Introduction
Inflammation is a complex defense mechanism that
involves specialized cells and soluble factors, which are initiated
to suppress and combat external threats to protect normal
physiological functions (1).
Analogous inflammatory progressions are considered to occur in the
brain and peripheral tissues. In the brain, microglia act as
important immune effector cells of the central nervous system
(CNS); microglia are sensitive to external environmental stimuli
and are the first cells to respond following damage (2). Under normal conditions, brain
microglia are in a quiescent state. However, following ischemia and
hypoxia, microglia rapidly transform from a resting to an active
state, increasing their production of inflammatory cytokines
(3). Chronic inflammation of the
brain gradually becomes noxious, which is associated with
progressive tissue damage in degenerative diseases.
Alzheimer's disease (AD) is a degenerative disease
of the CNS, the pathological characteristics of which were first
described by the German doctor Alios Alzheimer in 1906 (4). Among the various proposals for
mechanisms underlying pathogenesis of AD, inflammation is a theory
of interest. Previous studies have demonstrated the existence of
inflammatory markers in AD brains, including cytokine and chemokine
overproduction, and microgliosis within injured areas (5–8).
Additionally, it has been demonstrated that levels of various
proinflammatory factors were enhanced in patients with AD,
including interleukin (IL)-6, IL-1β and tumor necrosis factor-α
(TNF-α) (9). Activated microglia
have been revealed to exert neurotoxic effects by releasing IL-6,
IL-1β, TNF-α and inducible nitric oxide (NO) synthase (8), which is reported to be exacerbated in
neurodegenerative diseases, including AD (10), human immunodeficiency
virus-associated dementia (11)
and Parkinson's disease (12).
Toll-like receptors (TLRs) are crucial innate immune
receptors that have been reported to be involved in the development
and progression of AD (13).
Previous studies have demonstrated that TLRs serve critical roles
in anoxic and anoxemic injury (14–16).
It has been demonstrated that TLR4 knockout protected against
ischemic injury in mice (17). As
one of the key inflammatory-associated molecules, TNF
receptor-associated factor 6 (TRAF6) has been demonstrated to
induce nuclear factor-κB (NF-κB) activation (18). NF-κB is a key transcription factor
that participates in the regulation of inflammatory cytokine
expression. NF-κB may be transported into the nucleus following TLR
signaling pathway activation (19). Furthermore, it has been indicated
that suppressing the activation of NF-κB is associated with
neuroprotective effects (20,21).
It has been demonstrated that the TLR4 signaling pathway may be
implicated in the progression of AD and may therefore be a novel
therapeutic target (22). However,
the function and mechanism of the TLR4/TRAF6/NF-κB pathway in AD
has not yet been elucidated.
Statins, a class of
5-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase
inhibitors, were first isolated from the fungus Penicillium
citrinum in 1976 (23).
HMG-CoA reductase serves as a rate-limiting enzyme in the synthesis
of cholesterol in hepatic cells, and its activity may be
competitively suppressed by statins (24). In addition, a previous study
demonstrated that statins serve an indispensable role in the
protection of the CNS and the improvement of memory (25). Atorvastatin is a statin that, when
applied long-term, has been revealed to improve learning and memory
in AD rats (26). Atorvastatin has
also been demonstrated to downregulate the expression of IL-6,
IL-1β and TNF-α in the hippocampus, further protecting the CNS
(27). However, there is a lack of
research at present concerning atorvastatin and its protective
effects against neuronal inflammatory injury in AD.
The present study assessed whether atorvastatin may
have potential as a novel therapeutic agent in the suppression of
proinflammatory and neurotoxic factor release from oxygen-glucose
deprived (OGD) BV-2 microglia and the apoptosis of OGD hippocampal
neurons. Furthermore, the associated signaling pathway and
apoptosis-associated protein expression in OGD models treated with
atorvastatin were assessed.
Materials and methods
Ethical approval
Ethical approval for the experimental research on
animals in the present study was obtained from the Institutional
Review Board of Shaoxing Municipal Hospital (Shaoxing, China).
Reagents
DMEM medium and penicillin-streptomycin used in cell
culture were obtained from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The following antibodies were purchased from
Abcam (Cambridge, UK): Anti-TNF-α (1:1,000; cat. no. ab6671; rabbit
anti-mouse), anti-TLR4 (1:2,000; cat. no. ab83444; rabbit
anti-mouse), anti-TRAF6 (1:5,000; cat. no. ab33915; rabbit
anti-mouse), anti-NF-κB (1:5,000; cat. no. ab32360; rabbit
anti-mouse), anti-B-cell lymphoma 2 (Bcl-2)-associated X (Bax;
1:500; cat. no. ab32503; rabbit anti-mouse), anti-Bcl-2 (1:500;
cat. no. ab59348; rabbit anti-mouse), anti-caspase-3 (1:1,000; cat.
no. ab2302; rabbit anti-mouse), anti-GAPDH (1:10,000; cat. no.
ab181602; rabbit anti-mouse). anti-IL-6 (1:1,000; cat. no. 12912;
rabbit anti-mouse) and anti-IL-1β (1:1,000; cat. no. 12426; rabbit
anti-mouse) were purchase from CST Biological Reagents Co., Ltd.
(Shanghai, China). Atorvastatin was obtained from Pfizer, Inc. (New
York, NY, USA).
Cell culture
Mice (C57BL6; 7–9 weeks old; 1:2 male:female) were
purchased from Guangdong Medical Laboratory Animal Center. These
mice were bred to produce the newborn mice. Parental and newborn
mice were housed at 21±2°C with 60% relative humidity, a 12 h
light/dark cycle and free access to food and water. Six newborn
mice (born <24 h; weight, 1–2 g) were euthanized via cervical
dislocation and subsequently soaked and disinfected in 75% ethanol
for 3–5 min at room temperature. Brains were removed via
craniotomy. Brains were washed with cold D-Hanks solution (Beijing
Solarbio Science & Technology, Co., Ltd., Beijing, China) and
the meninges and blood vessels were removed. Hippocampus tissues
were sliced into 2 mm sections and washed in D-Hanks solution for
10 min at 37°C. Cells were subsequently digested with 0.125%
trypsin (Beyotime Institute of Biotechnology, Haimen, China) for 20
min at 37°C. A total of 20 ml Dulbecco's modified Eagle's medium
mixed 1:1 with Ham's F-12 (DMEM/F12; Gibco; Thermo Fisher
Scientific, Inc.) and 10% fetal bovine serum (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was added into cells via a pipette. The
suspension was then collected and filtrated with an 80 mesh nylon
filter (178 µm). The suspension was centrifuged at 800 × g for 15
min at 4°C, the supernatant was discarded and 10 ml DMEM/F12 was
added to the precipitate. Cell debris and residual tissues were
sunk through centrifugation at 800 × g for 10 min at 4°C and the
cell suspension was collected. Cells were seeded into a pre-coated
polylysine culture flask (75 cm2, 250 ml) and incubated
at 37°C overnight in 5% CO2. According to the metabolism
of the cells being assessed, fluid was changed every 2–3 days, as
described previously (28). After
2 weeks, the culture flask was agitated using a rotary shaker at
speed of 300 rpm overnight at 37°C. The supernatant cell suspension
was removed and inoculated into a second culture flask. A total of
2 h following the renewal of culture medium, the adherent cells was
BV-2 microglial cell line. The identification of microglia was
determined by detecting the expression of GFAP by western blot
analysis (data not shown) using an anti-GFAP antibody (cat. no.
ab7260; 1:10,000).
Preparation of microglial-conditioned
medium (MCM)
BV-2 microglial cells were used to establish a model
of activated microglia under OGD conditions as previously reported
(29). Cells (1×105)
were maintained in serum/glucose-free DMEM in an anaerobic
environment for 1.5 h at 37°C containing 5% CO2 and 95%
N2. Cells were then transferred into DMEM supplemented
with 1% B27, 2 mmol/l glutamine and 10% penicillin-streptomycin)
and incubated in atmosphere containing 95% air and 5%
CO2 at 37°C. Following a 48 h incubation, the MCM was
harvested. Centrifugation (800 × g for 1 min at room temperature)
was performed to purify the conditioned medium. The medium was
subsequently diluted with serum-free DMEM to 1:1. Following this,
the conditioned medium was used for the culture of OGD hippocampal
neurons in vitro.
Primary culture of hippocampal
neuronal cells
Primary hippocampal neuronal cell cultures were
performed as described previously (30). The hippocampal formations of
newborn mice (<24 h) were dissected and incubated with D-Hanks
solution. The tissues were digested using 0.125% trypsin and
grinded softly at 37°C. Subsequently, the dissociated cells were
scattered onto glass cover slips covered with poly-L-lysine
(molecular weight, 20,000–50,000; 0.1 mg/ml; Sigma-Aldrich; Merck
KGaA). Cells were then transferred into the MCM prepared in the
aforementioned experiment to establish the OGD model. Cells were
incubated under moist conditions at 37°C and 5% CO2 for
10–12 days. The medium was changed every 2–3 days.
Cell viability analysis
Cell viability was measured by performing an MTT
assay. A total of ~5×104 cells/ml in the logarithmic
phase were seeded into a 96-well plate and maintained for 12 h in
an incubator (37°C, 5% CO2). Atorvastatin at
five different concentrations (2.5, 5, 10, 20 and 50 µM) was added
and cells were incubated at 37°C and 5% CO2 for 12, 24
and 48 h. Cell viability was determined by adding 10 µl MTT (5
mg/ml; Beyotime Institute of Biotechnology) solution and incubating
at 37°C for 6 h. Following centrifugation at 800 × g for 1 min at
room temperature, the supernatant was discarded. Cells were treated
with 100 µl dimethyl sulfoxide under low-speed oscillation for 10
min. A microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) was used to determine the absorbance at 490 nm. Growth values
were calculated using optical density (OD) as follows: (OD treated
cells/OD untreated cells) ×100, as described in a previous study
(31).
Cell apoptosis analysis
Flow cytometry (FCM) was utilized for the analysis
of cell apoptosis. Cells were seeded at a density of
3×105 into a 6-well plate. Cells were treated with or
without 10 µM atorvastatin at 37°C for 24 h. Following OGD injury,
cells were fixed in 70% ethanol at 4°C for 2 h. Following this,
cells for assessment were washed with PBS (containing
KH2PO4, Na2HPO4, NaCl
and KCl; Beijing Solarbio Science & Technology, Co., Ltd.) and
resuspended in incubation buffer [10 mmol/l
4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid/NaOH (PH 7.4),
140 mmol/l NaCl, 5 mmol/l CaCl2] at a density of
1×106 cells/ml. Cells were then maintained with annexin
V fluorescein isothiocyanate (1 µg/ml) at 4°C for 15 min and
propidium iodide (1 µg/ml; Xilong Scientific Co., Ltd., Shenzhen,
Luogang, China) at 4°C for 5 min in the dark. A flow cytometer
(FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA) was used for
the analysis of cell apoptosis with CellQuest software version 5.1
(BD Biosciences).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from cells in three groups:
Control group; OGD only group; and OGD + 10 µM atorvastatin group.
Total RNA was extracted using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). A total of 2 µg RNA was used for cDNA
synthesis with a first strand cDNA kit (cat. no. 11483188001;
Sigma-Aldrich; Merck KGaA) with the temperature protocol: 25°C for
10 min, 42°C for 50 min and 70°C for 15 min. qPCR was performed
using the SYBR Green Premix reagent (Takara Bio, Inc., Otsu, Japan)
with an ABI 7300 Thermocycler (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The following thermocycling conditions were
utilized for qPCR: 10 min pretreatment at 94°C, 55°C for 40 sec and
70°C for 40 sec (20 cycles), 94°C for 30 sec, 65°C for 40 sec and
70°C for 40 sec (40 cycles), and a final extension at 70°C for 10
min. It was then held at 4°C. GAPDH was utilized as an internal
control. The method of 2−∆∆Cq was used for the
normalization of expression to GAPDH (32). The primers used for the
amplification in the present study are presented in Table I.
 | Table I.Sequences of primers utilized in
reverse transcription quantitative polymerase chain reaction. |
Table I.
Sequences of primers utilized in
reverse transcription quantitative polymerase chain reaction.
|
| Primer
sequence |
|---|
|
|
|
|---|
| Gene | Forward | Reverse |
|---|
| IL-6 |
CCAGTTGCCTTCTTGGGACT |
TCTGACAGTGCATCATCGCT |
| IL-1β |
ATCTCGCAGCAGCACATCAA |
ATGGGAACGTCACACACCAG |
| TNF-α |
CGGAAAGGACACCATGAGCA |
GGGAGCCCATTTGGGAACTT |
| TLR4 |
CCGCTCTGGCATCATCTTCA |
AGGTCCAAGTTGCCGTTTCT |
| TRAF6 |
CATGGACGCCAAACCAGAAC |
TACACCTCTCCCACTGCTTG |
| NF-κB |
CCTGCAACAGATGGGCTACA |
TTCCTCCTTTGGGACGATGC |
| Bax |
TCTCCGGCGAATTGGAGATG |
CTCACGGAGGAAGTCCAGTG |
| Bcl-2 |
TGCGTGAAGGCTTGAGATGT |
TCCCCCTTTCCTAGACCCAG |
| Caspase-3 |
TGGCTTGCCAGAAGATACCG |
CCGTTGCCACCTTCCTGTTA |
| GAPDH |
AATGGGCAGCCGTTAGGAAA |
GCGCCCAATACGACCAAATC |
Western blot analysis
Cultured cells were lysed on ice with
radioimmunoprecipitation buffer (pH 7.4; 0.1% SDS, 1 mM
MgCl2, 10 mM Tris-HCl, 1% Triton X-100). Debris was
removed and the supernatant was collected following centrifugation
at 800 × g for 1 min at room temperature. Protein concentration was
confirmed using a bicinchoninic acid protein assay (Bio-Rad
Laboratories, Inc.) following the manufacturer's protocol. An equal
quantity (50 µg) of protein from each sample was separated using 5%
SDS-PAGE gel and transferred to nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA) for 1.5 h. Following washing,
non-specific sites were blocked by immersing membranes into 5% low
fat dried milk at room temperature for 2 h. The membranes were
subsequently maintained overnight with primary antibodies
(anti-TNF-α, anti-TLR4, anti-TRAF6, anti-NF-κB, anti-Bax,
anti-Bcl-2, anti-caspase-3, anti-IL-6, anti-IL-1β and anti-GAPDH)
at 4°C. Following washing with PBS, corresponding horseradish
peroxidase-conjugated secondary antibodies (1:6,000; cat. no.
ab6721; goat anti-rabbit IgG H&L) were incubated with the
membranes at room temperature for 2 h. Signals were detected using
an Enhanced Chemiluminescent™ detection kit (EMD Millipore). Band
densities were quantified with Quantity One software version 4.6.9
(Bio-Rad Laboratories, Inc.).
ELISA
Quantikine ELISA kits for IL-6 (cat. no. SM6000B),
IL-1β (cat. no. SMLB00C) and TNF-α (cat. no. SMTA00B) was purchased
from R&D systems. All reagents mentioned in the following
section were included in the ELISA kits. In brief, plates were
removed from the sealed bag. A total of 100 µl standard products at
different concentrations (1,000, 500, 250, 125, 62.5, 31.2, 15.6
pg/ml) were added into their corresponding wells. Wells were
subsequently sealed with adhesive tape and incubated for 90 min at
37°C. The biotinylated antibody working fluid was prepared in
advance. All plates, excluding blank wells, were washed five times
with PBS. A total of 100 µl biotinylated antibody working fluid was
added to each well, sealed with adhesive tape and incubated for 60
min at 37°C. The enzyme solution was prepared in advance for 30 min
and then placed in the dark at room temperature. All plates,
excluding blank wells, were washed five times. Enzyme solution (100
µl) was then added to each well, sealed with adhesive tape and
maintained for 30 min at 37°C. The plates were washed five times
and the chromogenic substrate was added to each well and maintained
in the dark for 10–15 min at 37°C. A stop solution (100 µl) was
then added to wells and agitated immediately for 10 min. The OD 450
value was measured using a microplate reader (Bio-Rad Laboratories,
Inc.).
Statistical analysis
GraphPad Prism version 6.0 (GraphPad Software, Inc.,
La Jolla, CA, USA) was used to perform the statistical analysis.
All data are presented as the mean ± standard error. At least three
independent experiments were performed. The results of all assays
were analyzed using one-way analysis of variance followed by
Tukey's multiple comparisons test. P<0.05 was considered to
indicate a statistically significant result.
Results
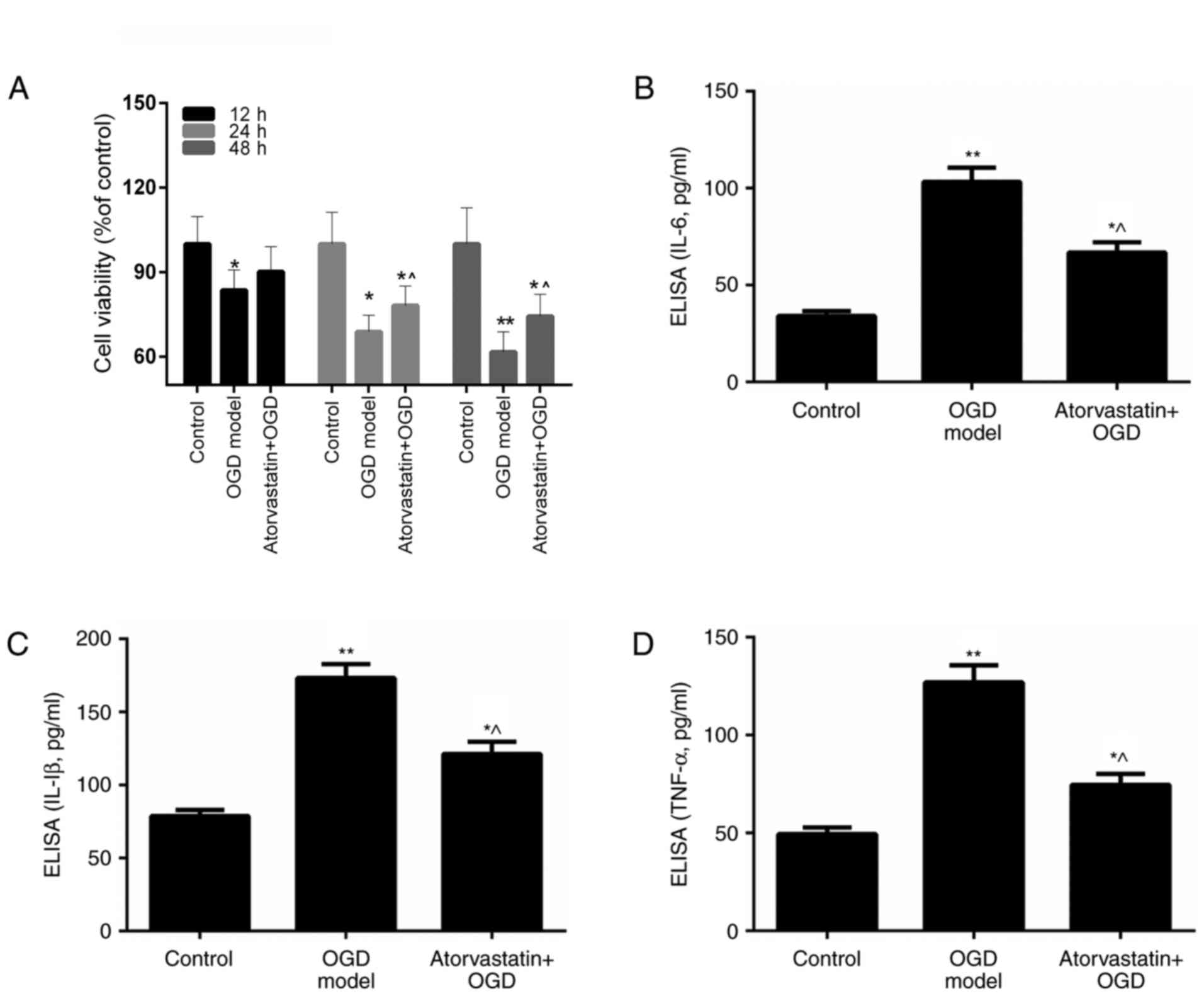
Atorvastatin suppresses OGD BV-2
microglial cell viability
Conditions of OGD have been previously demonstrated
to activate BV-2 microglia through the release of proinflammatory
and neurotoxic factors, including IL-1, TNF-α and NO, which lead to
the development of neuronal inflammatory injury (33,34).
In addition, the anti-inflammatory effect of atorvastatin has been
demonstrated to function in the protection of the CNS in AD
(35,36). The present study assessed whether
atorvastatin may affect the viability of OGD BV-2 microglia. Five
different concentrations (2.5, 5, 10, 20 and 50 µM) of atorvastatin
were applied to select the optimal concentration of atorvastatin
for the subsequent experiments (data not shown) and 10 µM was
selected. MTT results revealed that OGD led to significant decrease
in BV-2 microglia viability (P<0.05 vs. control). Following
atorvastatin treatment for 12, 24 and 48 h, it was revealed that
the viability of OGD BV-2 microglia was increased compared with the
OGD only group (Fig. 1A). These
results indicated that atorvastatin rescued the viability of
OGD-treated BV-2 microglia. Thus, it was hypothesized that
atorvastatin may exert its effects through regulation of
proinflammatory and neurotoxic factor release.
Atorvastatin reduces the expression of
IL-6, IL-1β and TNF-α in OGD BV-2 microglia
The present study assessed the mechanism by which
atorvastatin reduces the viability of OGD BV-2 microglia. The
present study investigated whether the suppressive effect of
atorvastatin on OGD BV-2 microglial cell viability may occur via
effects on the release of proinflammatory and neurotoxic factors.
Therefore, the expression of proinflammatory and neurotoxic factors
in OGD BV-2 microglia treated with atorvastatin was assessed. ELISA
results demonstrated that OGD significantly upregulated IL-6, IL-1β
and TNF-α expression in BV-2 microglia (P<0.01 vs. control;
Fig. 1B-D). However, IL-6, IL-1β
and TNF-α expression in OGD BV-2 microglia was significantly
decreased following atorvastatin treatment (P<0.05; Fig. 1B-D). In addition, RT-qPCR and
western blotting indicated that atorvastatin treatment markedly
reduced IL-6, IL-1β and TNF-α mRNA and protein expression in OGD
BV-2 microglia (P<0.01; Fig. 2A and
B). These results indicated that atorvastatin downregulated the
expression of IL-6, IL-1β and TNF-α in OGD BV-2 microglia.
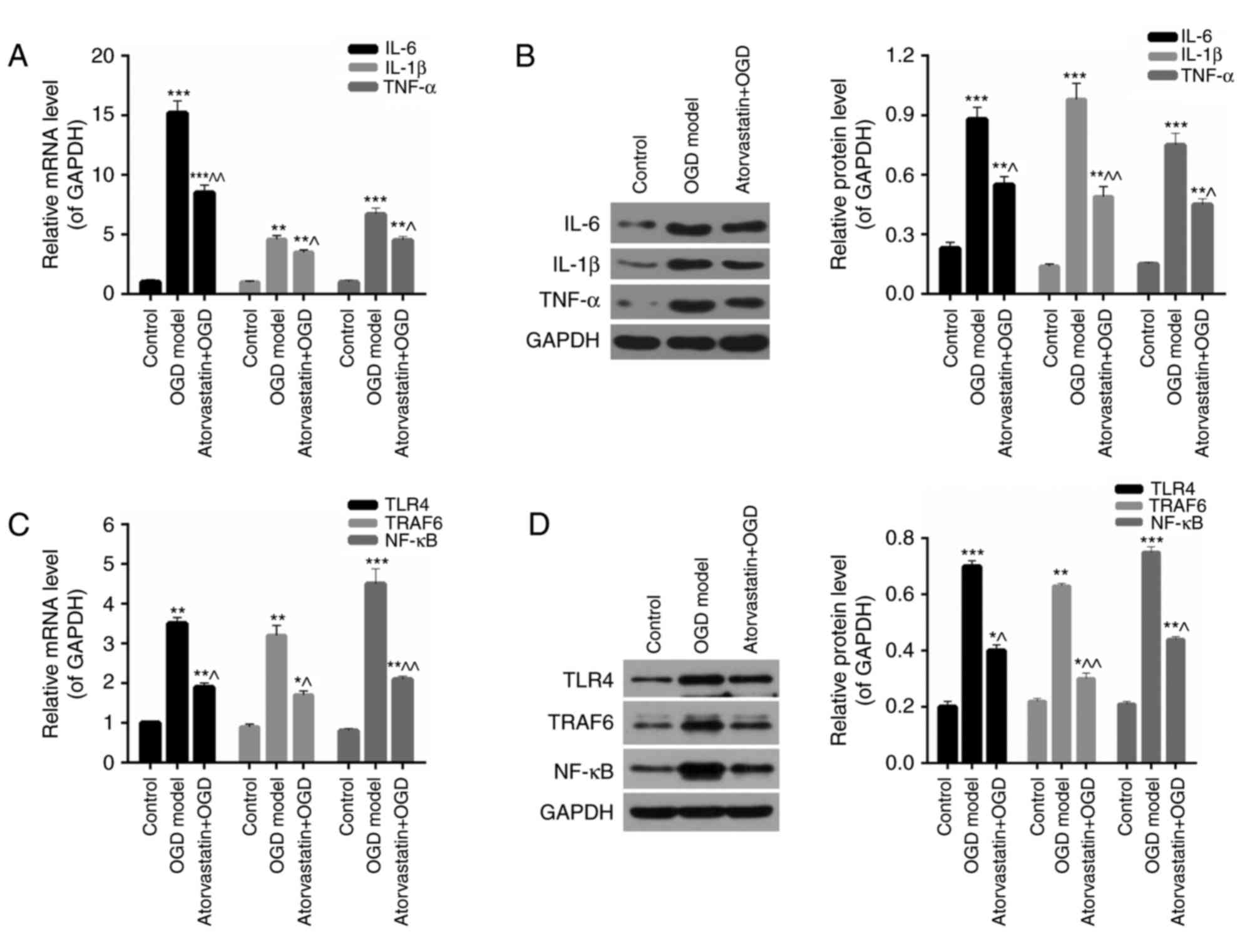 | Figure 2.Atorvastatin downregulates the
expression of inflammation factors and the TLR4/TRAF6/NF-κB pathway
in OGD BV-2 microglia. BV-2 microglial cells were subjected to OGD
conditions with or without treatment with 10 µM atorvastatin for 24
h. (A) RT-qPCR and (B) western blotting were performed to assess
the expression of IL-6, IL-1β and TNF-α in BV-2 microglia. (C)
RT-qPCR and (D) western blotting were performed to assess the
expression of TLR4, TRAF6 and NF-κB in BV-2 microglia. *P<0.05,
**P<0.01 and ***P<0.001 vs. control; ^P<0.05 and
^^P<0.01 vs. OGD model. TLR4, toll-like receptor 4; TRAF6, tumor
necrosis factor receptor-associated factor 6; NF-κB, nuclear
factor-κB; OGD, oxygen-glucose deprivation; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; IL,
interleukin; TNF-α, tumor necrosis factor-α. |
Atorvastatin downregulates the
TLR4/TRAF6/NF-κB pathway in OGD BV-2 microglia
Furthermore, the expression of the TLR4/TRAF6/NF-κB
pathway in OGD BV-2 microglia treated with atorvastatin was
assessed using RT-qPCR and western blotting. The results revealed
that OGD significantly increased the mRNA levels of TLR4, TRAF6 and
NF-κB in BV-2 microglia (P<0.01; Fig. 2C), while the mRNA expression of
these three genes was significantly reduced following treatment
with atorvastatin in OGD BV-2 microglia (P<0.05; Fig. 2C). Furthermore, western blot
analysis demonstrated that the OGD-induced overexpression of TLR4,
TRAF6 and NF-κB proteins in OGD BV-2 microglia was significantly
reduced following atorvastatin treatment, which was consistent with
the results obtained from RT-qPCR (P<0.05; Fig. 2D). The multiple protein bands
observed for TRAF6 and NF-κB presented Fig. 2D may have been caused by the
specificity of the antibodies utilized in western blotting.
Therefore, these results demonstrated that atorvastatin may
suppress the TLR4/TRAF6/NF-κB pathway in OGD BV-2 microglia.
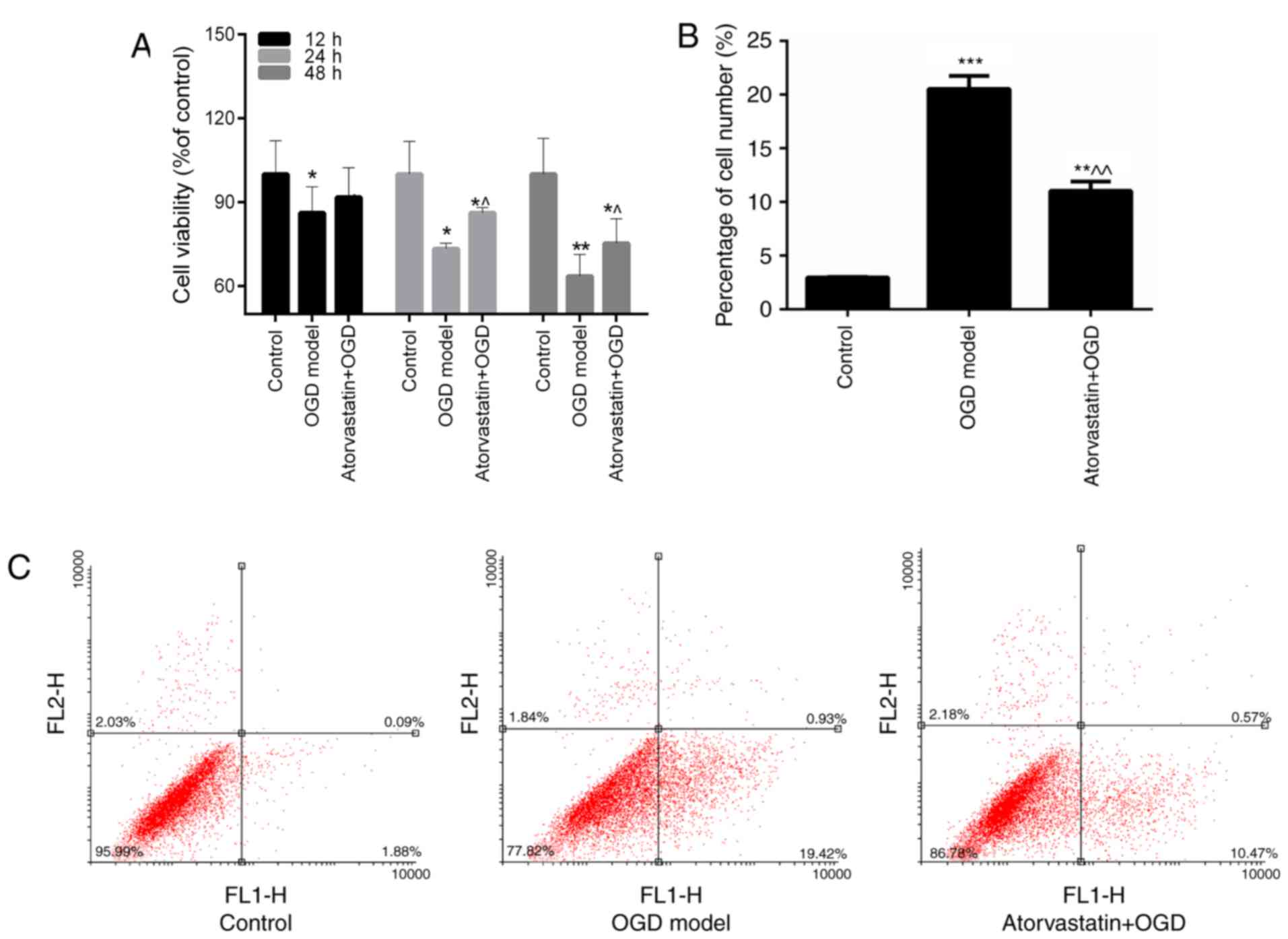
Atorvastatin improves cell viability
and inhibits apoptosis of hippocampal neuron
The viability and apoptosis of hippocampal neurons
treated with OGD and atorvastatin were also assessed in the current
study. Hippocampal neurons were cultured with MCM to create the OGD
model. MTT results indicated that OGD evidently depressed the
hippocampal neuron cell viability (P<0.05 vs. control), while
additional atorvastatin treatment enhanced the viability of OGD
hippocampal neurons (P<0.05 vs. OGD model; Fig. 3A). Furthermore, the anti-apoptosis
ability of atorvastatin treatment on OGD hippocampal neurons was
assessed. FCM data revealed that OGD led to a significant increase
in the apoptosis of hippocampal neurons (P<0.001 vs. control;
Fig. 3B and C). However,
atorvastatin markedly inhibited the apoptosis rate of OGD
hippocampal neurons (P<0.01; Fig.
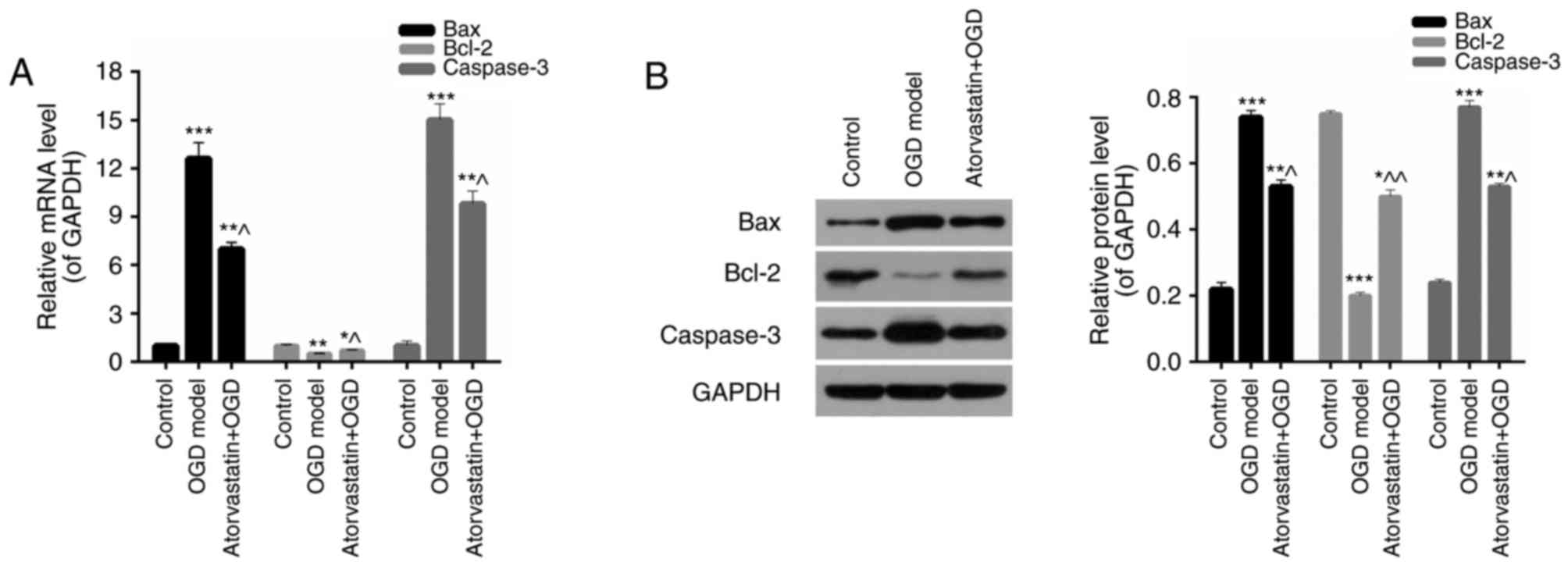
3B and C). Furthermore, the expression of apoptosis-associated
proteins in hippocampal neurons treated with OGD and atorvastatin
was assessed. The results of RT-qPCR and western blotting
demonstrated that the expression of Bax and caspase-3 in
hippocampal neurons was significantly increased under OGD
(P<0.001), while atorvastatin significantly downregulated Bax
and caspase-3 expression in OGD hippocampal neurons (P<0.05;
Fig. 4). However, Bcl-2 expression
decreased under OGD (P<0.01) and was significantly enhanced
following atorvastatin treatment (P<0.05; Fig. 4). These results verified the
hypothesis that atorvastatin treatment may inhibit OGD hippocampal
neuron apoptosis.
Discussion
AD is a chronic and polyfactoral neurodegenerative
disease of the elderly. Previous studies have demonstrated that
neuroinflammation induced by microglia is implicated in the
pathogenesis of AD (37,38). As one of the most numerous
immunocytes in the CNS, microglial cells have received attention in
patients with AD due to their conspicuous response to AD
pathophysiology (8,39). For example, it has been reported
that the proinflammatory cytokines released from activated
microglia, including IL-6, IL-1β and TNF-α, result in neuronal
damage and loss (8,9). Lovastatin was previously reported to
reduce the release of IL-6, IL-1β and TNF-α from rat primary
microglia (40). However, the
inhibitory effect of atorvastatin on the release of IL-6, IL-1β and
TNF-α from activated microglia is yet to be elucidated. In the
current study, mouse BV-2 microglia and hippocampal neurons induced
by OGD were utilized to establish a model of AD neuronal
inflammatory injury. The viability of BV-2 microglia treated with
OGD and atorvastatin was assessed. The results indicated that
atorvastatin treatment suppressed the viability of OGD BV-2
microglia. Due to these results, the present study further assessed
the expression of proinflammatory cytokines in BV-2 microglia
affected by OGD and treated with atorvastatin. The results
demonstrated that atorvastatin significantly reduced the expression
of IL-6, IL-1β and TNF-α in OGD BV-2 microglia. Therefore, it was
concluded that atorvastatin may improve the viability of OGD BV-2
microglia by downregulating the expression of IL-6, IL-1β and
TNF-α.
TLR4, TRAF6 and NF-κB are crucial factors that
participate in the regulation of inflammatory cytokine expression
(41). A previous study
demonstrated that suppression of the TLR4/NF-κB/signal transducer
and activator of transcription signaling cascade mitigated
microglial inflammation of the brain in AD (42). To the best of our knowledge, the
present study was the first to assess the effect of atorvastatin on
the TLR4/TRAF6/NF-κB pathway in neuronal inflammatory injury
associated with AD. In the present study, the expression of TLR4,
TRAF6 and NF-κB in BV-2 microglia with OGD and atorvastatin was
assessed. The results revealed that the expression of TLR4, TRAF6
and NF-κB in BV-2 microglia was significantly upregulated by OGD,
while atorvastatin treatment markedly reduced TLR4, TRAF6 and NF-κB
expression in OGD BV-2 microglia. These results demonstrated that
atorvastatin affected the TLR4/TRAF6/NF-κB pathway in OGD BV-2
microglia, indicating that atorvastatin may reduce the release of
proinflammatory cytokines from OGD BV-2 microglia by suppressing
the TLR4/TRAF6/NF-κB pathway. However, further investigation into
the exact regulatory mechanisms between atorvastatin and the
TLR4/TRAF6/NF-κB pathway are required.
It has been previously reported that cultured rat
hippocampal neurons can be used to establish OGD neuronal injury
models (43). Therefore,
hippocampal neurons from mice were selected to further assess the
protective effect of atorvastatin in OGD-induced neuronal
inflammatory injury. MTT results revealed that atorvastatin
increased the viability of OGD hippocampal neurons. According to
the cell apoptosis analysis conducted in the present study,
atorvastatin treatment significantly inhibited the apoptosis of OGD
hippocampal neurons. Furthermore, the expression of
apoptosis-associated proteins in hippocampal neurons treated with
OGD and atorvastatin were also assessed. It was determined that OGD
significantly increased the expression of Bax and caspase-3, and
reduced the expression of Bcl-2, in hippocampal neurons. However,
atorvastatin markedly downregulated the expression of Bax and
caspase-3, and upregulated the expression of Bcl-2, in OGD
hippocampal neurons. These results indicated that atorvastatin may
inhibit OGD hippocampal neuron apoptosis by regulating the
expression of Bax, Bcl-2 and caspase-3. In the current study, the
results indicated that atorvastatin may protect mouse BV-2
microglia and hippocampal neurons from OGD-induced neuronal
inflammatory injury by suppressing the TLR4/TRAF6/NF-κB pathway.
However, the present study used in vitro model only.
Therefore, to further elucidate the mechanism of neuronal
inflammatory injury, an in vivo model or co-culture between
microglia and neurons should be utilized.
In conclusion, the present study demonstrated that
atorvastatin treatment may protect BV-2 microglia and hippocampal
neurons from OGD-mediated neuronal inflammatory injury by
suppressing the TLR4/TRAF6/NF-κB pathway. This conclusion may
further support the utilization of atorvastatin in the treatment of
AD.
Acknowledgements
Not applicable.
Funding
The present study was supported by Shaoxing
Municipal Health Science and Technology Planning; Youth Science and
Technology 2016 Project (grant no. 2016QN005).
Availability of data and materials
All data generated and/or analyzed during this study
are included in this published article.
Authors' contributions
JH wrote the manuscript. JH, QY, YF and WS performed
the experiments. JH and FG conceptualized the study design. WS and
CZ analyzed the data. JH, QY and FG contributed to manuscript
revisions. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments in the present study were
approved by the Institutional Review Board of Shaoxing Municipal
Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brown KL, Cosseau C, Gardy JL and Hancock
RE: Complexities of targeting innate immunity to treat infection.
Trends Immunol. 28:260–266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Colton CA: Heterogeneity of microglial
activation in the innate immune response in the brain. J
Neuroimmune Pharmacol. 4:399–418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amantea D, Nappi G, Bernardi G, Bagetta G
and Corasaniti MT: Post-ischemic brain damage: Pathophysiology and
role of inflammatory mediators. Febs j. 276:13–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lage JM: 100 Years of Alzheimer's disease
(1906–2006). J Alzheimer Dis. 9 Suppl 3:S15–S26. 2006. View Article : Google Scholar
|
|
5
|
Aguzzi A, Barres BA and Bennett ML:
Microglia: Scapegoat, saboteur, or something else? Science.
339:156–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Czirr E and Wyss-Coray T: The immunology
of neurodegeneration. J Clin Invest. 122:1156–1163. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee CY and Landreth GE: The role of
microglia in amyloid clearance from the AD brain. J Neural Transm
(Vienna). 117:949–960. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perry VH, Nicoll JA and Holmes C:
Microglia in neurodegenerative disease. Nat Rev Neurol. 6:193–201.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Swardfager W, Lanctot K, Rothenburg L,
Wong A, Cappell J and Herrmann N: A meta-analysis of cytokines in
Alzheimer's disease. Biol psychiatry. 68:930–941. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eikelenboom P, Rozemuller AJ, Hoozemans
JJ, Veerhuis R and van Gool WA: Neuroinflammation and Alzheimer
disease: Clinical and therapeutic implications. Alzheimer Dis Assoc
Disord. 14 Suppl 1:S54–S61. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wesselingh SL, Takahashi K, Glass JD,
McArthur JC, Griffin JW and Griffin DE: Cellular localization of
tumor necrosis factor mRNA in neurological tissue from HIV-infected
patients by combined reverse transcriptase/polymerase chain
reaction in situ hybridization and immunohistochemistry. J
Neuroimmunol. 74:1–8. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matusevicius D, Navikas V, Söderström M,
Xiao BG, Haglund M, Fredrikson S and Link H: Multiple sclerosis:
The proinflammatory cytokines lymphotoxin-alpha and tumour necrosis
factor-alpha are upregulated in cerebrospinal fluid mononuclear
cells. J Neuroimmunol. 66:115–123. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Walter S, Letiembre M, Liu Y, Heine H,
Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W and
Fassbender K: Role of the toll-like receptor 4 in neuroinflammation
in Alzheimer's disease. Cell Physiol Biochem. 20:947–956. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kaczorowski DJ, Atsunori N, Mccurry KR and
Billiar TR: Toll-like receptors and myocardial
ischemia/reperfusion, inflammation, and injury. Curr Cardiol Rev.
5:196–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stevens SL and Stenzel-Poore MP: Toll-like
receptors and tolerance to ischaemic injury in the brain. Biochem
Soc Trans. 34:1352–1355. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gesuete R, Kohama SG and Stenzelpoore M:
Toll-Like receptors and ischemic brain injury. J Neuropathol Exp
Neurol. 73:378–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hyakkoku K, Hamanaka J, Tsuruma K,
Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H and
Hara H: Toll-like receptor 4 (TLR4), but not TLR3 or TLR9,
knock-out mice have neuroprotective effects against focal cerebral
ischemia. Neuroscience. 171:258–267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sato S, Sugiyama M, Yamamoto M, Watanabe
Y, Kawai T, Takeda K and Akira S: Toll/IL-1 receptor
domain-containing adaptor inducing IFN-beta (TRIF) associates with
TNF receptor-associated factor 6 and TANK-binding kinase 1, and
activates two distinct transcription factors, NF-kappa B and
IFN-regulatory factor-3, in the Toll-like receptor signaling. J
Immunol. 171:4304–4310. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kaisho T and Akira S: Toll-like receptor
function and signaling. J Allergy Clin Immunol. 117:979–987; quiz
988. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bi X, Yan B, Fang S, Yang Y, He J, Li XM
and Kong J: Quetiapine regulates neurogenesis in ischemic mice by
inhibiting NF-kappaB p65/p50 expression. Neurol Res. 31:159–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Valerio A, Dossena M, Bertolotti P, Boroni
F, Sarnico I, Faraco G, Chiarugi A, Frontini A, Giordano A, Liou
HC, et al: Leptin is induced in the ischemic cerebral cortex and
exerts neuroprotection through NF-kappaB/c-Rel-dependent
transcription. Stroke. 40:610–617. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin JJ, Kim HD, Maxwell JA, Li L and
Fukuchi K: Toll-like receptor 4-dependent upregulation of cytokines
in a transgenic mouse model of Alzheimer's disease. J
Neuroinflammation. 5:232008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Endo A, Kuroda M and Tanzawa K:
Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A
reductase by ML-236A and ML-236B fungal metabolites, having
hypocholesterolemic activity. FEBS Lett. 72:323–326. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dimmeler S, Aicher A, Vasa M, Mildner-Rihm
C, Adler K, Tiemann M, Rutten H, Fichtlscherer S, Martin H and
Zeiher AM: HMG-CoA reductase inhibitors (statins) increase
endothelial progenitor cells via the PI 3-kinase/Akt pathway. J
Clin Invest. 108:391–397. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Annu Rev Pharmacol Toxicol. 45:89–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Georgieva-Kotetarova MT and Kostadinova
II: Effect of atorvastatin and rosuvastatin on learning and memory
in rats with diazepam-induced amnesia. Folia Med (Plovdiv).
55:58–65. 2013.PubMed/NCBI
|
|
27
|
Zhang YY, Fan YC, Wang M, Wang D and Li
XH: Atorvastatin attenuates the production of IL-1beta, IL-6, and
TNF-alpha in the hippocampus of an amyloid beta1-42-induced rat
model of Alzheimer's disease. Clin Interv Aging. 8:103–110.
2013.PubMed/NCBI
|
|
28
|
Gordon R, Hogan CE, Neal ML, Anantharam V,
Kanthasamy AG and Kanthasamy A: A simple magnetic separation method
for high-yield isolation of pure primary microglia. J Neurosci
Methods. 194:287–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Badiola N, Malagelada C, Llecha N, Hidalgo
J, Comella JX, Sabriá J and Rodríguez-Alvarez J: Activation of
caspase-8 by tumour necrosis factor receptor 1 is necessary for
caspase-3 activation and apoptosis in oxygen-glucose deprived
cultured cortical cells. Neurobiol Dis. 35:438–447. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu B, Zhang H, Xu C, Yang G, Tao J, Huang
J, Wu J, Duan X, Cao Y and Dong J: Neuroprotective effects of
icariin on corticosterone-induced apoptosis in primary cultured rat
hippocampal neurons. Brain Res. 1375:59–67. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li W, Zhai L, Wang H, Liu C, Zhang J, Chen
W and Wei Q: Downregulation of LncRNA GAS5 causes trastuzumab
resistance in breast cancer. Oncotarget. 7:27778–27786.
2016.PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hoehn BD, Palmer TD and Steinberg GK:
Neurogenesis in rats after focal cerebral ischemia is enhanced by
indomethacin. Stroke. 36:2718–2724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yrjänheikki J, Tikka T, Keinänen R,
Goldsteins G, Chan PH and Koistinaho J: A tetracycline derivative,
minocycline, reduces inflammation and protects against focal
cerebral ischemia with a wide therapeutic window. Proc Natl Acad
Sci USA. 96:13496–13500. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Clarke RM, O'Connell F, Lyons A and Lynch
MA: The HMG-CoA reductase inhibitor, atorvastatin, attenuates the
effects of acute administration of amyloid-beta1-42 in the rat
hippocampus in vivo. Neuropharmacology. 52:136–145. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cordle A and Landreth G:
3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors
attenuate beta-amyloid-induced microglial inflammatory responses. J
Neurosci. 25:299–307. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alzheimer's Association: 2011 Alzheimer's
disease facts and figures. Alzheimer Dement. 7:208–244. 2011.
View Article : Google Scholar
|
|
38
|
Agostinho P, Cunha RA and Oliveira C:
Neuroinflammation, oxidative stress and the pathogenesis of
Alzheimer's disease. Curr Pharm Des. 16:2766–2778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Streit WJ, Mrak RE and Griffin WS:
Microglia and neuroinflammation: A pathological perspective. J
Neuroinflammation. 1:142004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pahan K, Sheikh FG, Namboodiri AM and
Singh I: Lovastatin and phenylacetate inhibit the induction of
nitric oxide synthase and cytokines in rat primary astrocytes,
microglia, and macrophages. J Clin Invest. 100:2671–2679. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tang L, Zhou XD, Wang Q, Zhang L, Wang Y,
Li XY and Huang DM: Expression of TRAF6 and pro-inflammatory
cytokines through activation of TLR2, TLR4, NOD1, and NOD2 in human
periodontal ligament fibroblasts. Arch Oral Biol. 56:1064–1072.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Capiralla H, Vingtdeux V, Zhao H,
Sankowski R, Al-Abed Y, Davies P and Marambaud P: Resveratrol
mitigates lipopolysaccharide- and Aβ-mediated microglial
inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J
Neurochem. 120:461–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Iijima T, Mishima T, Akagawa K and Iwao Y:
Mitochondrial hyperpolarization after transient oxygen-glucose
deprivation and subsequent apoptosis in cultured rat hippocampal
neurons. Brain Res. 993:140–145. 2003. View Article : Google Scholar : PubMed/NCBI
|