Introduction
Autophagy is a membrane trafficking process that
leads to the degradation of long-lived cytoplasmic proteins and
excess or damaged organelles by lysosomal hydrolyases. The hallmark
of autophagy is the emergence of double membrane vesicles called
autophagosomes, which begins with the elongation of crescent-shaped
double-membrane structures known as the phagophores, during which a
portion of the cytoplasm, including organelles, is sequestrated.
Matured autophagosomes eventually fuse with lysosomes, known as
autolysosome, for protein and pathogen degradation and contents
recycling (1). Macroautophagy
(hereafter referred to as autophagy) is a homeostatic process in
cells, whereby long-lived proteins and damaged organelles are
degraded within lysosomes (2).
Autophagy is an important physiological response that alters during
exercise and over aging (3–6). It
also serves as an important function in innate host defense by
eliminating intracellular pathogens (7,8).
Autophagy plays a key role in mediating ssRNA virus detection and
interferon-alpha secretion by plasmacytoid dendritic cells (pDCs)
(9,10), and contributes to the regulation
and function of innate and adaptive immune responses (11). Autophagy genes have been shown to
play a protective role in vivo against many of these
pathogens. Disturbances in autophagy are widely implicated in many
human diseases, including cancer, neurodegeneration and infectious
disease, as well as cardiovascular and pulmonary disease (7,12–14).
Autophagy also contributes to the regulation of innate and adaptive
immune responses (7,8,15–20),
and is implicated in clearance of pathogens and antigen
presentation, thereby playing a protective role during many viral
and bacterial infections (21–23).
The mechanisms by which intracellular bacteria and viruses are
targeted to autophagosomes for degradation have been extensively
investigated (24,25), with bacteria, virus and parasite
autophagy termed Xenophagy (15).
However, evolution has led to several viruses developing mechanisms
to evade their inhibition by autophagy, with some virus able to
subvert autophagy for their own replication (26). Coxsackieviruses B3 (CVB3) belongs
to the Enterovirus genus, within the Picornaviridae family. CVB3
has a positive-stranded RNA genome packaged in a nonenveloped
icosahedral viral capsid and infects cells via the coxsackievirus
and adenovirus receptor (CAR) or decay-accelerating factor (DAF).
CVB3 entry induces a direct cytopathic effect (CPE) and even cell
death in host cells (27,28). CVB3 can induce autophagic reaction
in vivo and in vitro (29–33),
although the processes linking CVB3-induced autophagy and its
impact on CVB3 replication still have to be determined.
Materials and methods
Western blot analysis
Western blotting was performed as described
previously (34). Polyclonal
antibody against LC3-I/II (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) and antibody against β-actin (Proteintech Group, Inc.,
Chicago, IL, USA) were used at a dilution of 1:1,000. Monoclonal
anti-enterovirus antibody (Dako Co.; Agilent Technologies, Inc.
Inc., Santa Clara, CA, USA) was used at a dilution of 1:100. Goat
anti-mouse IgG horseradish peroxidase-conjugated secondary antibody
(Cwbio) and goat anti-rabbit IgG horseradish peroxidase-Cwbio were
used at a dilution of 1:2,000. Rapamycin was used at a
concentration of 10 nM and ZSTK474 (both Selleck Chemicals,
Houston, TX, USA) was used at a concentration of 50 nM.
Virus infection
CVB3 was propagated in HeLa cells in DMEM medium
supplemented with 2% FBS, and the virus titer was routinely
determined by plaque assay. HeLa cells were maintained in DMEM
medium containing 10% FBS and infected with CVB3 at 10 tissue
culture infective dose (TCID50) values, then cells were harvested
at different post-infection (p.i.) time-points for subsequent
experiments. In drug intervention groups, HeLa cells were
pretreated with the Rapamycin or ZSTK474 for 2 h before viral
infection, and DMEM contained 10% FBS acted as a sham.
Cell transfection
HeLa cells that stably expressed GFP-LC3 were
produced by transfecting HeLa cells with pEGFP-LC3 followed by
Geneticin (G418) selection. 4 mg of expression plasmid combined
with 10 µl Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) were added to HeLa cells
according to the manufacturer's instructions. At 5 h
post-transfection, the cells were refreshed by the DMEM containing
10% FBS. 24 h later, G418 was added at concentrations of 1,000 and
500 µg/ml for maintenance of transfection.
Semi-quantitative PCR
Extraction of total RNA reverse transcription was
performed following the method of the RNA Extraction kit (Omega
Bio-Tek, Inc., Norcross, GA, USA) and the Revert Aid First Strand
cDNA Synthesis kit (Thermo Fisher Scientific, Inc.). cDNA was
subjected to Semi-quantitative PCR with the following primers: CVB3
[referring to Li et al (34)] forward, 5′-TGGTGGGCTATGGAGTATGG-3′
and reverse, 5′-CACTGGATGGGGTGTTGTCT-3′ and β-actin forward,
5′-CTAAGGCCAACCGTGAAAAGATGAC-3′ and reverse,
5′-TGGGTACATGGTGGTGCCACCAGAC-3′. The amplification profile was 2
min and 30 sec at 95°C, 40 sec at 94°C, 40 sec at 59°C and 72°C at
40 sec for 35 cycles, 5 min at 72°C.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD) and were analyzed using one-way analysis of variance
followed Duncan's post hoc analysis. A P<0.05 was considered to
be indicate a statistically significant difference.
Results
Rapamycin and ZSTK474 promote
CVB3-induced CPE
The mTOR signal pathway modulates viral replication
(29). In order to elucidate the
role of the mTOR pathway in CVB3-induced CPE, HeLa cells were
pretreated with Rapamycin (10 nM) or ZSTK474 (50 nM) or 0.1% DMSO
for 2 h before viral infection, with DMEM containing 10% FBS acting
as a sham. 2 h later, CBV3 infected cells at 10 TCID50, were
observed by microscope for CPE at 6 h p.i., 9 h p.i., 12 h p.i.,
and 24 h p.i. At 12 h p.i, the infected cells changed in
morphology, as indicated by cell rounding, cell shrinkage, cell
floating and cell detachment, which was more obvious in cells
pretreated with Rapamycin or ZSTK474 (Fig. 1).
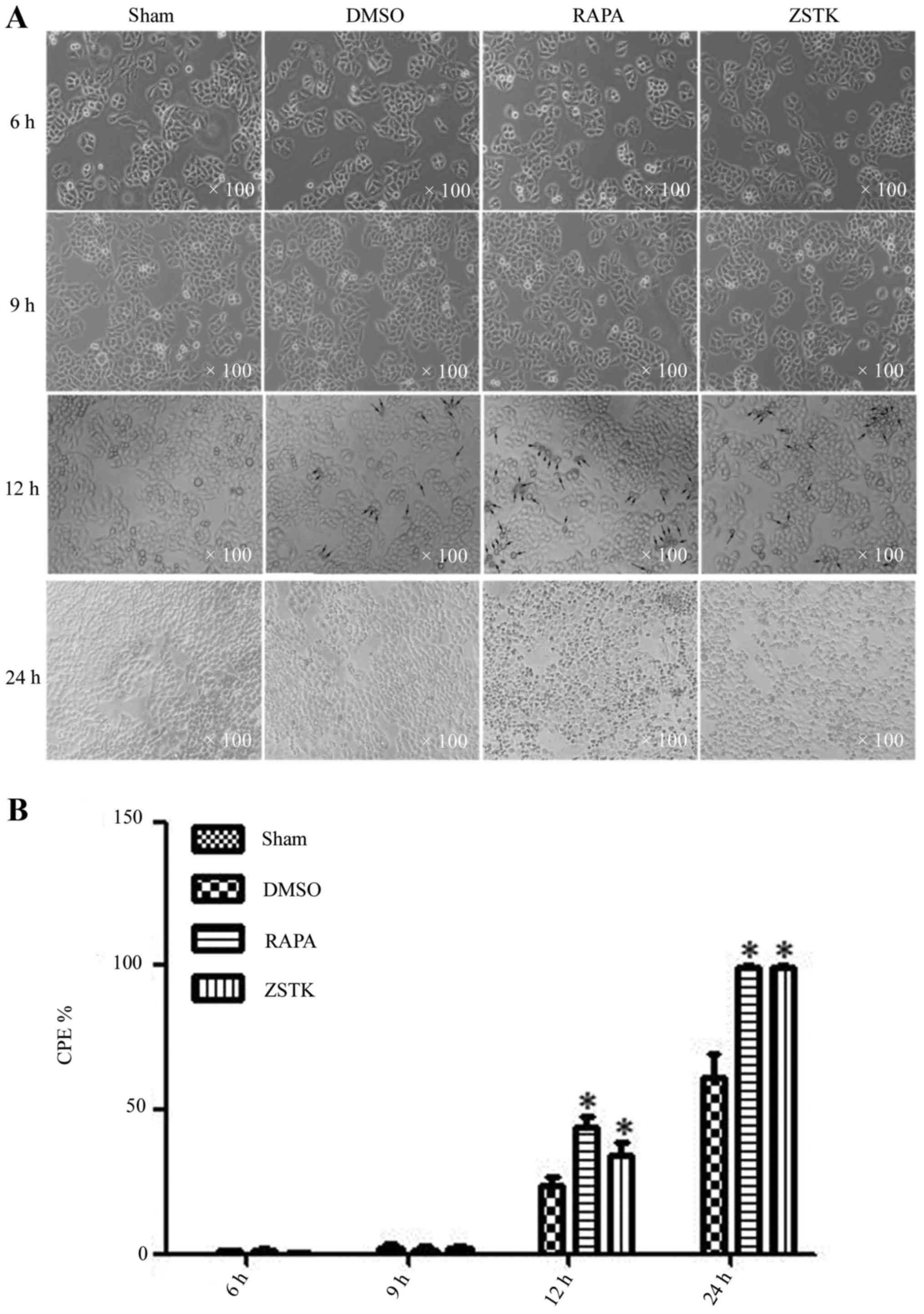 | Figure 1.Rapamycin and ZSTK474 promote
CVB3-induced CPE. Rapamycin and ZSTK474 have impacts on GFP-LC3
dots formation after CVB3 infection. HeLa cells were divided into
sham, Rapamycin (10 nM), ZSTK474 (50 nM), and DMSO (0.1%) groups,
then infected with CVB3 at 10 TCID50. (A) Collected at 6, 9, 12,
and 24 h p.i. CPE was determined by cellular change under
microscopy. (B) There was no obvious CPE at 6 and 9 h p.i., while
it could be seen at 12 and 24 h p.i. Compared with the control/DMSO
group, CPE at 12 and 24 h p.i was more evident in the Rapamycin and
ZSTK474 group. *P<0.05 compared with the control group. CVB3,
coxsackievirus B3; CPE, cytopathic effect; TCID50, tissue
culture infective dose. |
Role of Rapamycin and ZSTK474 in
CVB3-induced autophagy
Our previous work has demonstrated that pretreating
the virus-infected HeLa cells with Rapamycin and ZSTK474 impacts on
CVB3-induced autophagy. In order to investigate any temporal
aspects to this, we dealt with the HeLa cells with Rapamycin (10
nM) or ZSTK474 (50 nM) or 0.1% DMSO following the method described
in paragraph 3.1. 2 h later, we infected the cells with CVB3 at 10
TCID50. At 6, 9, 12 and 24 h p.i, cells were harvested for western
blotting. GFP-LC3 dots formation was also observed with fluorescent
microscopy. At 6 h p.i., we observed an obvious LC3II/LC3I ratio
increase and GFP-LC3 dots formation in CVB3-infected groups.
Rapamycin increased CVB3-induced autophagy over all time-points, as
indicated by increased LC3II/LC3I ratio and the green fluorescent
dots formation vs. the DMSO group (P<0.05). At 6 and 9 h p.i.,
ZSTK474 also increased CVB3-induced LC3II/LC3I ratio and increased
the GFP-LC3 dots formation. However, at 12 and 24 h p.i., ZSTK474
invert this effect on virus infected cells (Fig. 2).
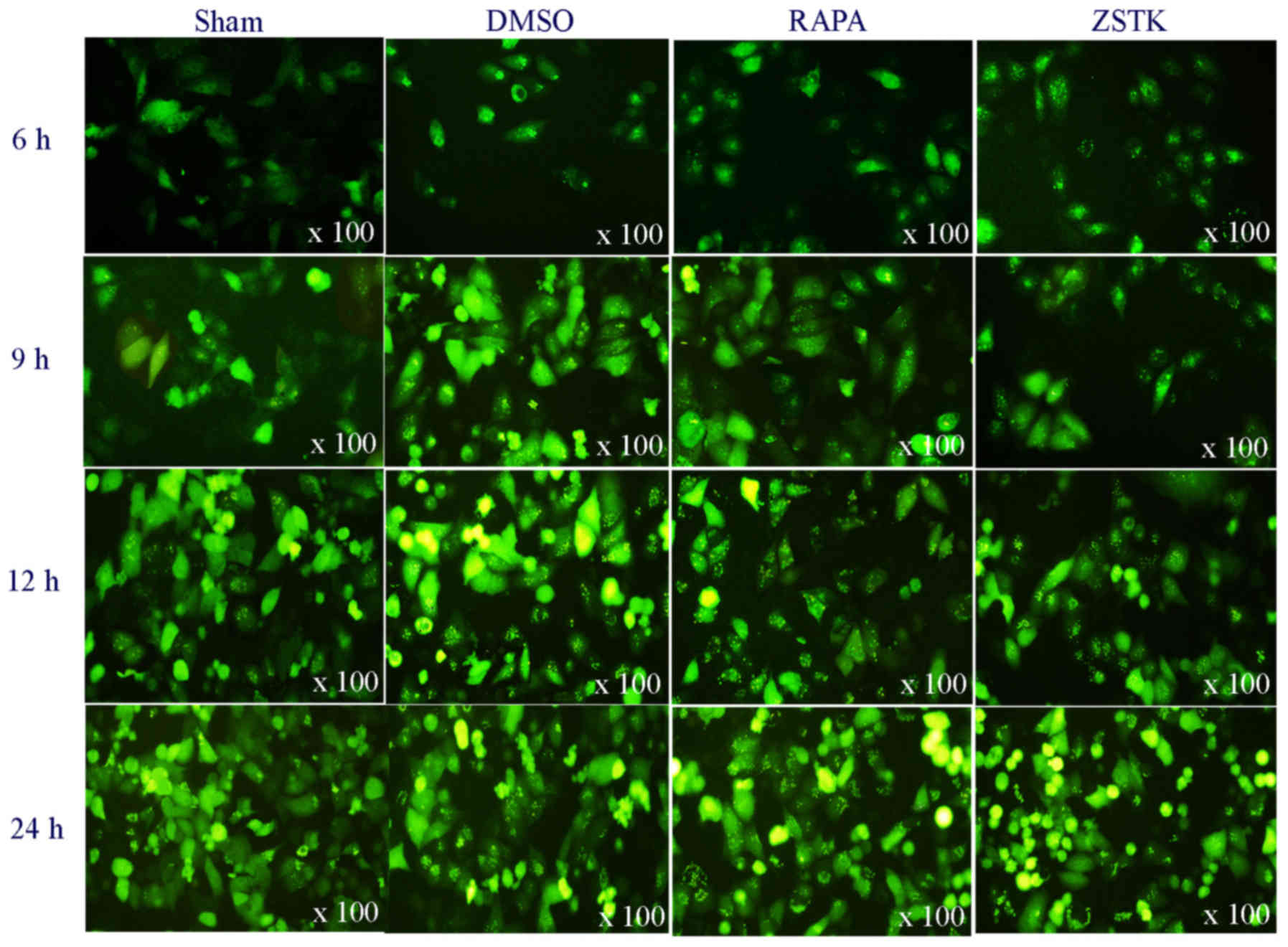 | Figure 2.Rapamycin and ZSTK474 have impacts on
GFP-LC3 dots formation after CVB3 infection. HeLa cells were
divided into sham, Rapamycin (10 nM), ZSTK474 (50 nM), and DMSO
(0.1%) groups, then infected with CVB3 at 10 TCID50. Collected at
6, 9, 12, and 24 h p.i, GFP-LC3 dots formation was observed under
fluorescent microscopy. At 6 h p.i, GFP-LC3 dots formed in virus
infected cells. When compared with control/DMSO group, the dots
were more obvious in the Rapamycin group, and at 6 and 9 h p.i, the
GFP-LC3 dot numbers increased in ZSTK474 group, vs. the control
group. At 12 and 24 h p.i, the GFP-LC3 dot numbers decreased in the
ZSTK474 group, vs. the control group. CVB3, coxsackievirus B3; CPE,
cytopathic effect; TCID50, tissue culture infective dose. |
Role of Rapamycin and ZSTK474 in virus
replication
To further observe the association of autophagy and
virus replication at different infected time, HeLa cells were
pretreated with Rapamycin or ZSTK474 or DMSO for 2 h just as
described in paragraph 3.1, then the cells were infected with CVB3
at 10 TCID50, and harvested at 6, 9, 12 and 24 h p.i, for CVB3 mRNA
and viral capsid protein VP-1 measurement by semiquantitative PCR
and Western blot analysis (Fig.
3). At 6 and 9 h p.i, Rapamycin and ZSTK474 increased
CVB3-induced autophagy as well as decreasing VP-1 protein
expression and CVB3 mRNA synthesis (P<0.05). At 12 and 24 h p.i,
ZSTK474 mitigated CVB3-induced autophagy as well as decreasing VP-1
protein expression and CVB3 mRNA synthesis (P<0.05). At 12 and
24 h p.i., Rapamycin increased CVB3-induced autophagy and promoted
CVB3 mRNA synthesis (P<0.05), whilst at 12 h (P<0.05), but
not 24 h (P>0.05), p.i., Rapamycin increased VP-1 protein
expression.
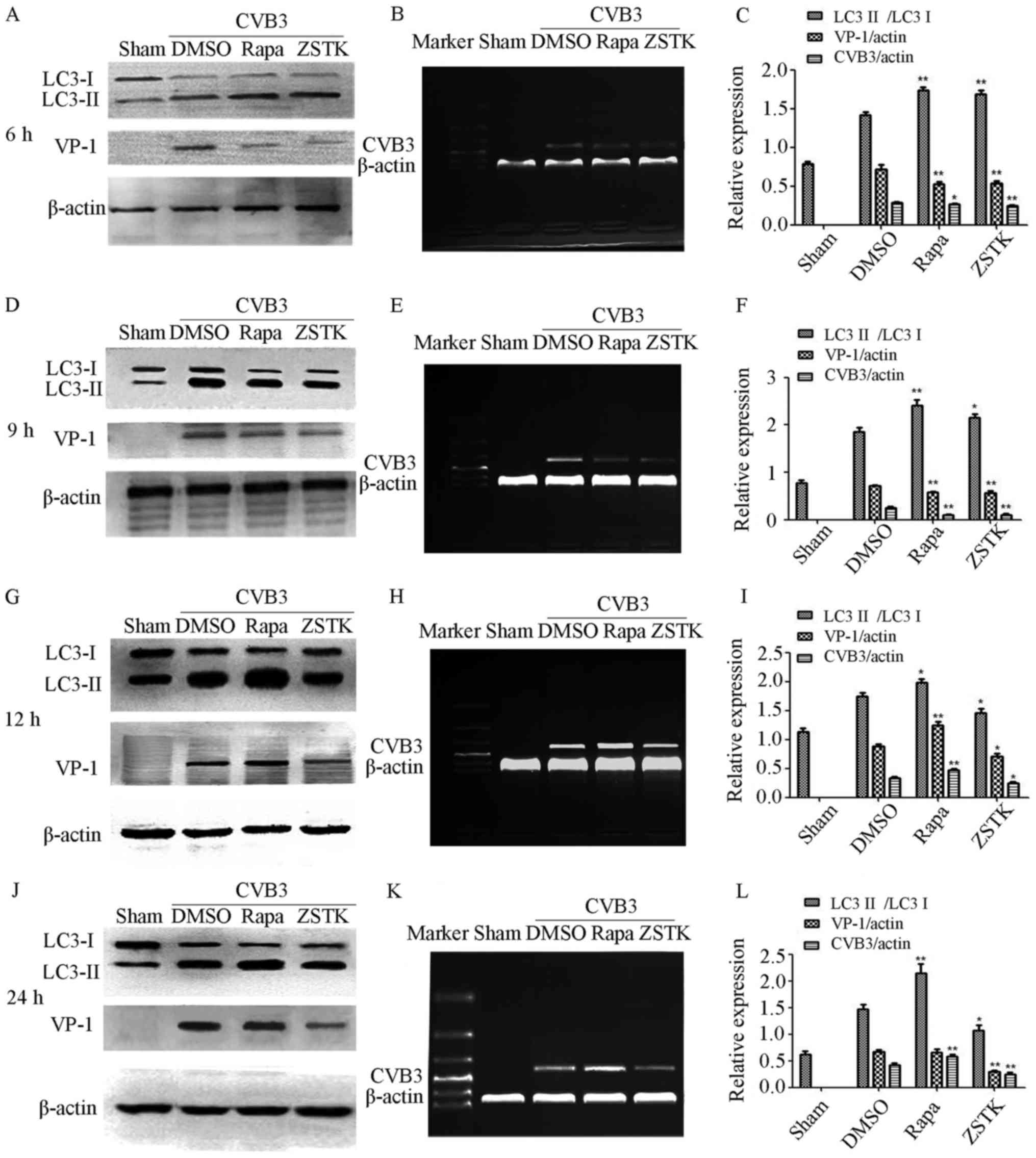 | Figure 3.Role of autophagy in CVB3 replication.
HeLa cells were divided into sham, Rapamycin (10 nM), ZSTK474 (50
nM), and DMSO (0.1%) groups, then infected with CVB3 at 10 TCID50
and harvested at 6, 9, 12 and 24 h p.i for CVB3 mRNA and viral
capsid protein VP-1 analysis. (A) The protein change at 6 h p.i in
different group, shows the LC3II/LC3I ratio increased in all virus
infected group, Rapamycin and ZSTK474 increased the LC3II/LC3I
ratio further, accompanied by decreased VP-1 protein expression.
(B) Rapamycin and ZSTK474 decreased CVB3 mRNA synthesis at 6 h p.i.
(C) The statistical outcome at 6 h p.i. in the different groups.
(D) The protein change at 9 h p.i in different groups, LC3II/LC3I
ratio increased in all virus infected group. Rapamycin and ZSTK474
increased the LC3II/LC3I ratio further, accompanied by decreased
VP-1 protein expression. (E) Rapamycin and ZSTK474 could decrease
CVB3 mRNA synthesis at 9 h p.i. (F) The statistical outcome at 9 h
p.i. in the different groups. (G) The protein change at 12 h p.i in
the different groups, shows the LC3II/LC3I ratio increased in all
virus infected group. Rapamycin increase the LC3II/LC3I ratio and
promoted VP-1 protein expression, in contrast to ZSTK474, which
decreased the LC3II/LC3I ratio and VP-1 protein expression. (H)
Rapamycin promoted CVB3 mRNA synthesis but ZSTK474 weaken it at 12
h p.i. (I) The statistical outcome at 12 h p.i. in the different
groups. (J) The protein change at 24 h p.i. Rapamycin increased the
LC3II/LC3I ratio, in contrast to ZSTK474, which decreased the
LC3II/LC3I ratio and VP-1 protein expression. (K) When compared
with control/DMSO group, Rapamycin promoted CVB3 mRNA synthesis,
with ZSTK474 weakening it at 24 h p.i. (L) The statistical outcome
at 24 h p.i. in the different groups. *P<0.05 compared with the
control group. **P<0.05 compared with the control group. CVB3,
coxsackievirus B3; TCID50, tissue culture infective dose. |
Discussion
Since autophagy serves as an important function in
innate host defense by eliminating intracellular pathogens,
Predictably, evolution has led to several viruses developing
mechanisms by which to evade the inhibitory effects of the
autophagy. Investigating the course of autophagy is therefore of
some importance. The present study demonstrates differential
temporal effects of important intracellular pathways in
CVB3-induced autophagy and virus replication. Firstly, autophagy
was determined by the microscopic observation of the cytopathetic
effect, the calculation of the LC3II/LC3I ratio and the counting of
GFP-LC3 dots. CVB3 replication was analysed by measuring VP-1
protein expression and CVB3 mRNA synthesis. CVB3 clearly provoked
autophagy, as indicated by an increased LC3II/LC3I ratio and
GFP-LC3 dots count after infection. Rapamycin promoted CVB3-induced
autophagy during all p.i. time-points, whilst ZSTK474 also
increased CVB3-induced autophagy at 6 and 9 h p.i, but not at 12
and 24 h p.i. CVB3 virus infection can lead to the cleaving of the
RasGAP protein, in turn driving activation of the
Ras/Raf/mitogen/extracellular signal-regulated kinase (MEK)/ERK
pathway, in association with increasing the levels of cytoplasmic
calcium, resulting in mitochondrial damage, a decrease in ATP
concentration and the activation of the AMP-activated protein
kinase (AMPK)/MEK/ERK pathway (35). As our previous work indicated that
the phosphoinositide 3-kinase (PI3K)/AKT/mTOR signal pathway is
involved in CVB3-induced autophagy, it seems likely that
CVB3-driven autophagy involves the interaction of multiple
signaling pathways.
Previous research indicates that both the mTOR
inhibitor, Rapamycin, and PI3K inhibitor, LY294002, can promote
CVB3-induced CPE (29). The
MEK/ERK pathway and the PI3K/Akt pathway are known to interact in
some circumstances, with the inhibition of one leading to the
activation of the other (36).
Rapamycin can provoke autophagy by the modulation of rapamycin
complex (mTORC)1, which also plays a role in the regulation of
mTORC2. Being upstream of mTORC1, the inhibition of mTORC2 could
inhibit mTORC1, thereby further aggravating the autophagic reaction
induced by CVB3 infection. What's more, it may be speculated that
the inhibition of mTORC1 with Rapamycin may drive feedback on the
MEK/ERK signaling pathway, thereby further promoting autophagy
following CVB3 infection. However, at 24 h p.i., we did not find
Rapamycin to affect VP-1 protein expression, possibly as a
consequence of energy and amino acid loss following virus
infection, with mTOR being further inhibited by Rapamycin,
resulting in the decreased phosphorylation of 4EBP1 and the
inhibition of cap-dependent translation. ZSTK474 aggravated
autophagy by inhibiting the PI3K signaling pathway initially.
However, as infection time increased, the continuous consumption of
energy and cellular content may lead to changes in the signaling
pathways involved in CVB3-induced autophagy. ZSTK474 may block PI3K
completely so that CVB3 can not trigger autophagy through the
PI3K/AKT/mTOR signal pathway, under energy deprivation or amino
acid loss, although PI3KC3 can trigger autophagy directly.
Consequently, future research should determine as to whether the
PI3KC3 pathway takes part in the later CVB3-induced autophagy, as
well as to whether ZSTK474 can also have effects on PI3KC3.
In the present study, we showed that ZSTK474 and
Rapamycin could promote CVB3-induced autophagy, decrease CVB3 mRNA
replication and VP1 expression at 6 and 9 h p.i. When combined with
our previous work showing that p62 protein expression decreases at
the beginning of CVB3 infection, this may be taken to indicate that
CVB3-induced autophagy can help host cells to initially clear the
virus, and therefore to be initially protective. As infection time
increases, we found changes in the processes linked to autophagy,
with CVB3 mRNA replication and VP1 expression at 12 and 24 h.p.i
altered. When combined with our previous work showing that the p62
protein expression increased gradually following CVB3 infection, we
speculate that following CBV3 virus infection, the accumulated
virus and damaged organelles exceed the ability of lysosome to
cope, coupled to the direct damage of the virus to lysosome, all
leading to a change of autophagic flux, resulting in an incomplete
autophagy process. Subsequently, autophagy can be detrimental,
which might be exploited for viral replication. Our findings may
supply a clue that autophagy has been implicated as a mechanism for
the CVB3 induced myocarditis during the virus infection. The role
of autophagy during this process may be of great importance to the
resultant of the disease. At the beginning, the role of autophagy
may be protective helping host cells clear the virus, alternations
in autophagy following CVB3 infection may play a role in the viral
persistence and pathogenesis in the host cells.
In conclusion, we demonstrate that two important
intracellular pathways can have differential effects at different
time-points in CVB3-induced autophagy, contributing to autophagy
initially helping host cells clear the virus, but being detrimental
at later time-points, which may be exploited for viral
replication.
Our research indicates Rapamycin and ZSTK474 can
have differential effects at different p.i. time-points regarding
CVB3 replication and CVB3-induced autophagy. Early during the
course of infection, autophagy may help host cells clear the virus,
thereby affording protection, whereas when infection time
increases, autophagy may be exploited for viral replication.
Acknowledgements
The authors appreciate the Center Laboratory at the
Third Xiangya Hospital of the Central South University provided us
with the experimental equipment and technical guidance necessary to
complete our work.
Funding
The present study was supported by grants from
National Natural Scientific Foundation of China (grant no.
81570346).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article and its supplementary
information files.
Authors' contributions
HC and ZY designed the study, analyzed the data and
wrote the manuscript. HC, LT, JC, AT, CL, ZL and ZY were involved
in data interpretation, discussion and preparation of the final
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CPE
|
cytopathic effect
|
|
CVB3
|
Coxsackievirus B3
|
|
PI3K
|
Phosphatidylinositol 3-kinase
|
|
mTOR
|
mammalian target of rapamycin
|
References
|
1
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dice JF: Chaperone-mediated autophagy.
Autophagy. 3:295–299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pauly M, Assense A, Rondon A, Thomas A,
Dubouchaud H, Freyssenet D, Benoit H, Castells J and Flore P: High
intensity aerobic exercise training improves chronic intermittent
hypoxia-induced insulin resistance without basal autophagy
modulation. Sci Rep. 7:436632017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eisenstein M: Molecular biology: Remove,
reuse, recycle. Nature. 514:S2–S4. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garber K: Autophagy. Explaining exercise.
Science. 335:2812012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
García-Prat L, Martínez-Vicente M,
Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla
V, Gutarra S, Ballestar E, Serrano AL, et al: Autophagy maintains
stemness by preventing senescence. Nature. 529:37–42. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Randow F and Münz C: Autophagy in the
regulation of pathogen replication and adaptive immunity. Trends
Immunol. 33:475–487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee HK, Lund JM, Ramanathan B, Mizushima N
and Iwasaki A: Autophagy-dependent viral recognition by
plasmacytoid dendritic cells. Science. 315:1398–1401. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Su C, Zhan G and Zheng C: Evasion of host
antiviral innate immunity by HSV-1, an update. Virol J. 13:382016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kudchodkar SB and Levine B: Viruses and
autophagy. Rev Med Virol. 19:359–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao YH, Xia N, Zhou SF, Tang TT, Yan XX,
Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, et al: Interleukin-17A
contributes to myocardial ischemia/reperfusion injury by regulating
cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll
Cardiol. 59:420–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Connell D and Liang C: Autophagy
interaction with herpes simplex virus type-1 infection. Autophagy.
12:451–459. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Doria A, Gatto M and Punzi L: Autophagy in
human health and disease. N Engl J Med. 368:18452013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deretic V and Levine B: Autophagy,
immunity, and microbial adaptations. Cell Host Microbe. 5:527–549.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Virgin HW and Levine B: Autophagy genes in
immunity. Nat Immunol. 10:461–470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fader CM, Aguilera MO and Colombo MI:
Autophagy response: Manipulating the mTOR-controlled machinery by
amino acids and pathogens. Amino Acids. 47:2101–2112. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou XJ and Zhang H: Autophagy in
immunity: Implications in etiology of autoimmune/autoinflammatory
diseases. Autophagy. 8:1286–1299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scharl M and Rogler G: Inflammatory bowel
disease: Dysfunction of autophagy? Dig Dis. 30 Suppl 3:S12–S19.
2012. View Article : Google Scholar
|
|
20
|
Puri P and Chandra A: Autophagy modulation
as a potential therapeutic target for liver diseases. J Clin Exp
Hepatol. 4:51–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhong L, Hu J, Shu W, Gao B and Xiong S:
Epigallocatechin-3-gallate opposes HBV-induced incomplete autophagy
by enhancing lysosomal acidification, which is unfavorable for HBV
replication. Cell Death Dis. 6:e17702015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Leo A, Colavita F, Ciccosanti F, Fimia
GM, Lieberman PM and Mattia E: Inhibition of autophagy in
EBV-positive Burkitt's lymphoma cells enhances EBV lytic genes
expression and replication. Cell Death Dis. 6:e18762015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J,
Ding H and Yuan Z: Subversion of cellular autophagy machinery by
hepatitis B virus for viral envelopment. J Virol. 85:6319–6333.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Watson RO, Manzanillo PS and Cox JS:
Extracellular M. tuberculosis DNA targets bacteria for autophagy by
activating the host DNA-sensing pathway. Cell. 150:803–815. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Orvedahl A, Sumpter R Jr, Xiao G, Ng A,
Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al:
Image-based genome-wide siRNA screen identifies selective autophagy
factors. Nature. 480:113–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu J, Wang H, Gu J, Deng T, Yuan Z, Hu B,
Xu Y, Yan Y, Zan J, Liao M, et al: BECN1-dependent CASP2 incomplete
autophagy induction by binding to rabies virus phosphoprotein.
Autophagy. 13:739–753. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garmaroudi FS, Marchant D, Hendry R, Luo
H, Yang D, Ye X, Shi J and McManus BM: Coxsackievirus B3
replication and pathogenesis. Future Microbiol. 10:629–653. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sin J, Mangale V, Thienphrapa W, Gottlieb
RA and Feuer R: Recent progress in understanding coxsackievirus
replication, dissemination, and pathogenesis. Virology.
484:288–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Z, Yang L, Liu Y, Tang A, Li X, Zhang
J and Yang Z: LY294002 and Rapamycin promote coxsackievirus-induced
cytopathic effect and apoptosis via inhibition of PI3K/AKT/mTOR
signaling pathway. Mol Cell Biochem. 385:169–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang HM, Ye X, Su Y, Yuan J, Liu Z, Stein
DA and Yang D: Coxsackievirus B3 infection activates the unfolded
protein response and induces apoptosis through downregulation of
p58IPK and activation of CHOP and SREBP1. J Virol. 84:8446–8459.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhai X, Bai B, Yu B, Wang T, Wang H, Wang
Y, Li H, Tong L, Wang Y, Zhang F, et al: Coxsackievirus B3 induces
autophagic response in cardiac myocytes in vivo. Biochemistry
(Mosc). 80:1001–1009. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xin L, Xiao Z, Ma X, He F, Yao H and Liu
Z: Coxsackievirus B3 induces crosstalk between autophagy and
apoptosis to benefit its release after replicating in
autophagosomes through a mechanism involving caspase cleavage of
autophagy-related proteins. Infect Genet Evol. 26:95–102. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Robinson SM, Tsueng G, Sin J, Mangale V,
Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, et
al: Coxsackievirus B exits the host cell in shed microvesicles
displaying autophagosomal markers. PLoS Pathog. 10:e10040452014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li X, Li Z, Zhou W, Xing X, Huang L, Tian
L, Chen J, Chen C, Ma X and Yang Z: Overexpression of 4EBP1,
p70S6K, Akt1 or Akt2 differentially promotes Coxsackievirus
B3-induced apoptosis in HeLa cells. Cell Death Dis. 4:e803–e809.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xin L, Ma X, Xiao Z, Yao H and Liu Z:
Coxsackievirus B3 induces autophagy in HeLa cells via the
AMPK/MEK/ERK and Ras/Raf/MEK/ERK signaling pathways. Infect Genet
Evol. 36:46–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hoeflich KP, O'Brien C, Boyd Z, Cavet G,
Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, et
al: In vivo antitumor activity of MEK and phosphatidylinositol
3-kinase inhibitors in basal-like breast cancer models. Clin Cancer
Res. 15:4649–4664. 2009. View Article : Google Scholar : PubMed/NCBI
|














