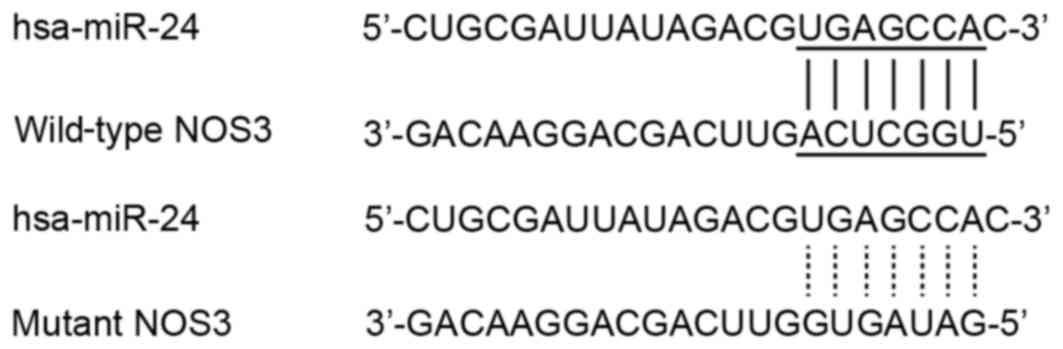Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a severe
and life-threatening disease, which results in fatality in 27–44%
of patients; 46% of survivors live with severe functional and
cognitive impairment (1,2). The response to SAH involves securing
the cerebral aneurysms and treating the cerebral vasospasm, which
occurs in 70% of SAH patients between 3 and 14 days following SAH
(3). Cerebral vasospasm results in
delayed cerebral ischemia and is primarily responsible for
unfavorable results and even mortality in SAH patients (1). However, currently no definitive
therapy is available to address this complication.
The endothelial nitric oxide synthase (NOS3) gene in
vessel endothelium continuously produces nitric oxide (NO) to
maintain basal vascular tone (4).
As a result of aneurysmal SAH, the expression of the NOS3 gene is
altered and the balanced modulation of cerebral vascular tone is
interrupted (5,6). Previous studies have reported
aberrant cerebrospinal fluid NO levels in humans following SAH
(7,8). In addition, adenovirus gene transfer
of NOS3 in dogs and in ex vivo human arteries has
demonstrated protective effects in SAH (6,9). NO
serves a role in the inhibition of inflammation and proliferation
of smooth muscle, pathological alterations which are observed in
cerebral vasospasm (10,11). Previous studies have suggested a
correlation between NOS3 polymorphisms and the development of
intracranial aneurysms as well as coronary spasm, which has
physiological similarity to cerebral vasospasm (12,13).
MicroRNAs (miRNAs) are non-coding RNAs consisting of
~22 nucleotides. They act as meta-regulators of gene expression and
are pivotal for cellular alterations that are required for the
process of development. miRNAs in the brain serve important roles
in the formation and function of the dendritic spine, and in the
synaptic plasticity that is necessary for normal cognitive
function. A thorough understanding of the mechanisms underlying the
regulation of miRNA expression is important, as abnormal regulation
of miRNAs has been associated with an array of neurological
disorders (14).
miRNAs additionally serve as regulators of vascular
phenotype via suppression or maintenance of differentiation
(15). In SAH, the identification
of blood borne biomarkers is required to estimate the risk of late
cerebral ischemia (LCI) and cerebral vasospasm. Previous studies
have demonstrated numerous alterations in gene expression in
cerebral arteries in response to various types of strokes including
focal cerebral ischemia and SAH, and following cardiac arrest
(16–18). It has been demonstrated that
stimulation of certain signal transduction pathways in the cerebral
vasculature following stroke results in transcriptional regulation
of proteins, inflammatory mediators and vasoconstrictor receptors
engaged in the maintenance of the integrity of the blood-brain
barrier (19). However, regulation
may occur at other levels, and SAH may cause alterations in miRNA
expression in cerebral arteries, which following release into serum
may as act as biomarkers to estimate the risk of LCI and cerebral
vasospasm.
Differential expression of miRNA (miR)-24 has been
identified in vascular tissue from SAH patients with vasospasm
(20), and dysregulation of NOS3
has been revealed to be involved in the underlying molecular
mechanism of vasospasm following SAH (21,22).
Using an online miRNA database, NOS3 was identified as a potential
target of miR-24. The present study validated NOS3 as a target gene
of miR-24 and investigated the involvement of miR-24 and NOS3 in
the development of vasospasm following SAH.
Materials and methods
Sample collection
The present study was approved by the Human Ethics
Committee of Qingdao University (Qingdao, China). A total of 29 SAH
patients with (n=13) or without (n=16) vasospasm (age, 21–75) were
recruited from the Department of Neurosurgery, The Affiliated
Hospital of Qingdao University (Qingdao, China) between December
2013 and December 2014. Diagnosis was performed by cerebral
angiogram, computed tomography scan (Fisher grade ≥2) and computed
tomographic angiogram. Participants or their first-degree relatives
signed informed consent forms prior to samples being obtained.
Patients underwent surgical clipping, endovascular coiling or a
combination of the two. The blood samples were obtained on day 7
after the onset of SAH. Leukocytes were isolated from the blood
using Human Peripheral Blood Leukocytes Isolation kit (Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China)
followed by centrifugation for 1 h at 700 × g and 37°C. The
isolated leukocytes were subsequently used for RNA extraction and
functional experiments.
Cell culture and transfection
Vascular smooth muscle cells (VSMCs; American Type
Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's
modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), supplemented with 1%
penicillin/streptomycin, 2 mM glutamine and 10% fetal bovine serum
(FBS; Invitrogen; Thermo Fisher Scientific, Inc.), at 37°C in 5%
CO2. A total of 12 h later, VSMCs in DMEM (Invitrogen;
Thermo Fisher Scientific, Inc.) without antibiotics were seeded
into 6-well plates at a density of 3–6×105 cells/well. A
total of 24 h following this, at a confluence of 80%,
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to transfect: i) miR-24 mimic
(UGGCUCAGUUCAGCAGGAACAG; cat. no. AM17100; Ambion; Thermo Fisher
Scientific, Inc.); ii) miR-24 inhibitor oligonucleotide
(GAGCUUCCAGGUAGCAGGUAGCAGGGCUGCUGUUCUGAGCUGUGGAUUGGACCCGCCCU
CCGGUGCCUACUGAGCUGAUAACAGUUCUGAUUUUACACACUGGCUCAGUUCAGCAGGAACAGGAGUCGAGCCCGAGAGCAAAAAAGACUA;
cat. no. AM17000; Ambion, Thermo Fisher Scientific, Inc.); iii)
scrambled oligonucleotide (ACUGUUCUGAUUUUACACACUGGCUC; Cy3
dye-labeled anti-miR negative control; cat. no. AM17011; Ambion;
Thermo Fisher Scientific, Inc.); and iv) NOS3 siRNA (forward,
5′-GAAGAGGAAGGAGUCCAGUAACACA-3′ and reverse,
5′-UGUGUUACUGGACUCCUUCCUCUUC-3′; cat. no. AM17000; Ambion; Thermo
Fisher Scientific, Inc.) into VSMCs according to the manufacturer's
protocol. In brief, cells at ~50% confluence on an 100-mm culture
dish were treated with a mixture of 30 nM siRNA and 1.25 µl/ml
Lipofectamine™ RNAiMAX (Thermo Fisher Scientific, Inc.) in 5 ml of
Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.) for 3 h at
37°C.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
To analyze the expression levels of NOS3 mRNA and
miR-24, a NucleoSpin miRNA kit (Machery-Nagel GmbH, Düren, Germany)
was used to isolate total RNA from VSMCs or patient samples
according to the manufacturer's protocol. The Agilent RNA 6000 Nano
kit and a 2100 Bioanalyzer (Agilent Technologies, Inc., Santa
Clara, CA, USA) were used to determine the integrity of the total
RNA and a NanoDrop 2000c (Thermo Fisher Scientific, Inc.) was used
to measure RNA concentration. A Universal cDNA Synthesis kit
(Machery-Nagel GmbH) was used to reverse-transcribe RNA to cDNA
according to the manufacturer's protocol. A SYBR® Green
Master mix (Thermo Fisher Scientific, Inc.) was used to perform
qPCR analysis of NOS3 mRNA expression, using 4 µl diluted cDNA in a
final reaction volume of 10 µl. The thermocycling conditions were
as follows: 20 sec at 95°C, 3 sec at 95°C, followed by 40 cycles of
3 sec at 95°C and 30 sec at 60°C. A TaqMan® miRNA assay
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used to
detect the expression of miR-24. The following thermocycling
conditions were used: 5 min at 95°C; followed by 45 cycles of 5 sec
at 95°C, 5 sec at 60°C and 10 sec at 72°C. The 2−ΔΔCq
method was used to normalize the data (23). U6 was used for normalization of
expression of miR-24 and GAPDH for NOS3. Each experiment was
performed in triplicate. The sequences of PCR primers were: NOS3
(forward: 5′-TGCTGGCATACAGGACTCAG-3′; reverse:
5′-TAGGTCTTGGGGTTGTCAGG-3′); miR-24 (forward:
5′-ACACTCCAGCTGGGTGGCTCAGTTCAGCAG-3′; reverse:
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAG-3′); GAPDH (forward:
5′-GCCAAAAGGGTCATCATCTC-3′; reverse: 5′-GTAGAGGCAGGGATGATGTTC-3′;
U6 (forward: 5′-CTCGCTTCGGCAGCACA-3′, reverse:
5′-AACGCTTCACGAATTTGCGT-3′).
Luciferase assay
To confirm whether NOS3 expression was directly
regulated by miR-24, the 3′ untranslated region (UTR) of NOS3 was
amplified by PCR, and the PCR product was inserted into a pLUC
reporter vector (Promega Corporation, Madison, WI, USA). As a
control, a mutant 3′UTR was inserted. The sequences of the
wild-type and mutant NOS3 3′UTRs are presented in Fig. 1. Lipofectamine 2000 was used to
co-transfect the VSMCs with miR-24 at a final concentration of 100
nM/well, using 0.4 mg firefly luciferase reporter vector containing
the 3′UTR sequences and 0.02 mg control pRL-CMV plasmid containing
Renilla luciferase (Promega Corporation). Cells were cultured for
48 h, following which the Dual-Luciferase Reporter assay system
(Promega Corporation) was used to assess the luciferase activity
according to the manufacturer's protocol. Renilla luciferase
activity was used to normalize the values. Each experiment was
performed at least 3 times.
Western blot analysis
VSMCs or patient samples were lysed with
radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany) containing protease inhibitors
(Roche Applied Science, Madison, WI, USA). Lysates were obtained by
centrifugation for 15 min 12,000 × g at 4°C. A Bio-Rad Protein
microassay (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was
performed to estimate the protein concentration, and boiling water
was used to heat the samples. A 12% SDS-PAGE was used to separate
the target protein (35 µg/lane) according to manufacturer's
protocol. The proteins were transferred from the gel to a
polyvinylidene difluoride (PVDF) membrane (PerkinElmer, Inc.,
Waltham, MA, USA) by electroblotting for 2 h at 90 V. The membrane
was incubated with 5% non-fat milk powder in TBS containing 0.1%
Tween 20 in the dark at 4°C for 2 h to prevent nonspecific binding.
Membranes were subsequently incubated with a mouse anti-human NOS3
antibody (1:1,000; cat. no. sc-376751; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) or a mouse anti-human β-actin antibody
(1:1,000; cat. no. sc-418965; Santa Cruz Biotechnology, Inc.) in 5%
non-fat milk for 16 h at 4°C, following which membranes were washed
twice with PBS. PVDF membranes were incubated with a rabbit
anti-mouse horseradish peroxidase-conjugated secondary antibody
(1:3,000; sc-2364, Cell Signaling Technology, Inc., Danvers, MA,
USA) for 2 h at room temperature. Visualization of the protein
bands was performed by incubating the membranes with an Enhanced
Chemiluminescence reagent (Thermo Fisher Scientific, Inc.) for 2
min. Analysis Software (VisionWorks® LS, 97-0186-02
Single; UVP, LLC, Phoenix, AZ, USA) was used to analyze the
intensity of the protein bands, and ImageJ software (National
Institutes of Health, Bethesda, MD, USA) was used to determine the
optical density. Three independent experiments were performed.
Statistical analysis
SPSS software version 10.0 (SPSS, Inc., Chicago, IL,
USA) was used to perform statistical analyses. One-way analysis of
variance was used to compare multiple groups. Data are presented as
the mean ± standard deviation of at least three independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
NOS3 is a predicted target gene of
miR-24
miR-24 has been reported to be associated with a
variety of diseases by acting on different signaling pathways
(24). The present study aimed to
determine the association between miR-24 expression levels in
vasospasm following SAH and its underlying mechanism. It has
previously been reported that miR-24 is downregulated in the
vasculature of SAH patients with vasospasm (20). Computational analysis was performed
using an online miRNA database (www.mirdb.org)
to identify the possible target gene of miR-24, and NOS3 was
predicted as a target of miR-24, with a potential binding site in
the 3′UTR of NOS3 and was selected for further analysis based on
its-physiological and pathological functions (Fig. 1).
NOS3 is a direct target of miR-24 in
VSMCs
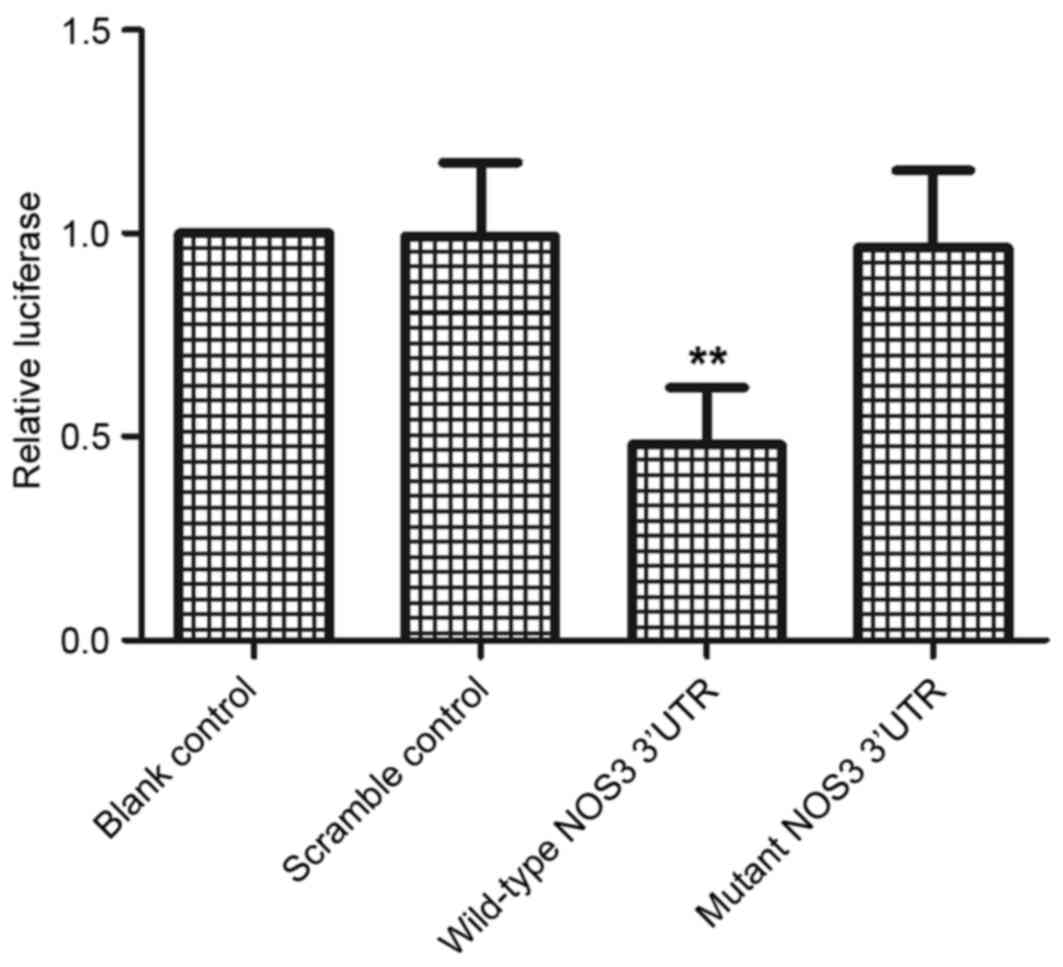
To confirm whether miR-24 and NOS3 interact, a
vector containing the 3′UTR of NOS3 was constructed and a
luciferase assay was performed. Only the luciferase activity from
the cells co-transfected with miR-24 and wild-type NOS3 3′UTR was
significantly reduced compared with the scramble control
(P<0.01; Fig. 2). The
observation that mutations in the predicted binding site abolished
the inhibitory effect of miR-24 on luciferase activity indicated
that the predicted binding site is the real ‘seed sequence’ in the
3′UTR of NOS3. Therefore, the luciferase assay suggested that
miR-24 binds to the NOS3 3′UTR, resulting in a significant decrease
in luciferase activity compared with the scramble control.
Negative regulatory association
between miR-24 and NOS3
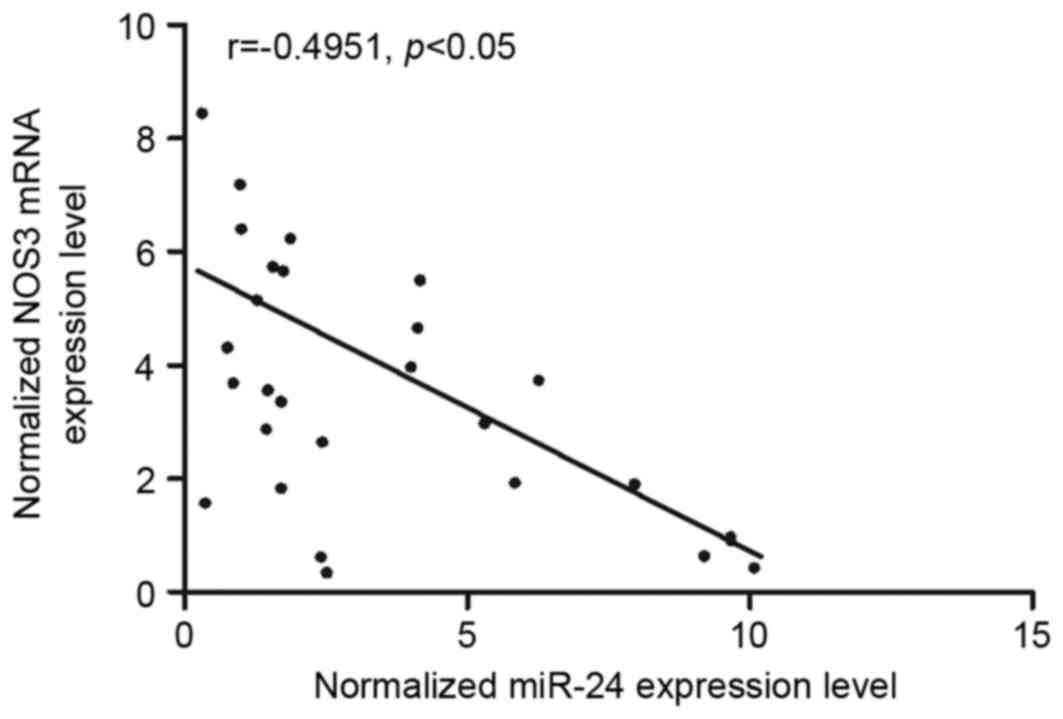
Vascular tissues were collected from SAH patients
with (n=13) or without (n-16) vasospasm and used used to
investigate the interaction between miR-24 and NOS3. Analysis of
the expression levels of miR-24 and NOS3 mRNA in the 29 tissues
demonstrated that the two were negatively correlated (Fig. 3), confirming the negative
regulatory association between miR-24 and NOS3.
Comparison of expression levels of
miR-24 and NOS3 in SAH patients with or without vasospasm
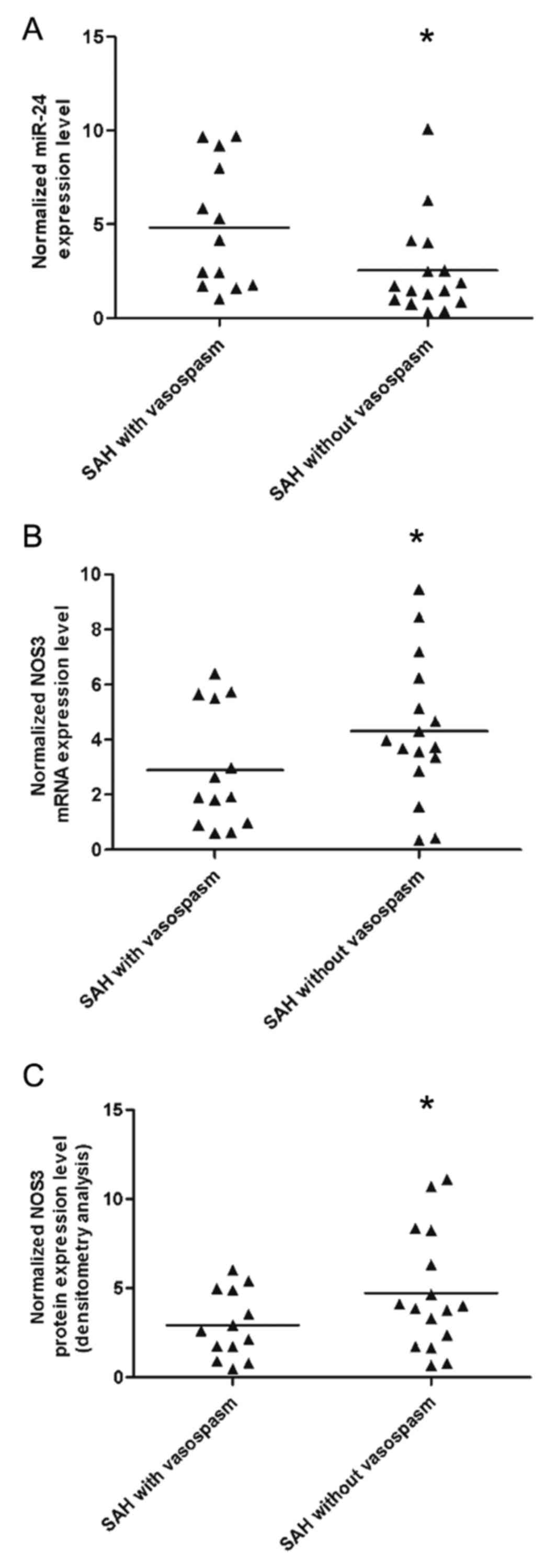
RT-qPCR and western blot analysis were performed to
detect and compare the expression levels of miR-24 and the mRNA and
protein expression levels of NOS3 between vascular tissues from SAH
patients with or without vasospasm. miR-24 expression levels were
increased in SAH patients with vasospasm (Fig. 4A) compared with those without
vasospasm (P<0.01), whereas the mRNA (Fig. 4B) and protein (Fig. 4C) expression levels of NOS3 were
decreased in SAH patients with vasospasm compared with those
without vasospasm (P<0.01). These findings indicated that
downregualtion of NOS3 caused by the upregulation of miR-24 may be
responsible for the occurrence of vasospasm in patients with
SAH.
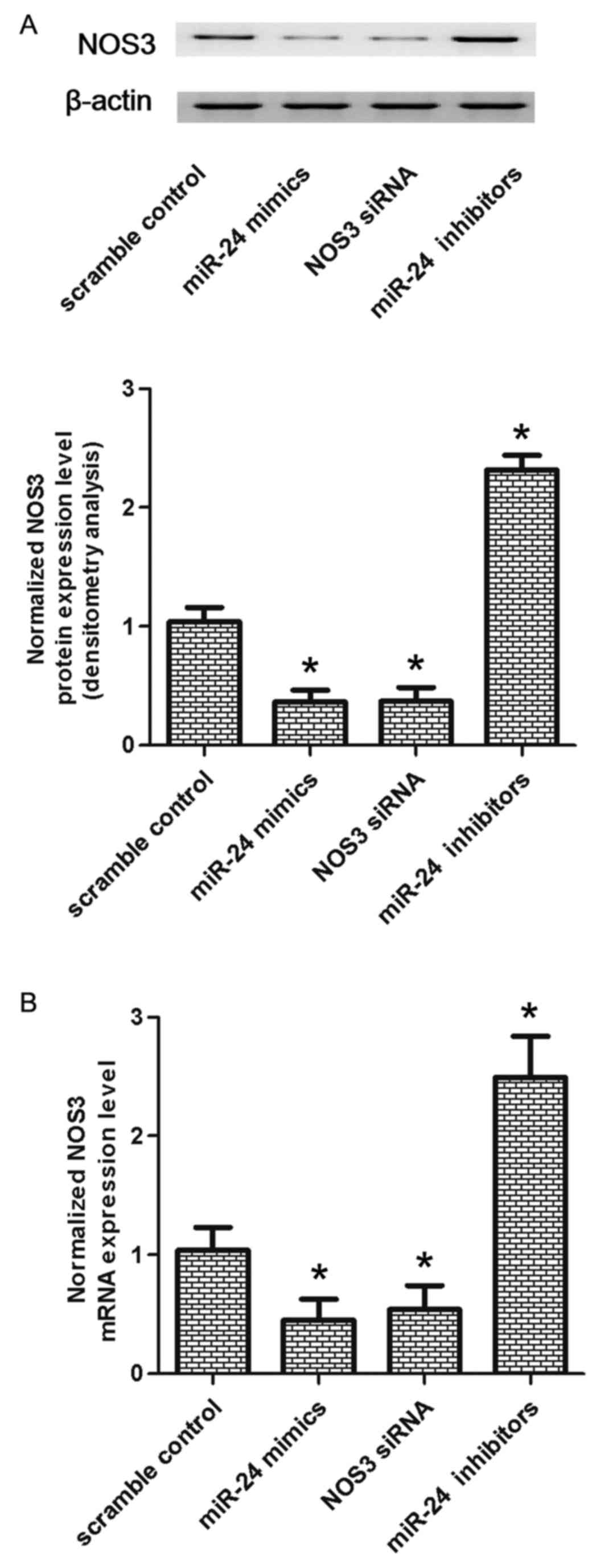
Effect of miR-24 on the expression of
NOS3
To further confirm whether NOS3 was negatively
correlated with miR-24, the mRNA and protein expression levels of
NOS3 were analyzed in VSMCs transfected with scramble control, an
miR-24 mimic, NOS3 siRNA or an miR-24 inhibitor. As presented in
Fig. 5, compared with scramble
control, cells transfected with an miR-24 inhibitor exhibited
greater expression levels of NOS3 mRNA and protein (P<0.05).
VSMCs transfected with a miR-24 mimic or NOS3 siRNA had reduced
expression levels of NOS3 compared with the scramble control
(P<0.01). These observations indicated a negative regulatory
association between miR-24 and NOS3.
Discussion
Previous studies have suggested prominent roles for
miRNAs in a variety of diseases, including cancer and
cardiovascular disease. A growing list of miRNAs, known as
‘angiomiR’, has been demonstrated to serve cell-independent and
cell-dependent regulatory roles in angiogenesis (25,26).
The two clusters of miR-23~27~24 include an intergenic
miR-23a~27a~24-2 cluster and an intronic miR-23b~27b~24-1 cluster
present in the genome of vertebrates (27). Previous studies have demonstrated
that in vitro and in vivo angiogenesis via Sprouty2
and Semaphorin 6A requires miR-23 and miR-27; miR-24 inhibited
angiogenesis through simultaneous regulation of multiple components
of the actin cytoskeleton pathways (28,29).
A variety of genes downstream of Rho signaling, including LIM
domain kinase 2 (Limk2), p21-activating kinase 4 (Pak4) and
diaphanous homolog 1 are direct targets of miR-24. Angiogenesis is
inhibited by silencing of PAK4 or LIMK2, simulating the phenotype
of in vitro miR-24 overexpression, whereas the tube
formation defects in miR-24 overexpressing ECs may be partially
rescued by overexpression of PAK4 and LIMK2 by adenoviruses
(30). Furthermore, subretinal
administration of miR-24 inhibits laser-mediated choroidal
neovascularization in vivo (30). These results indicated a novel
underlying mechanism via which miR-24 regulates angiogenesis and
actin cytoskeleton dynamics, and suggested miR-24 may be a
potential novel therapeutic agent for the treatment of aberrant
angiogenesis via regulation of actin cytoskeleton pathways. The
present study performed computational analysis to search for the
possible target gene of miR-24, and predicted NOS3 as a target of
miR-24 with a potential binding site in the 3′UTR of NOS3. A
luciferase assay revealed that miR-24 binds to the NOS3 3′UTR,
resulting in a significant decrease in luciferase activity compared
with the scramble control. Analysis of tissue samples from SAH
patients revealed a negative correlation between expression levels
of miR-24 and NOS3 mRNA, confirming the negative regulatory
association between miR-24 and NOS3.
A recent study has suggested that NO acts as an
activator of vasomotor tone that may affect development of cerebral
vasospasm following SAH (31). NO,
as an endogenous vasodilator, is generated by its cleavage from
L-arginine by NOS. The generation of NOS, and subsequent NO, is
stimulated by gene transcription mediated by numerous intra- and
extracellular stimuli. The NO level is reduced following SAH
(32). The production of NOS is
performed by endothelial cells, neurons and other cells. The
reduced level of NO following SAH may be associated with reductions
in NO via disruption or scavenging, and/or NOS generation and/or
activity (33). Following SAH, the
function of NOS3 is damaged in cerebral vessels, which limits
vessel relaxation caused by the NOS3-associated elevation in NO
generation. Delivery of NOS metabolites and NO donors have
demonstrated effectiveness in reducing angiographic cerebral
vasospasm (34,35). However, the short half-life of NO,
potential toxicity and side effects limit the application of these
studies to the clinic (34,35).
The present study revealed that miR-24 expression levels increased
in SAH patients with vasospasm compared with those without
vasospasm, whereas the expression levels of NOS3 decreased in SAH
patients with vasospasm compared with those without vasospasm. The
findings indicated that downregualtion of NOS3 caused by
upregulation of miR-24 may be responsible for the occurrence of
vasospasm in patients with SAH.
The excessive production of or depletion of the
effective vasodilator NO has been investigated with regard to the
involvement of secondary complications in the induction of cerebral
infarction and ischemia. NO is synthesized non-enzymatically via a
range of nitrate-nitrite reduction-oxidation reactions or
enzymatically by 3 principal isoforms of NOS including inducible,
endothelial and neuronal NOS (35). Physiological levels of NO
generation provide a variety of benefits, including preservation of
normal vascular tone and vasodilation of the microcirculation,
prevention of excessive platelet aggregation and adhesion,
antithrombotic effects, VSMC hyperplasia, and suppression of
endothelial apoptosis (36).
Previous study revealed an association between SAH in mice and
oxidative stress in the brain parenchyma, and with NOS3 dysfunction
(homodimeric uncoupling) (37).
NOS3 uncoupling promoted NO depletion and increased oxidative
stress, and was associated with a range of secondary complications
including reactive oxygen species release, neuronal apoptosis and
microthrombosis (38). The present
study investigated the mRNA and protein expression levels of NOS3
in VSMCs transfected with scramble control, an miR-24 mimic, NOS3
siRNA or an miR-24 inhibitor. Transfection with an miR-24 inhibitor
increased the expression levels of NOS3, whereas transfection with
an miR-24 mimic or NOS3 siRNA decreased NOS3 expression levels.
These observations indicated that there was a negative regulatory
association between miR-24 and NOS3.
In conclusion, the results of the present study
demonstrated that miR-24 directly targets NOS3, and downregulation
of NOS3 may induce vasospasm following SAH. This may be caused by
the upregualtion of miR-24 in VSMCs. The present study indicated
that miR-24 is a potential therapeutic target for the treatment of
SAH/vasospasm.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HTL and JW were responsible for study planning, data
collection, data analysis and interpretation, preparation of the
manuscript and literature analysis. SFL performed data collection,
data analysis and interpretation. LC performed data analysis and
interpretation and preparation of the manuscript. WZT was
responsible for data analysis and interpretation and literature
analysis; and YGF for data collection, data analysis and
interpretation.
Ethics approval and consent to
participate
The present study was approved by the Human Ethics
Committee of Qingdao University (Qingdao, China). Participants or
their first-degree relatives signed informed consent forms prior to
samples being obtained.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Connolly ES Jr, Rabinstein AA, Carhuapoma
JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech
AM, Ogilvy CS, et al: Guidelines for the management of aneurysmal
subarachnoid hemorrhage: A guideline for healthcare professionals
from the American Heart Association/american Stroke Association.
Stroke. 43:1711–1737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Suarez JI, Tarr RW and Selman WR:
Aneurysmal subarachnoid hemorrhage. N Engl J Med. 354:387–396.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sarrafzadeh A, Haux D, Sakowitz O,
Benndorf G, Herzog H, Kuechler I and Unterberg A: Acute focal
neurological deficits in aneurysmal subarachnoid hemorrhage:
Relation of clinical course, CT findings, and metabolite
abnormalities monitored with bedside microdialysis. Stroke.
34:1382–1388. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Quyyumi AA, Dakak N, Andrews NP, Gilligan
DM, Panza JA and Cannon RO III: Contribution of nitric oxide to
metabolic coronary vasodilation in the human heart. Circulation.
92:320–326. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weir B and MacDonald L: Cerebral
vasospasm. Clin Neurosurg. 40:40–55. 1993.PubMed/NCBI
|
|
6
|
Khurana VG, Smith LA, Baker TA, Eguchi D,
O'Brien T and Katusic ZS: Protective vasomotor effects of in vivo
recombinant endothelial nitric oxide synthase gene expression in a
canine model of cerebral vasospasm. Stroke. 33:782–789. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Woszczyk A, Deinsberger W and Böker DK:
Nitric oxide metabolites in cisternal CSF correlate with cerebral
vasospasm in patients with a subarachnoid haemorrhage. Acta
Neurochir (Wien). 145:257–263; discussion 263–644. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sadamitsu D, Kuroda Y, Nagamitsu T,
Tsuruta R, Inoue T, Ueda T, Nakashima K, Ito H and Maekawa T:
Cerebrospinal fluid and plasma concentrations of nitric oxide
metabolites in postoperative patients with subarachnoid hemorrhage.
Crit Care Med. 29:77–79. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khurana VG, Smith LA, Weiler DA, Springett
MJ, Parisi JE, Meyer FB, Marsh WR, O'Brien T and Katusic ZS:
Adenovirus-mediated gene transfer to human cerebral arteries. J
Cereb Blood Flow Metab. 20:1360–1371. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moncada S, Palmer RM and Higgs EA: Nitric
oxide: Physiology, pathophysiology, and pharmacology. Pharmacol
Rev. 43:109–142. 1991.PubMed/NCBI
|
|
11
|
Dumont AS, Dumont RJ, Chow MM, Lin CL,
Calisaneller T, Ley KF, Kassell NF and Lee KS: Cerebral vasospasm
after subarachnoid hemorrhage: Putative role of inflammation.
Neurosurgery. 53:123–133; discussion 133–135. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Khurana VG, Sohni YR, Mangrum WI,
McClelland RL, O'Kane DJ, Meyer FB and Meissner I: Endothelial
nitric oxide synthase gene polymorphisms predict susceptibility to
aneurysmal subarachnoid hemorrhage and cerebral vasospasm. J Cereb
Blood Flow Metab. 24:291–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshimura M, Nakayama M, Shimasaki Y,
Ogawa H, Kugiyama K, Nakamura S, Ito T, Mizuno Y, Harada E, Yasue
H, et al: A T-786->C mutation in the 5′-flanking region of the
endothelial nitric oxide synthase gene and coronary arterial
vasomotility. Am J Cardiol. 85:710–714. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saugstad JA: MicroRNAs as effectors of
brain function with roles in ischemia and injury, neuroprotection,
and neurodegeneration. J Cereb Blood Flow Metab. 30:1564–1576.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cordes KR, Sheehy NT, White MP, Berry EC,
Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN and Srivastava D:
miR-145 and miR-143 regulate smooth muscle cell fate and
plasticity. Nature. 460:705–710. 2009.PubMed/NCBI
|
|
16
|
Vikman P, Ansar S, Henriksson M, Stenman E
and Edvinsson L: Cerebral ischemia induces transcription of
inflammatory and extracellular-matrix-related genes in rat cerebral
arteries. Exp Brain Res. 183:499–510. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vikman P, Beg S, Khurana TS,
Hansen-Schwartz J and Edvinsson L: Gene expression and molecular
changes in cerebral arteries following subarachnoid hemorrhage in
the rat. J Neurosurg. 105:438–444. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johansson S, Povlsen GK and Edvinsson L:
Expressional changes in cerebrovascular receptors after
experimental transient forebrain ischemia. PLoS One. 7:e418522012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Edvinsson LI and Povlsen GK: Vascular
plasticity in cerebrovascular disorders. J Cereb Blood Flow Metab.
31:1554–1571. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu D, Han L, Wu X, Yang X, Zhang Q and
Jiang F: Genome-wide microRNA changes in human intracranial
aneurysms. BMC Neurol. 14:1882014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Starke RM, Kim GH, Komotar RJ, Hickman ZL,
Black EM, Rosales MB, Kellner CP, Hahn DK, Otten ML, Edwards J, et
al: Endothelial nitric oxide synthase gene single-nucleotide
polymorphism predicts cerebral vasospasm after aneurysmal
subarachnoid hemorrhage. J Cereb Blood Flow Metab. 28:1204–1211.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khurana VG, Sohni YR, Mangrum WI,
McClelland RL, O'Kane DJ, Meyer FB and Meissner I: Section on
cerebrovascular surgery: Galbraith Award: Endothelial nitric oxide
synthase (eNOS) and heme oxygenase-1 (HO-1) gene polymorphisms
predict susceptibility to aneurysmal subarachnoid hemorrhage (SAH)
and post-SAH cerebral vasospasm. Clin Neurosurg. 51:343–350.
2004.PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lang B, Shang C and Meng L: Targeted
Silencing of S100A8 Gene by miR-24 to Increase Chemotherapy
Sensitivity of Endometrial Carcinoma Cells to Paclitaxel. Med Sci
Monit. 22:1953–1958. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Garofalo M and Croce CM: microRNAs: Master
regulators as potential therapeutics in cancer. Annu Rev Pharmacol
Toxicol. 51:25–43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang S and Olson EN: AngiomiRs-key
regulators of angiogenesis. Curr Opin Genet Dev. 19:205–211. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou Q, Gallagher R, Ufret-Vincenty R, Li
X, Olson EN and Wang S: Regulation of angiogenesis and choroidal
neovascularization by members of microRNA-23~27~24 clusters. Proc
Natl Acad Sci USA. 108:8287–8292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Urbich C, Kaluza D, Frömel T, Knau A,
Bennewitz K, Boon RA, Bonauer A, Doebele C, Boeckel JN,
Hergenreider E, et al: MicroRNA-27a/b controls endothelial cell
repulsion and angiogenesis by targeting semaphorin 6A. Blood.
119:1607–1616. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou Q, Anderson C, Zhang H, Li X, Inglis
F, Jayagopal A and Wang S: Repression of choroidal
neovascularization through actin cytoskeleton pathways by
microRNA-24. Mol Ther. 22:378–389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Siuta M, Zuckerman SL and Mocco J: Nitric
oxide in cerebral vasospasm: Theories, measurement, and treatment.
Neurol Res Int. 2013:9724172013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jung CS, Iuliano BA, Harvey-White J, Espey
MG, Oldfield EH and Pluta RM: Association between cerebrospinal
fluid levels of asymmetric dimethyl-L-arginine, an endogenous
inhibitor of endothelial nitric oxide synthase, and cerebral
vasospasm in a primate model of subarachnoid hemorrhage. J
Neurosurg. 101:836–842. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim P, Schini VB, Sundt TM Jr and
Vanhoutte PM: Reduced production of cGMP underlies the loss of
endothelium-dependent relaxations in the canine basilar artery
after subarachnoid hemorrhage. Circ Res. 70:248–256. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hino A, Tokuyama Y, Weir B, Takeda J, Yano
H, Bell GI and Macdonald RL: Changes in endothelial nitric oxide
synthase mRNA during vasospasm after subarachnoid hemorrhage in
monkeys. Neurosurgery. 39:562–567; discussion 567–568. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Afshar JK, Pluta RM, Boock RJ, Thompson BG
and Oldfield EH: Effect of intracarotid nitric oxide on primate
cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg.
83:118–122. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Faassen EE, Bahrami S, Feelisch M,
Hogg N, Kelm M, Kim-Shapiro DB, Kozlov AV, Li H, Lundberg JO, Mason
R, et al: Nitrite as regulator of hypoxic signaling in mammalian
physiology. Med Res Rev. 29:683–741. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Förstermann U and Münzel T: Endothelial
nitric oxide synthase in vascular disease: From marvel to menace.
Circulation. 113:1708–1714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sabri M, Ai J, Knight B, Tariq A, Jeon H,
Shang X, Marsden PA and Macdonald Loch R: Uncoupling of endothelial
nitric oxide synthase after experimental subarachnoid hemorrhage. J
Cereb Blood Flow Metab. 31:190–199. 2011. View Article : Google Scholar : PubMed/NCBI
|