nerve injury. Cell Rep.
16:605–614. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Battaglia AA, Sehayek K, Grist J, McMahon
SB and Gavazzi I: EphB receptors and ephrin-B ligands regulate
spinal sensory connectivity and modulate pain processing. Nat
Neurosci. 6:339–340. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Epstein JB, Wilkie DJ, Fischer DJ, Kim YO
and Villines D: Neuropathic and nociceptive pain in head and neck
cancer patients receiving radiation therapy. Head Neck Oncol.
1:262009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ruscheweyh R, Wilder-Smith O, Drdla R, Liu
XG and Sandkühler J: Long-term potentiation in spinal nociceptive
pathways as a novel target for pain therapy. Mol Pain. 7:202011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nakai K, Nakae A, Oba S, Mashimo T and
Ueda K: P2X4 receptor expression in a rat model of trigeminal
neuropathic pain. Neuroreport. 21:559–563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Attal N: Neuropathic Pain: Mechanisms,
therapeutic approach, and interpretation of clinical trials.
Continuum (Minneap Minn). 18:161–175. 2012.PubMed/NCBI
|