Introduction
According to World Health Organization (WHO) 2008
Classification of tumours of haematopoietic and lymphoid tissues
and 2016 revision, myeloproliferative neoplasms (MPNs) can be
classified into two major groups, chronic myeloid leukemia (CML)
and Philadelphia-negative MPNs (PN-MPNs), such as polycythemia vera
(PV), essential thrombocythemia (ET) and primary myelofibrosis
(PMF) (1). These disorders are
more frequently found in elderly patients, mostly in men (1).
One of the major genetic insights into the
pathogenesis of the PN-MPNs included the identification of the
somatic point gain-of-function mutations in Janus kinase 2 gene
(JAK2), leading to the activation of the JAK/STAT signaling
pathway (signal transducer and activator of transcription),
culminating in exacerbated cellular proliferation, resistance to
apoptosis and evolution to MPNs (2–4). On
the other hand, the identification of Philadelphia chromosome (Ph),
a translocation involving chromosomes 9 and 22 that results in the
formation of the BCR-ABL fusion gene, constitutes the
defining leukemogenic event in CML (5,6). ET
is characterized by a high platelet count, often associated with
thrombotic and hemorrhagic events, and the presence of JAK2
mutation in about 50–60% of cases (7–9).
As far as we know from literature revision, the
frequency of concurrent presence of JAK2V617F mutation and
BCR-ABL translocation in a single individual with a MPN is a
rare event, independently of what phenotype expresses earlier,
PN-MPN or CML (10–13).
Although ET and CML are considered to be mutually
exclusive, rare cases of concomitant presence of BCR-ABL
translocation positive CML and JAK2V617F mutation positive
ET have been reported in the literature (10,13).
We report here the case of two patients initially
included in a data base population of 58 patients with the
diagnosis of ET in the presence of JAK2V617F mutation, with
the suspicion of coexistence with BCR-ABL translocation.
Patient anonymity was guaranteed and consent was provided, in
agreement with the Declaration of Helsinki. The Institutional
Ethics Board of the Hospital of São Francisco Xavier, West Lisbon
Hospital Centre (Lisbon, Portugal) approved the present study.
Case report
Case report 1
A 75-year-old man with a medical history of
dyslipidemia, hypertension, acute myocardial infarction, and
ischemic stroke in August 2013. In December 2013 this patient was
hospitalized with his second ischemic stroke. Although he had
confirmed poor adherence to the prescribed therapy for
cardiovascular risk patients, in January 2014 he was referred to
the hematology consultation for maintained thrombocytosis and
leukocytosis, since at least August 2013 (as there was no previous
laboratory data available).
Evaluation revealed platelet count of
1,405×109/l, leukocytosis (15×109/l), with
normal formula, and without immature precursor cells as well as
normal hemoglobin (Table I).
 | Table I.Results over time and therapy
prescribed for case study 1. |
Table I.
Results over time and therapy
prescribed for case study 1.
|
| Time point |
|---|
|
|
|
|---|
|
| 2014 | 2016 | 2017 |
|---|
|
|
|
|
|
|---|
| Characteristic | January | February | June | September | August | January |
|---|
| Platelets
(×109/l) | 1,405 | 698 | 375 | 547 | 199 | 1,596 |
| Hemoglobin
(g/dl) | 14.3 | 13.4 | 12.3 | 12.8 | 13.9 |
7.9 |
| Leucocytes
(×109/l) | 15.1 |
6.9 |
6.2 |
6.3 |
3.9 | 18.9 |
| JAK2V617F
mutation | Positive (25%) | – | – |
| Positive | – |
| BCR/ABL
t(9;22) (FISH) | Positive 16%
(atypical pattern) | – | – | Positive 21%
(atypical pattern) | – | – |
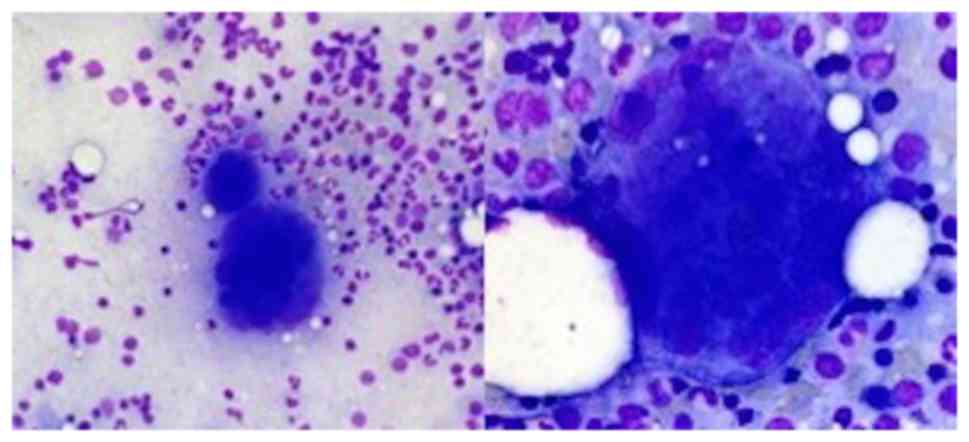
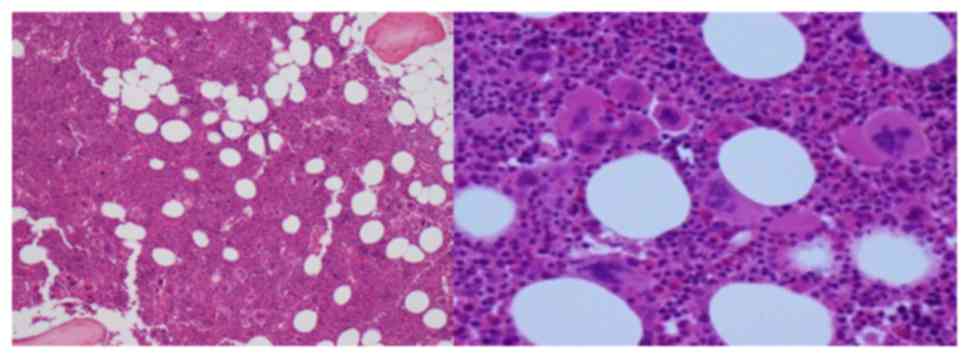
Abdominal ultrasound confirmed absent splenomegaly
and Bone marrow (BM) aspirate showed megakaryocytic hyperplasia and
enlarged megakaryocytes, with no abnormalities of the myeloid and
erythroid series (Fig. 1). BM
biopsy showed a hypercellular marrow (80%), megakaryocytic and
granulocytic (slight) hyperplasia (Fig. 2).
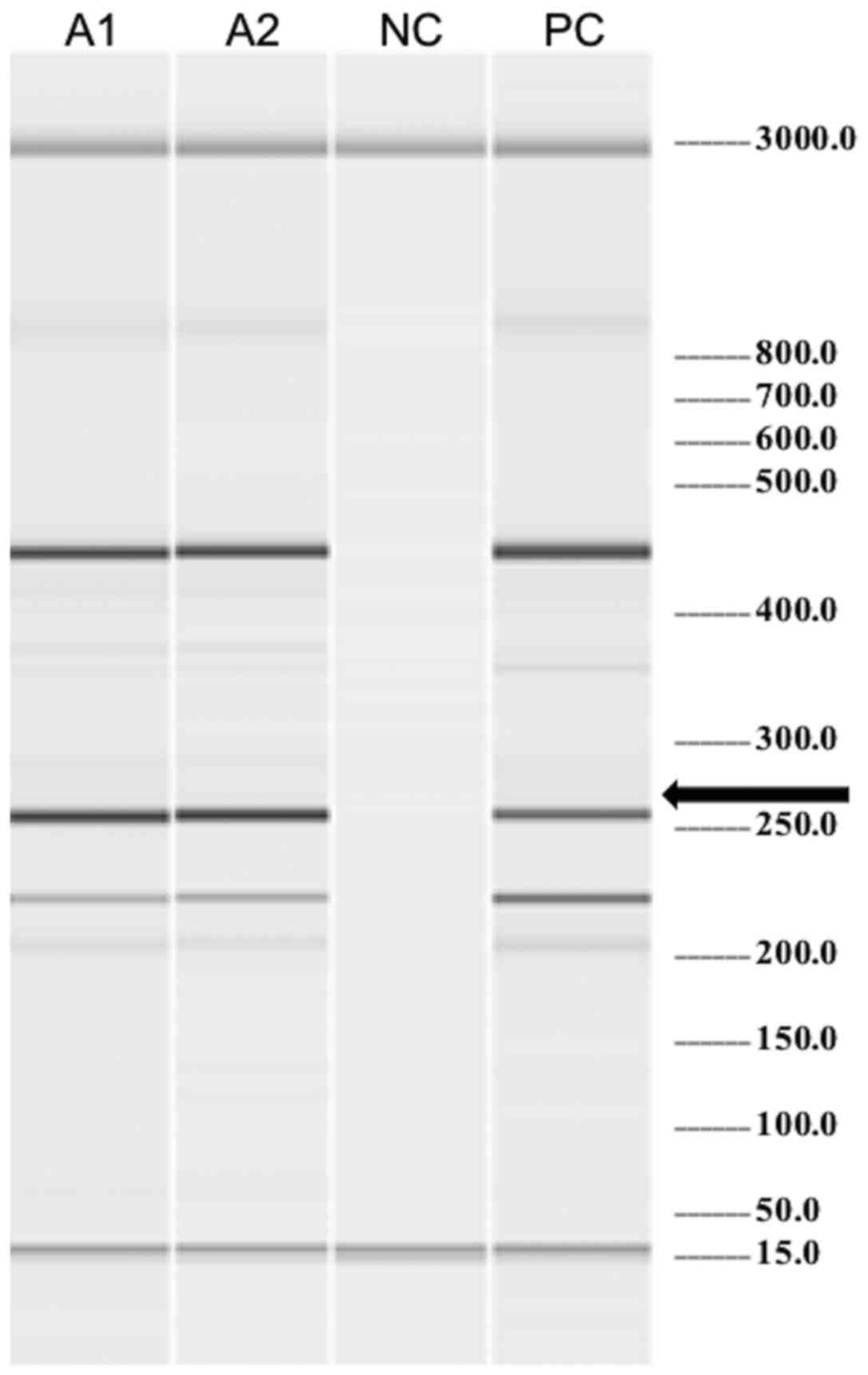
Molecular biology (Fig.
3) and cytogenetic tests were performed in peripheral blood and
the results revealed positivity for the JAK2V617F mutation
and a karyotype of 45,X,-Y[5]/46,XY[15]. The fluorescence in
situ hybridization (FISH) was positive for the BCR-ABL
translocation in 16% with an atypical pattern. The BCR-ABL
transcript was not detected by the conventional reverse
transcriptase-polymerase chain reaction (RT-PCR) method (specific
for p190 and p210 transcripts). This high risk patient received a
daily hydroxyurea (HU), and low dose aspirin regimen as secondary
thrombotic prevention. A good response to treatment was achieved,
with normalization of leukocytes and platelets reduction of greater
than 50% after one month and normalization of platelets after five
months of therapy (Table I). This
patient had very poor compliance to the therapy and hospital
check-ups, so tyrosine kinase inhibitor (TKI) that was planned to
be introduced, was never started since the patient did not come to
collect the medication at the hospital. His clinical and laboratory
situation has worsened and the patient died by the beginning of
2017, from infectious complications.
Case report 2
A 76 years old man, with previous history of
cardiovascular risk factors, namely Diabetes mellitus, dyslipidemia
and Ischemic cardiomiopathy submitted to cardiac bypass due to
myocardial infarction in 2001.
Presented with isolated thrombocytosis
(1,022×109/l) in 2005, which led the patient to the
Hematology Department to study the etiology behind the maintained
increased level of platelet count (Table II).
| Table II.Results over time and therapy
prescribed for case study 2. |
Table II.
Results over time and therapy
prescribed for case study 2.
|
| Time point |
|---|
|
|
|
|---|
|
| 2005 | 2009 | 2013 | 2016 | 2017 |
|---|
|
|
|
|
|
|
|
|---|
| Characteristic | March | March | September | February | January | August |
|---|
| Platelets
(×109/l) | 1022 | 478 | 684 | 909 | 413 | 252 |
| Hemoglobin
(g/dl) | 14.6 | 14.7 | 12.2 | 13.1 | 14.1 | 11.8 |
| Leucocytes
(×109/l) |
9.4 |
6.9 | 22.4 | 30.1 | 38.4 |
7.3 |
| JAK2V617F
mutation | – | – | – | – | Positive | – |
| BCR/ABL t(9;22)
(FISH) | – | – | – | – | Positive 17%
(atypical pattern) | – |
In a patient with previous history of thrombotic
event, it was imperative to understand the etiology of such
abnormal changes in blood analysis, since it might have been in
close relation to the previous cardiac event described.
At this time, high platelet count was asymptomatic,
and there was neither clinical nor analytical blood data for
detecting an associated inflammatory process or any recent
surgeries explaining this finding.
Abdominal ultrasound showed normal spleen
morphology, and there were no Howell-Jolly bodies nor pitted
erythrocytes found in blood smear analysis, that could be
interpreted as reactive thrombocytosis due to functional
hyposplenism.
Blood sideremia and iron stores were between normal
ranges, and no history of hemorrhage was present. Excluded
secondary causes of thrombocytosis and based on an indolent
clinical course, a primary cause was admitted. The MPNs are the
most common responsible entities and so cytogenetics and molecular
biology tests on JAK2V617F mutation and BCR-ABL
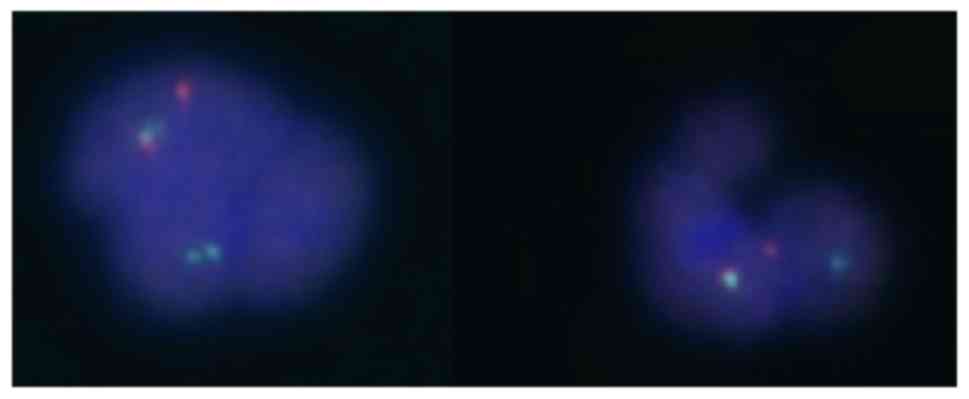
t(9;22) were performed in peripheral blood. JAK2V617F was
positive and, once again, the FISH test was positive for the
BCR-ABL translocation in 17% with an atypical pattern
(Fig. 4), but BCR-ABL
transcript was not detected by the RT-PCR method. No metaphases
were observed in the karyotype for evaluation. Having this data
discussed, and taking into account the presence of these mutations,
the diagnosis of ET JAK2V617F and BCR-ABL positive
was admitted. The patient started on HU 500 mg (alternate day
progressing to 1 g/alternate day) and TKI (Imatinib, 400 mg/day). A
few months later, TKI was suspended and the patient remained under
treatment with HU, actually with well controlled disease.
Regarding the methodology used for genetic study,
extraction of whole DNA from peripheral blood was accomplished by
cell lysis followed by ethanol precipitation and recovery of the
DNA by elution in a buffer solution (QIAamp® DNA Mini
kit; Qiagen GmbH, Hilden, Germany). The presence/absence of
JAK2V617F mutation was determined by amplification
refractory mutation system (ARMS)-PCR (in-house), based on
amplification of a genomic fragment which includes the region
corresponding to amino acid 617 of the JAK2 protein, and on the
differential detection on agarose gel of the normal or mutated
alleles through the use of allele-specific primers. The test result
is qualitative and the test sensitivity is 1%. Quantification of
JAK2 was obtained by high resolution melting PCR (HRM-PCR)
(LightCycler® 480 Instrument; Roche Molecular
Diagnostics, Pleasanton, CA, USA), with a sensitivity of about 10%
of mutated cells. Conventional RT-PCR was performed for the
identification of BCR-ABL transcripts (specific for p190 and
p210), after RNA extraction, according to the methodology described
by van Dongen et al (14).
Results are analyzed on agarose gel electrophoresis. FISH analysis
was done on 100 nuclei after hybridization with specific probes for
t(9;22) BCR-ABL (Vysis LSI BCR-ABL Dual Color, Dual
Fusion Translocation Probe).
Discussion
Several authors have investigated the relationship
between JAK2V617F and BCR-ABL anomalies and many
theories have been postulated in the last years, especially after
the identification of JAK2V617F mutation in 2005.
The Janus kinase 2 gene (JAK2; cytogenetic
location: 9p24.1) provides instructions for making a protein that
promotes the growth and division (proliferation) of cells. This
protein is part of a signaling pathway called the JAK/STAT pathway,
which transmits chemical signals from outside the cell to the
cell's nucleus. The JAK2 protein is especially important for
controlling the production of blood cells from hematopoietic stem
cells. These stem cells are located within the bone marrow and have
the potential to develop into red blood cells, white blood cells,
and platelets.
The Philadelphia chromosome (chromosome 22) results
from the reciprocal translocation of genetic material between
chromosome 9 and chromosome 22, and contains the fusion gene
BCR-ABL, which codes for a tyrosine kinase signaling protein
that causes the cells to divide uncontrollably (particularly CML
cells).
From 2007 to 2015, at least 42 patients with this
double mutated phenotype were reported in the literature (10,12,15,16).
Moreover, the italian group of Pieri et al (17) studied 314 patients with CML and
identified 8 cases (2.55%) with concomitant JAK2V617F
mutation. Pagnano et al detected only one case with
JAK2V617F mutation among 55 cases of CML analyzed (13).
Among these different studies reported, several
patterns were described: i) Initially diagnosed with CML and
treated with imatinib that proceeded to a JAK2V617F
myelopro-liferative phenotype; ii) initially diagnosed with CML
coexisting with JAK2V617F mutation positive PV, ET or PMF;
or iii) initially diagnosed with JAK2V617F mutation positive
PN-MPN, ET more rarely, evolving years later to CML (10). Commonly, men above 50 years old
were the most frequently affected (10).
A question still has to be clarified: Which is the
first anomaly to occur? Several working groups reported that in
some cases of PN-MPNs that evolved to CML, JAK2V617F
mutation was the first leukemogenic event and BCR-ABL the second
positive clone (10). Moreover, it
was also speculated that JAK2V617F mutations are present in
hematopoietic stem cells, with an additional BCR-ABL
translocation being subsequently acquired in a sub-clone (10,18).
However, other groups didn't confirm these results, and postulate
that the two anomalies are present since the beginning of the
process (10). Indeed, about the
amount of cellular clones involved, there are reports that state
that two different clones are involved, with the phenotypic
expression depending on which one of the aberrations is
‘dominating’, as a result of therapy targeted to the other anomaly
(10,11,19,20).
On the other hand, there are some authors evidencing that all the
myeloid cells bear JAK2V617F mutation, including
granulocytic and erythroid colonies, while BCR-ABL
translocation is confined to a small compartment of myeloid
progenitor cells, only in granulocytic colonies (10). In contrast, other reports showed
the simultaneous presence of both BCR-ABL transcript and
JAK2V617F mutation in the majority of granulocytic and
erythroid colonies at the time CML diagnosis was established,
corroborating the hypothesis that only one cellular clone is
bearing concomitantly the two anomalies (10,11).
Therefore, the phenotypic heterogeneity can be the
result of the expression of a pre-existing mutated clone previously
‘silent’ or of the accumulation of several genetic events
conferring genetic instability and leading to a ‘new’ anomaly
(12).
As far as we know from literature revision, there
are no other reports of positivity for JAK2 mutations, other
than V617F, with the concomitant presence of BCR-ABL
translocation.
One of the diagnostic criteria for ET, is the
absence of the Ph chromosome. BCR-ABL positive ET without
features of CML in blood and bone marrow is a rare entity and
constitute less than 5% of ET diagnosis. Some authors have proposed
to consider those cases as CML associated with a rather poor
prognosis because of the high tendency to progress to myelofibrosis
and blastic transformation after a few to several years (16,20,21).
An important difference between BCR-ABL
positive ET and BCR-ABL positive CML at time of presentation
is the absence of splenomegaly in the first situation (16).
The bone marrow in BCR-ABL positive ET is
featured by predominant smaller than normal and hypo/mononucleated
megakaryocytes caused by BCR-ABL gene and protein induced
maturation defect of the hematopoietic stem cells. This contrasts
with clustered enlarged megakaryocytes in BCR-ABL negative
ET due to growth advantage and proliferation of constitutively
activated JAK2 or MPL somatic mutated megakaryocytes
(16).
The first patient reported had diagnostic features
that matched CML and ET. However, his overall clinical presentation
including bone marrow features was more commonly suggestive of ET.
Since the t(9;22) was positive in FISH, according to the results,
there should have been found a positive result in molecular biology
tests as well. Moreover, no Ph chromosome was detected by
karyotype. The second patient reported was more suggestive of ET
and did not have typical clinical, nor morphologic findings for
CML. The t(9;22) was also positive only by FISH, with a negative
result in molecular biology tests. In this case it was not possible
to evaluate karyotype due to metaphases absence.
In both patients, search for JAK2V617F
mutation and BCR-ABL was concomitant, making very difficult
to know if both mutations were present from the beginning or the
order of appearance of each one of them. The fact that the study
has been performed before therapy institution, excludes the
possible inhibitory effect of it over one of the altered clones,
making the other more expressive.
Given the above, several questions have to be
raised: Are these genomic alterations found in these two patients
and their atypical pattern really true and clinically significant
or are they false positive results? May those be new/distinct
clinical entities? Should we consider Ph positive ET as distinct
entity, separate from Ph negative ET and Ph positive CML?
As mentioned above, studies describing cases
initially diagnosed with JAK2V617F mutation positive PN-MPN,
evolving later to CML, were rarely ET patients (10), in contrast to our report.
Although, the concomitant presence of these two
anomalies in these patients didn't seemed to exclude the diagnosis
of ET, at diagnosis or in some point of their clinical course, both
patients evidenced a distinct clinical (thrombocytosis with
associated leukocytosis) or morpho-histological (megakaryocytic)
phenotype from what was expected for ET with isolated
JAK2V617F positive or Ph positive CML, but apparently not
influencing the course of the disease.
Both patients showed an atypical pattern for
BCR-ABL translocation search by FISH, said to be atypical
because only one fusion signal was observed, instead of the two
signals expected, with a percentage of BCR-ABL translocation
of approximately 20%. RT-PCR was performed using only a single
primer pair, failing the identification of p190 and p210
transcripts of BCR-ABL fusion gene, and making the presence
of BCR-ABL tyrosine kinase activity questionable. Real time PCR was
a distinct possible technique to be used for the identification and
quantification of BCR-ABL p210 (mainly b3a2 and b2a2 types)
transcripts, however it was not performed.
Since no BCR-ABL transcripts were detected by
RT-PCR, one hypothesis is that the unique fusion signal detected by
FISH could correspond only to der(9), and not to Ph chromosome with
associated tyrosine kinase activity (on chromosome 22).
Confirmation could be achieved doing FISH in metaphases, which was
only possible in the first case, since the second patient had no
metaphases to allow it. This way, we were not able to be sure of
the localization of break points and consequent fusion.
On the other hand, a missense on the primer site or
the probe pairing region could also explain such RT-PCR result, but
there is a vast experience with the used primers, internationally
designed and certified.
Regarding clarification of the possible mechanism of
association of JAK2V617F mutation and a ‘true’
BCR-ABL translocation involved in our patients, it would be
useful to analyze JAK2V617F mutation and BCR-ABL gene
in each colony of BFU-E or CFU-C.
Given the above, probably these cases correspond to
two patients with a variant ET, in which we possibly can
hypothesize that the presence of der(9) chromosome might be
involved in those phenotypic differences. As far as we are aware,
no other studies describing these two ‘truly’ genomic alterations
have found a BCR-ABL aberrant pattern similar to our cases.
However, Larsen et al (22). Described the case of four patients
JAK2V617F positive with associated distinct karyotypic
aberrations [including der(9;18)], presenting with a distinct
clinical and prognostic profile. Likewise, another study also
reported the association of der(9) chromosome and acute
lymphoblastic leukemia (23), with
prognostic impact.
Moreover, WHO does not currently address the
classification of MPNs that have more than one genetic abnormality,
but it is well established that the presence of additional
co-operating mutations in myeloid genes (along with other important
risk factors) has a straight relationship with phenotype and
clinical outcome definition (24,25).
Cytogenetic analysis allows to identify subgroups of patients with
a distinct phenotype and prognostic profile, and should be
performed in conjunction with JAK2 mutation analysis PN-MPNs
patients (22).
Furthermore, the concomitant presence of two
molecular markers is well defined for certain diseases, and raises
several issues, including the best therapeutic strategy to adopt.
But, therapeutic decisions should not be based only on molecular
biology test results (18).
CML can express on the background of a
JAK2V617F positive PN-MPN, and treatment with TKI might
reveal/make more expressive the PN-phenotype. It is of great
importance to recognize and investigate the association of both
anomalies, especially in CML patients who have an unusual
clinical/laboratorial course, with hemoglobin and/or platelets
count increase, or when they do not respond to therapy, making the
diagnosis of other MPNs to have practical therapeutic
consequences.
It seems that for these complex patients the most
efficient therapeutic choice is to associate a TKI with a
JAK2 inhibitor (10,11).
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FM and APA contributed to the conception and design
of the work, acquisition, analysis and interpretation of data;
drafted and wrote the manuscript, revising it critically for
important intellectual content; were accountable for all aspects of
the work in ensuring that questions related to the accuracy and
integrity of any part of the work were appropriately investigated
and resolved. TM, PSS, RC, SM, SS, JFV and FL analyzed the data and
revised the paper. SR provided the histological images and revised
the paper. All authors approved the final manuscript.
Ethics approval and consent to
participate
Patient anonymity and consent was guaranteed, in
agreement with the Declaration of Helsinki. the Institutional Ethic
Board of Hospital of São Francisco Xavier, West Lisbon Hospital
Centre (ref. no. 120/CE_2009) approved this study.
Consent for publication
Patient anonymity and consent was guaranteed, in
agreement with the Declaration of Helsinki.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kralovics R, Passamonti F, Buser AS, Teo
SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M and Skoda RC: A
gain-of-function mutation of JAK2 in myeloproliferative disorders.
N Engl J Med. 352:1779–1790. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levine RL, Wadleigh M, Cools J, Ebert BL,
Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et
al: Activating mutation in the tyrosine kinase JAK2 in polycythemia
vera, essential thrombocythemia, and myeloid metaplasia with
myelofibrosis. Cancer Cell. 7:387–397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baxter EJ, Scott LM, Campbell PJ, East C,
Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N,
et al: Acquired mutation of the tyrosine kinase JAK2 in human
myeloproliferative disorders. Lancet. 365:1054–1061. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nowell PC: The minute chromosome (Phl) in
chronic granulocytic leukemia. Blut. 8:65–66. 1962. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rowley JD: Letter: A new consistent
chromosomal abnormality in chronic myelogenous leukaemia identified
by quinacrine fluorescence and Giemsa staining. Nature.
243:290–293. 1973. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tefferi A and Pardanani A:
Myeloproliferative, neoplasms: A contemporary review. JAMA Oncol.
1:97–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine RL: Mechanisms of mutations in
myeloproliferative neoplasms. Best Pract Res Clin Haematol.
22:489–494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hinds DA, Barnholt KE, Mesa RA, Kiefer AK,
Do CB, Eriksson N, Mountain JL, Francke U, Tung JY, Nguyen HM, et
al: Germ line variants predispose to both JAK2 V617F clonal
hematopoiesis and myeloproliferative neoplasms. Blood.
128:1121–1128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qin YW, Yang YN, Li S and Wang C:
Coexistence of JAK2V617F mutation and BCR-ABL translocation in a
pregnant woman with essential thrombocythemia. Indian J Hematol
Blood Transfus. 30 Suppl 1:S331–S334. 2014. View Article : Google Scholar
|
|
11
|
Zhou A, Knoche EM, Engle EK, Fisher DA and
Oh ST: Concomitant JAK2 V617F-positive polycythemia vera and
BCR-ABL-positive chronic myelogenous leukemia treated with
ruxolitinib and dasatinib. Blood Cancer J. 5:e3512015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ursuleac I, Colita A, Adam T, Jardan C,
Ilea A and Coriu D: The concomitant occurrence of JAK2V617F
mutation and BCR/ABL transcript with phenotypic expression-an
overlapping myeloproliferative disorder or two distinct diseases?
-case report. J Med Life. 6:34–37. 2013.PubMed/NCBI
|
|
13
|
Pagnano KB, Delamain MT, Magnus MM,
Vassallo J, DE Souza CA, DE Almeida D and Lorand-Metze I:
Concomitant essential thrombocythemia with JAK2 V617F mutation in a
patient with chronic myeloid leukemia with major molecular response
with imatinib and long-term follow-up. Oncol Lett. 12:485–487.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Dongen JJ, Macintyre EA, Gabert JA,
Delabesse E, Rossi V, Saglio G, Gottardi E, Rambaldi A, Dotti G,
Griesinger F, et al: Standardized RT-PCR analysis of fusion gene
transcripts from chromosome aberrations in acute leukemia for
detection of minimal residual disease. Report of the BIOMED-1
concerted action: Investigation of minimal residual disease in
acute leukemia. Leukemia. 13:1901–1928. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hummel JM, Kletecka MC, Sanks JK,
Chiselite MD, Roulston D, Smith LB, Czuchlewski DR,
Elenitoba-Johnson KS and Lim MS: Concomitant BCR-ABL1 translocation
and JAK2(V617F) mutation in three patients with myeloproliferative
neoplasms. Diagn Mol Pathol. 21:176–183. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michiels JJ, Ten Kate FWJ, De Raeve H and
Gadisseur A: Bone marrow features and natural history of
BCR/ABL-positive thrombocythemia and chronic myeloid leukemia
compared to BCR/ABL-negative thrombocythemia in essential
thrombocythemia and polycythemia vera. J Hematol Thromboembolic
Dis. 3:2015.
|
|
17
|
Pieri L, Spolverini A, Scappini B, Occhini
U, Birtolo S, Bosi A, Albano F, Fava C and Vannucchi AM:
Concomitant occurrence of BCR-ABL and JAK2V617F mutation. Blood.
118:3445–3446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heller P, Kornblihtt LI, Cuello MT,
Larripa I, Najfeld V and Molinas FC: BCR-ABL transcripts may be
detected in essential thrombocythemia but lack clinical
significance. Blood. 98:19902001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bee PC, Gan GG, Nadarajan VS, Latiff NA
and Menaka N: A man with concomitant polycythaemia vera and chronic
myeloid leukemia: The dynamics of the two disorders. Int J Hematol.
91:136–139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kwong YL, Chiu EK, Liang RH, Chan V and
Chan TK: Essential thrombocythemia with BCR/ABL rearrangement.
Cancer Genet Cytogenet. 89:74–76. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
LeBrun DP, Pinkerton PH, Sheridan BL,
Chen-Lai J, Dubé ID and Poldre PA: Essential thrombocythemia with
the Philadelphia chromosome and BCR-ABL gene rearrangement. An
entity distinct from chronic myeloid leukemia and Philadelphia
chromosome-negative essential thrombocythemia. Cancer Genet
Cytogenet. 54:21–25. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Larsen TS, Hasselbalch HC, Pallisgaard N
and Kerndrup GB: A der(18)t(9;18)(p13;p11) and a der(9;18)(p10;q10)
in polycythemia vera associated with a hyperproliferative phenotype
in transformation to postpolycythemic myelofibrosis. Cancer Genet
Cytogenet. 172:107–112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Specchia G, Albano F, Anelli L, Storlazzi
CT, Zagaria A, Mancini M, Cuneo A, Pane F, Foà R, Manolelli F, et
al: Deletions on der(9) chromosome in adult Ph-positive acute
lymphoblastic leukemia occur with a frequency similar to that
observed in chronic myeloid leukemia. Leukemia. 17:528–531. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rumi E and Cazzola M: Diagnosis, risk
stratification, and response evaluation in classical
myeloproliferative neoplasms. Blood. 129:680–692. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tefferi A: Myeloproliferative neoplasms: A
decade of discoveries and treatment advances. Am J Hematol.
91:50–58. 2016. View Article : Google Scholar : PubMed/NCBI
|