Introduction
Lung cancer is one of the most common malignant
tumors in the world, exhibiting a high mortality rate due to
abnormal cell proliferation and a high metastasis rate (1). Small-molecule epidermal growth factor
receptor tyrosine kinase inhibitors (EGFR-TKI), including
erlotinib, exert a marked clinical effect in lung cancer with
EGFR-activating mutations, and have been used as a standard therapy
in the patients with lung cancer (2,3).
However, the majority of patients develop drug resistance following
a period of treatment with EGFR-TKI, which markedly limits the
therapeutic efficacy of EGFR-TKI (4). Therefore, investigating the
resistance-associated mechanism is required to explore an effective
therapeutic approach to enhance the sensitivity of lung cancer to
EGFR-TKI.
Epithelial-mesenchymal transition (EMT) is known to
be a molecular mechanism underlying the acquisition of TKI
resistance (5). Cells undergo
morphological alterations from an epithelial phenotype to a
mesenchymal phenotype during EMT, lose epithelial cell-cell
adhesion and are able to move through the extracellular matrix,
leading to increased proliferation, invasion and metastasis
(6). EMT has been frequently
reported to be activated during cancer metastasis in multiple types
of human cancer (7,8). EMT is characterized by the loss of
cell adhesion molecules, including catenin and E-cadherin, and the
acquisition of mesenchymal marker proteins including zinc finger
protein SNAI1, fibronectin, type I collagen, and vimentin (9). EMT has been demonstrated to be
involved in the sensitivity of cancer cells to conventional
chemotherapies (10,11). In addition, sensitivity to EGFR-TKI
is additionally regulated by EMT in lung cancer cells (12). TGF-β1 is frequently used to drive
the EMT process and induce resistance to EGFR-TKI in lung cancer
cells (13). Examining novel
factors affecting EMT is important to prevent the development of
EGFR-TKI resistance.
Napsin A has been identified to be a novel member of
the aspartate protease family (14), and was observed to be correlated
with the maturation of the spleen, kidney and lung, in addition to
surfactant synthesis in the lung (15–17).
Napsin A was demonstrated to be expressed in normal lung tissue and
was detected in lung adenocarcinoma (18–20).
It was observed that napsin A was negatively associated with the
degree of transformation of cancer cells (20–22).
Additionally, cells with low napsin A expression or without napsin
A expression appear to be susceptible to EMT (23). The present study hypothesized that
the expression of napsin A may affect EMT-mediated TKI resistance
in lung cancer cells.
The present study employed a number of lung cancer
cell lines, differentially-sensitive to the EGFR-TKI erlotinib. The
expression of E-cadherin and vimentin, associated with EMT, in
addition to napsin A was detected in lung cancer cells prior to and
following the induction of TGF-β1. Lung cancer H322 cells with high
napsin A expression were selected for the investigation of the
effect of napsin A silencing on the sensitivity of lung cancer
cells to erlotinib, through TGF-β1 induction. It was observed that
napsin A-silenced cells exhibited increased resistance to erlotinib
under the conditions of TGF-β1 induction compared with napsin
A-expression cells, suggesting that napsin A served an important
role in the sensitivity of EMT-mediated resistant lung cancer cells
to TKI.
Materials and methods
Cell culture and reagents
The lung cancer cell lines (H358, H322, H441, A549
and HCC827) were obtained from the American Type Culture Collection
(Manassas, VA, USA)and were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (Lonza Group, Ltd., Basel, Switzerland), 5
mmol/l L-glutamine, 5 mmol/l non-essential amino acids and 100 U/ml
penicillin and streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.), in a humidified 5% CO2 incubator at 37°C.
Erlotinib (Tarceva®) was purchased from Cayman Chemical
Company (Ann Arbor, MI, USA). A 10-mmol/l erlotinib stock solution
was prepared in dimethyl sulfoxide (DMSO).
RNA interference for napsin A
siRNA against napsin A and the negative control were
designed and chemically synthesized by Shanghai GenePharma Co.,
Ltd. (Shanghai, China). The target sequence of siRNA against napsin
A was as follows: AAT CTT AAA CCC ACT GAA TGG. The small
interference RNA of negative control (siCtrl): Sense,
5′-UUCUCCGAACGUGUCACGUTT-3′; Antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. A total of 2×104 H322 cells
were seeded into each well of a 12-well plate and were cultured to
80% confluence. Cell transfections were performed using 100 nmol
siRNA and 5 µl Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
Cells were further cultured for 48 h following transfection, and
cells were subsequently lysed and analyzed for the protein
expression of napsin A by western blotting. In addition, cells were
treated with 0, 1, 5, 10 and 15 ng/ml TGF-β1 for 60 h, and then
cell proliferation and the EMT-associated protein levels were
detected.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted using an RNA isolation kit
(A&A Biotechnology, Gdynia, Poland), according to the
manufacturer's protocol. cDNA was obtained by RT using the
RevertAid™ First Strand cDNA synthesis kit (Fermentas,
Thermo Fisher Scientific, Inc., Pittsburgh, PA, USA) and was
amplified using a TaqMan® Gene Expression Assay (Applied
biosystems, Thermo Fisher Scientific, Inc.) with fluorogenic
Carboxyfluorescein-labeled probes using specific primers for target
proteins. The specific primers for PCR were forward,
5′-GGATTGCAAATTCCTGCCATTC-3′ and reverse, 5′-AACGTTGTCCCGGGTGTCA-3′
for E-cadherin; forward, 5′-GGAAGGCGAGGAGAGCAGGATT-3′ and reverse,
5′-TTCAAGGTCATCGTGATGCTGAGAAG-3′ for vimentin; forward,
5′-GGAGCCTGAGGAGGCC-3′ and reverse, 5′-GGACTTGGGATTAATGCG-3′ for
napsin A; and forward, 5′-GATCCCTCCAAAATCAAGTG-3′ and reverse,
5′-GAGTCCTTCCACGATACCAA-3′ for GAPDH. Real-time fluorescence
detection was performed with the ABI PRISM 7700 Sequence Detector
(Applied Biosystems, Thermo Fisher Scientific, Inc.). PCR involved
40 amplification cycles of 94°C for 10 sec, 53°C for 30 sec and
72°C for 40 sec, followed by final extension at 72°C for 10 min.
mRNA expression of target proteins was calculated using the formula
2∆∆Cq (24) and was
normalized to the level of GAPDH. The relative mRNA level was
presented as a percentage of the control.
Western blot analysis
Cells were cultured to 80% confluence. The cells
were washed twice with PBS and proteins were extracted using M-PER
Mammalian Protein Extraction Reagent (Pierce; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
Following centrifugation at 12,000 × g for 10 min, the supernatant
was collected and quantified using a Bicinchoninic acid (BCA)
quantification kit (Beyotime Institute of Biotechnology, Haimen,
China). The proteins (50 µg) were separated by SDS-PAGE on a 12%
gel (Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) and transferred to polyvinylidene fluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
non-fat dried milk in TBS-Tween 20 for 1 h, and incubated with
specific primary antibodies overnight at 4°C. Mouse monoclonal
anti-E-cadherin (cat. no. sc-21791; 1:2,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), mouse monoclonal
anti-vimentin (cat. no. sc-6260; 1:2,000; Santa Cruz Biotechnology,
Inc.), mouse monoclonal anti-N-cadherin (cat no. sc-8424; 1:2,000;
Santa Cruz Biotechnology, Inc.), anti-GAPDH antibody (cat. no.
sc-365062; 1:3,000; Santa Cruz Biotechnology, Inc.) and rabbit
monoclonal anti-napsin A antibody (cat. no. ab133249; 1:10,000;
Abcam, Cambridge, UK) were used, followed by horseradish
peroxidase-conjugated secondary antibodies goat anti-mouse (cat no.
sc-2005; 1:2,000; Santa Cruz Biotechnology, Inc.) and anti-rabbit
immunoglobulin G (cat no. sc-2004; 1:2,000; Santa Cruz
Biotechnology, Inc.) for 2 h at room temperature. Development was
performed using an enhanced chemiluminescence detecting reagent (GE
Healthcare Life Sciences, Little Chalfont, UK). The protein blots
were quantified by densitometry using QuantityOne software version
4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA, USA), and the
amounts were expressed relative to the internal reference
GAPDH.
Proliferation assay
Cell proliferation was evaluated using MTT
(Sigma-Aldrich, Merck KGaA, Darmstadt, Germany). A total of 2,000
cells were seeded into each well of a 96-well plate in 100 µl
medium and incubated with or without varying concentrations of
erlotinib for different times, at 37°C in a 5% CO2
incubator. Subsequently, cells were incubated with 20 µl 5 mg/ml
MTT for 4 h, and cells were lysed for 10 min by the addition of 200
µl DMSO (OriGen Biomedical, Inc., Austin, Texas, USA). Absorbance
was measured at 490 nm using a Rainbow microplate reader (Tecan
Group, Ltd., Mannedorf, Switzerland). Cell proliferation was
expressed as a percentage of the untreated control.
Apoptosis assay
Cells were cultured to 80% confluence and treated
with 1 µmol/l erlotinib for 48 h. Apoptosis was analyzed using
annexin V-fluorescein isothiocyanate/propidium iodide (PI) assay
following the manufacturer's instructions. The amount of
phosphatidylserine on the outer surface of the plasma membrane (a
biochemical alteration unique to the membranes of apoptotic cells)
and the amount of PI, a dye that readily enters dead cells or cells
in the late stages of apoptosis and binds DNA, although it does not
bind to the plasma membrane of viable cells, were detected.
Fluorescence was detected using a FACSCalibur flow cytometer by
fluorescence activated cell sorting analysis, and data were
analyzed using CellQuest version 3.2 software (BD Biosciences, San
Jose, CA, USA). Cells with phosphatidylserine on the surface were
considered to be apoptotic.
Statistical analysis
Data were obtained from at least three experiments.
Values are expressed as the mean ± standard error of the mean.
Statistical analysis was preformed using SPSS version 16.0 for
MicroSoft™ Windows. One-way analysis of variance was
used to assess differences between groups. Duncan method was
employed for pairwise comparison followed by Bonferroni correction.
P<0.05 was considered to indicate a statistically significant
difference.
Results
EMT and napsin A are associated with
the sensitivity of lung cancer cells to the EGFR-TKI erlotinib
Lung cancer cells H322, H358, H441, A549 (wild-type
EGFR) cells (25) and HCC827 (EGFR
exon 19 deletion) were cultured to 80% confluence and exposed to
different concentrations of erlotinib for 48 h. Analysis of
erlotinib sensitivity was performed by cell growth inhibition
evaluation using an MTT assay and cell growth curves were drawn.
The half-maximal inhibitory concentration (IC50) values
of these cells were respectively calculated to be 1.0, 1.71, 1.72,
5.65 and 13.6 µmol/l (Fig. 1A).
Sensitivity was defined as >50% in vitro growth
inhibition at an erlotinib concentration of <5 µmol/l; moderate
sensitivity was defined as the IC50 at an erlotinib
concentration between 5 and 10 µmol/l; insensitivity was defined as
the IC50 at an erlotinib concentration of >10 µmol/l.
The results indicated that lung cancer cells H322, H358 and H441
(wild-type EGFR) were sensitive to erlotinib, A549 (wild-type EGFR)
cells were moderately sensitive, and HCC827 (EGFR exon19 deletion)
cells exhibited lower sensitivity to erlotinib. Additionally, cells
were treated with 1 µmol/l erlotinib for 48 h, and the expression
of EMT-associated proteins, including E-cadherin and vimentin, was
detected. RT-qPCR analysis and western blotting demonstrated that
erlotinib-sensitive H322, H358 and H441 cells exhibited increased
E-cadherin mRNA and protein expression levels compared with
erlotinib-moderately sensitive A549 cells and erlotinib-insensitive
HCC827 cells. However, vimentin exhibited opposite expression
(Fig. 1B and C). These data
suggested that EMT may be associated with the sensitivity of lung
cancer cells to erlotinib. Additionally, napsin A mRNA and protein
expression in erlotinib-sensitive H322, H358 and H441 cells was
demonstrated to be increased compared with erlotinib-moderately
sensitive A549 cells, and erlotinib-insensitive HCC827 cells
exhibited the lowest napsin A level, suggesting that napsin A maybe
positively associated with the sensitivity of lung cancer cells to
erlotinib.
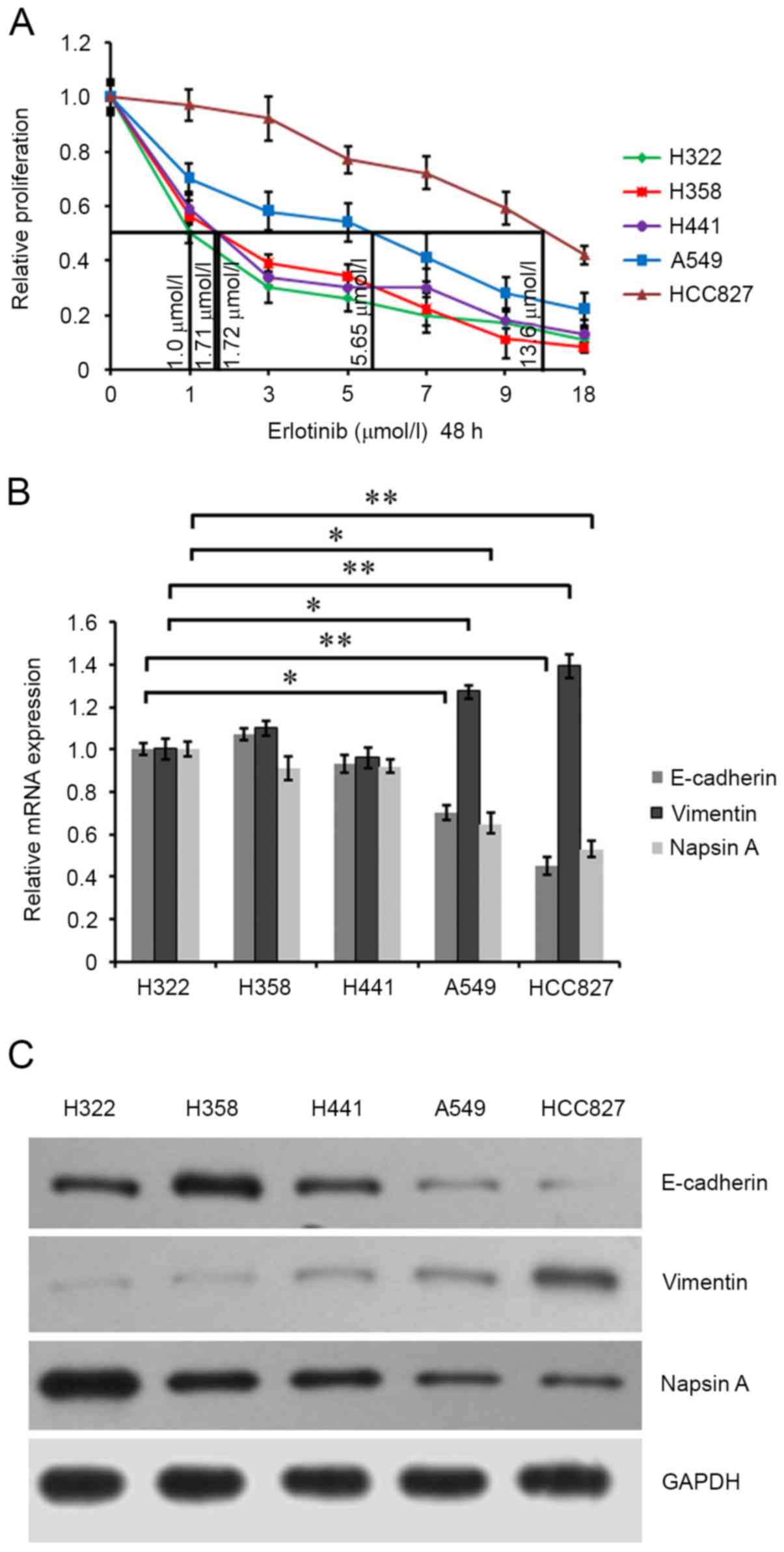 | Figure 1.Characterization of lung cancer H322,
H358, H441, A549 and HCC827 cells. (A) Cells were maintained in 100
µl medium in 96-well plates for 24 h and exposed to 0, 1, 3, 5, 7,
9 and 18 µmol/l erlotinib for 48 h. Cell proliferation was
evaluated by MTT assay. Growth curves of the cells were drawn.
Half-maximal inhibitory concentration values of the cells were
calculated. (B) The mRNA expression of E-cadherin, vimentin and
napsin A in these lung cancer cells were assessed by reverse
transcription-quantitative polymerase chain reaction analysis using
the corresponding primers. GAPDH was detected as an internal
standard. (C) The protein expression of E-cadherin, vimentin and
napsin A in cells was assessed by western blot analysis using
anti-E-cadherin, anti-vimentin and anti-napsin A antibodies. GAPDH
was detected as an internal standard. *P<0.05; **P<0.01. |
Napsin A silencing enhances
TGF-β1-induced EMT
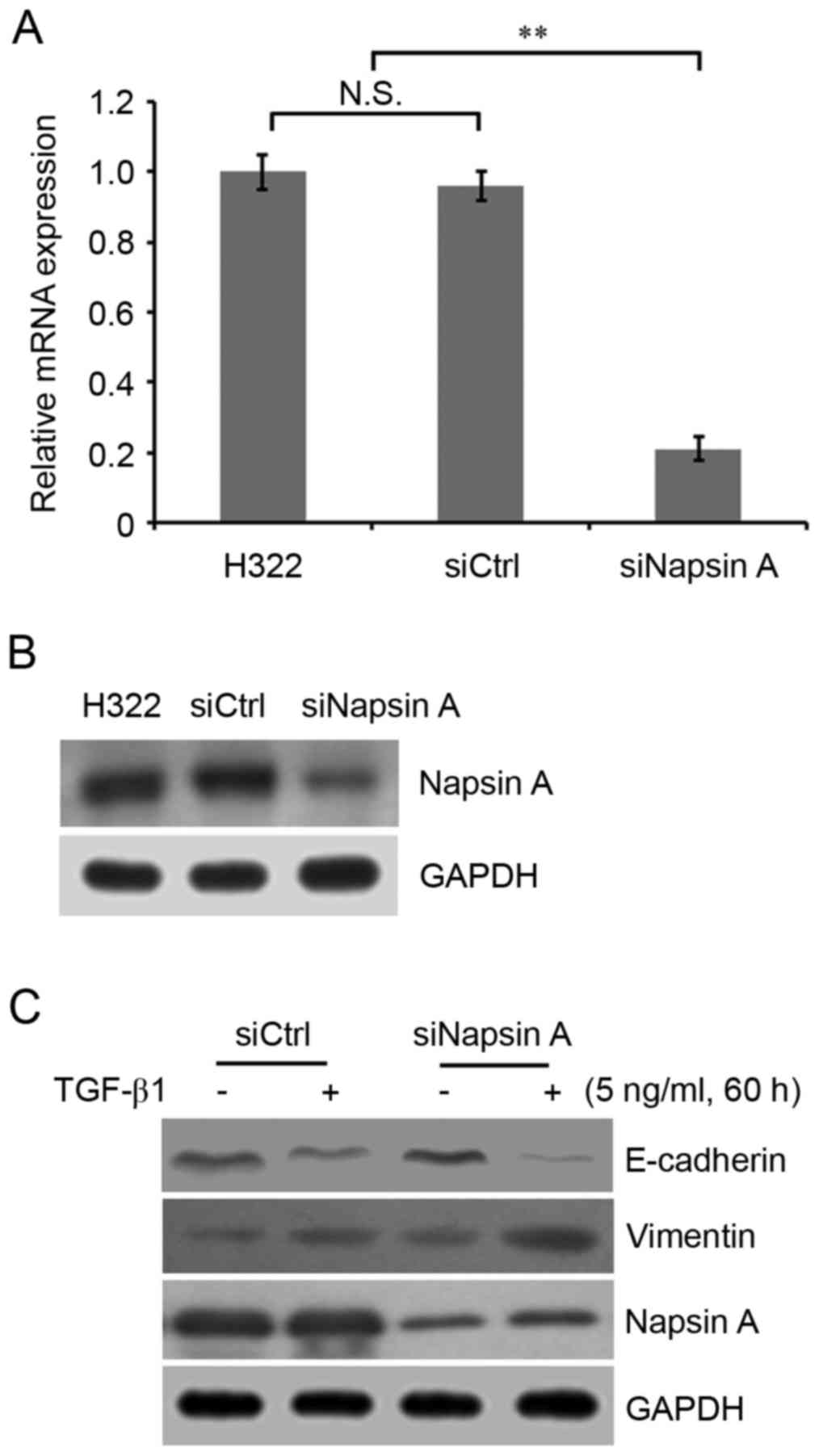
To investigate the correlation between napsin A
expression and EMT in lung cancer cells, the napsin A-expressing
H322 cells were used and napsin A was knocked down using siRNA
technology. The mRNA and protein expression of napsin A was
evaluated by RT-qPCR analysis and western blotting. The results
demonstrated that napsin A expression in napsin A-silenced cells
was significantly decreased compared with non-silenced control
cells (Fig. 2A and B).
Subsequently, cells were treated with 0, 1, 5, 10 and 15 ng/ml
TGF-β1 for 60 h, and a concentration response curve for TGF-β1was
performed. It was observed that cellular morphology began to alter
when the TGF-β1 concentration reached 5 ng/ml. In addition, of the
five concentrations, 5 ng/ml TGF-β1 stimulated cell proliferation
most rapidly and the EMT-associated protein levels were altered
(data not shown). Therefore, 5 ng/ml TGF-β1 was selected to induce
EMT. It was demonstrated that napsin A silencing significantly
enhanced the TGF-β1-induced EMT phenotype characterized by
decreased E-cadherin and increased vimentin expression in napsin
A-silenced H322 cells compared with highly napsin A-expressing
control H322 cells (Fig. 2C).
N-cadherin expression in lung cancer cells was additionally
detected to be positively associated with vimentin expression in
this study (data not shown).
Napsin A silencing promotes
EMT-mediated erlotinib resistance
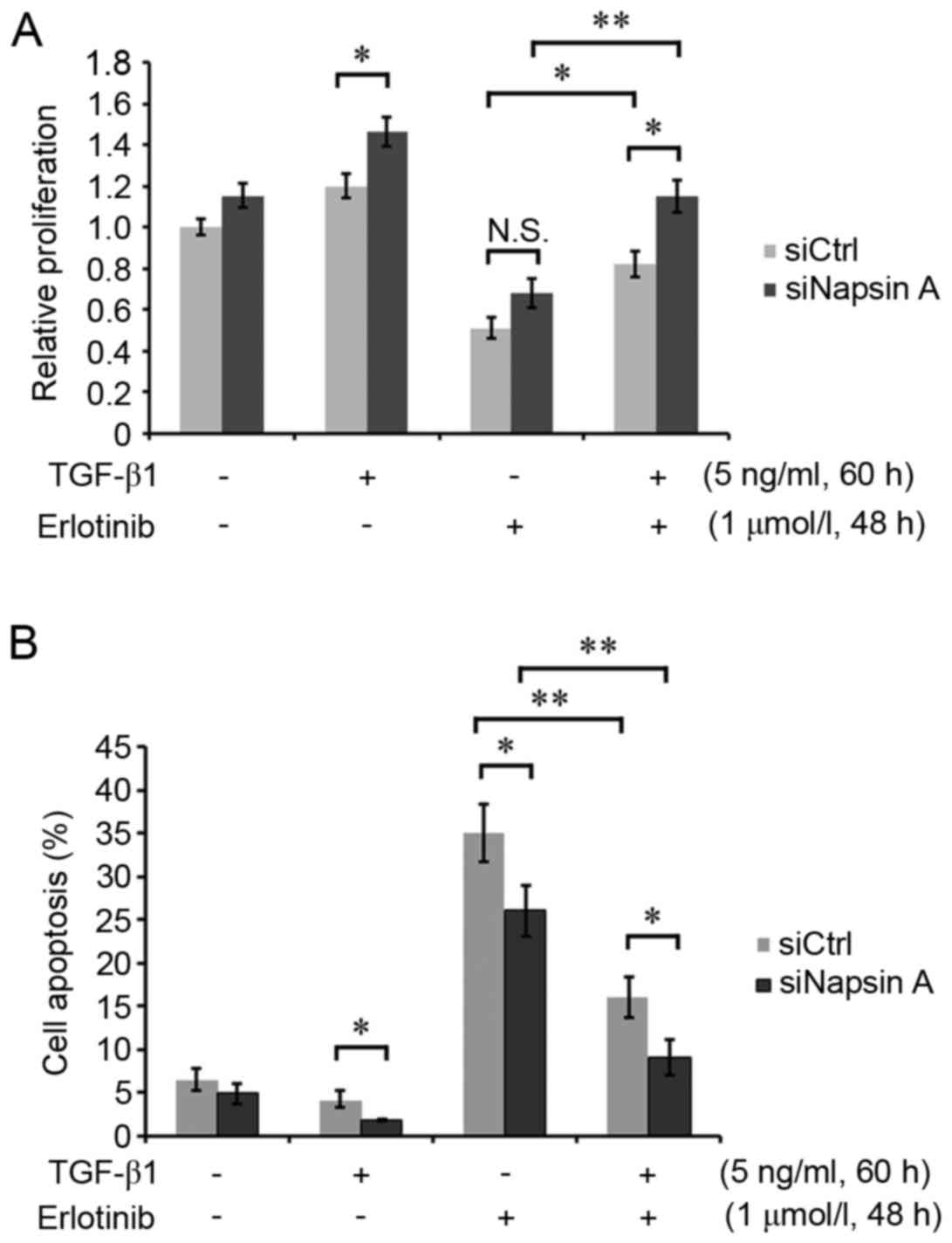
Napsin A-silenced H322 cells and control cells were
respectively induced with or without 5 ng/ml TGF-β1 for 60 h, and
subsequently treated with 1 µmol/l erlotinib for 48 h. The cell
proliferation assay demonstrated that TGF-β1-induced EMT mediated
the increased cell growth rate and resistance to erlotinib in H322
cells (Fig. 3A). However, napsin A
silencing enhanced the EMT-mediated erlotinib resistance of H322
cells (Fig. 3A). In addition, the
cellular apoptosis assay demonstrated that napsin A silencing
attenuated the inducing effect of erlotinib on apoptosis in
TGF-β1-treated cells compared with napsin A-expressing control
cells (Fig. 3B), suggesting that
the expression of napsin A may inhibit EMT-mediated erlotinib
resistance. These data indicated that napsin A was a potential
target for improving the sensitivity of EMT-induced resistant lung
cancer cells to the EGFR-TKI erlotinib.
Discussion
Lung cancer is a common malignancy with a high
mortality rate, which is a severe threat to human health (1). EGFR-TKIs, including Gefitinib and
erlotinib, have been used as the standard therapy in lung cancer
with EGFR-activating mutations (2,3).
However, the majority of patients eventually succumb to recurrence
due to drug resistance; thus, therapeutic efficacy is markedly
limited. Therefore, elucidating effective therapeutic strategies is
required to overcome the acquisition of EGFR-TKI resistance. The
present study demonstrated that the expression of napsin A was able
to increase the sensitivity of EMT-mediated resistant lung cancer
cells to erlotinib compared with napsin A-silenced cells.
EMT, defined by the combined loss of E-cadherin and
the gain of mesenchymal lineage marker expression, negatively
affected cellular responses to EGFR inhibitors (26). In the present study, three EGFR-TKI
erlotinib-sensitive lung cancer cell lines, H358, H322 and H441,
erlotinib-moderately sensitive A549 cells, and
erlotinib-insensitive HCC827 cells were used and the expression of
the EMT-associated proteins E-cadherin and vimentin, and the
expression of napsin A, which was reported to inhibit EMT in lung
cancer A549 cells (27), was
detected. It was observed that E-cadherin mRNA and protein
expression levels were positively associated with the sensitivity
of lung cancer cells to erlotinib, while vimentin exhibited a
negative association with erlotinib sensitivity. Like vimentin,
N-cadherin is an important marker of EMT, commonly expressed in
mesenchymal cells (28). The
downregulation of E-cadherin and the upregulation of vimentin and
N-cadherin are the characteristics of EMT (9) and EMT is the underlying mechanism of
acquisition of TKI resistance (5).
Therefore, it was hypothesized that EMT may be involved in the
development of TKI resistance in lung cancer cells. In addition,
the expression level of napsin A was demonstrated to be positively
associated with the sensitivity of lung cancer cells to erlotinib,
suggesting that napsin A may serve an adverse role in EMT-mediated
drug resistance. Therefore, highly napsin A-expressingH322 cells
were used to construct a napsin A-silenced cell line using siRNA
technology. Subsequently, TGF-β1 was used to induce cellular EMT,
and it was observed that TGF-β1-treated H322 cells were more
resistant to erlotinib compared with TGF-β1-untreated cells. In
addition, it was observed that napsin A silencing enhanced
TGF-β1-induced cellular resistance to erlotinib via proliferation
and apoptosis assays, verifying the aforementioned hypothesis.
Napsin A has been reported to contain an Arg-Gly-Asp
(RGD) sequence at the carboxyl terminal. The sequence is able to
recognize and bind integrins on the cell surface (27). Integrins are able to mediate cell
adhesion and signal transduction between cells and the ECM
(29), and is an important
regulator of cell proliferation, apoptosis, migration and
metastasis (30,31). Napsin A may suppress the
interaction between Integrins and the ECM by binding Integrins, and
thus inhibit the integrin-mediated signaling pathway. We have found
that focal adhesion kinase 1 (FAK) was inhibited by napsin A
expression in Gefitinib-resistant A549 cells (Zhou et al,
unpublished data). FAK serves an important role in integrin
signaling (27,32–36),
and may be activated by integrin signaling and modulate a number of
signaling pathways, including phosphatidylinositol 3-kinase/RAC-α
serine/threonine-protein kinase, signal transducer and activator of
transcription 1, and Ras-mitogen-activated protein kinase signaling
(32,37,38),
and thus triggers cell growth and transformation. It was
hypothesized that napsin A may repress the interaction between
Integrins and ECM through RGD sequence-mediated interaction with
integrin, and further inhibit the integrin signaling pathway, and
cell proliferation and transformation, by downregulating FAK
expression in lung cancer cells. Whether the mechanism is
implicated in other lung cancer cell lines requires further
investigation. Additionally, napsin A has been demonstrated to be
able to suppress cell growth in 293T cells (23), and napsin A expression in systemic
anaplastic large cell lymphoma (ALCL) was associated with an
increased international prognostic index in malignant lymphoma;
napsin A expression predicted a poor prognosis in patients with
ALCL and diffuse large B-cell lymphoma (39). Therefore, these data, combined with
the present finding that the expression of napsin A augmented the
effect of erlotinib on TGF-β1-induced TKI-resistant lung cancer
cells, suggested that napsin A may be a promising target for
improving the sensitivity of drug resistant cells, and may exhibit
clinical potential.
In conclusion, the results of the present study
demonstrated that napsin A served an important role in the
development of EMT-mediated resistance in lung cancer cells to
EGFR-TKI, and napsin A combined with EGFR-TKI may be a more
effective way of improving the sensitivity of lung cancer cells to
the TKI erlotinib. In order to verify the underlying
resistance-associated mechanism to EGFR-TKI in vivo in human
lung cancer tissues, in vivo xenograft models may be
constructed by injecting different lung cancer cell lines with or
without napsin A silencing into mice, and a napsin A-targeted gene
treatment maybe employed in order to further assess the clinical
importance and significance of the present study.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ, XL and ZW were major contributors in the
conception and design of the research and revision of the
manuscript for important intellectual content. Acquisition of data
was performed by JY and YZ. TX was the major contributor in the
analysis and interpretation of data and statistical analysis.
Drafting of the manuscript was performed by ZW.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
PDQ Adult Treatment Editorial Board, .
Non-small cell lung cancer treatment (PDQ®): Patient
version. NCI. May 12–2002–2015.
|
|
2
|
Soria JC, Mok TS, Cappuzzo F and Jänne PA:
EGFR-mutated oncogene addicted non-small cell lung cancer: Current
trends and future prospects. Cancer Treat Rev. 38:416–430. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nguyen KS and Neal JW: First-line
treatment of EGFR-mutant non-small cell lung cancer: The role of
erlotinib and other tyrosine kinase inhibitors. Biologics.
6:337–345. 2012.PubMed/NCBI
|
|
4
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to Gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu PF, Zhu YP, Yang CH, Wang YF and Wang
GH: The mechanism and countermeasures on the secondary resistance
of epidermal growth factor receptor tyrosine kinase inhibitor
(EGFR-TKI). Anti Tumor Pharmacy. 5:42015.
|
|
6
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guarino M, Rubino B and Ballabio G: The
role of epithelial-mesenchymal transition in cancer pathology.
Pathology. 39:305–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robert G, Gaggioli C, Bailet O, Chavey C,
Abbe P, Aberdam E, Sabatié E, Cano A, de Herreros Garcia A,
Ballotti R and Tartare-Deckert S: SPARC represses E-cadherin and
induces mesenchymal transition during melanoma development. Cancer
Res. 66:7516–7523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: Mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.PubMed/NCBI
|
|
11
|
Neel DS and Bivona TG: Secrets of drug
resistance in NSCLC exposed by new molecular definition of EMT.
Clin Cancer Res. 19:3–5. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Uramoto H, Iwata T, Onitsuka T, Shimokawa
H, Hanagiri T and Oyama T: Epithelial-mesenchymal transition in
EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res.
30:2513–2517. 2010.PubMed/NCBI
|
|
13
|
Yao Z, Fenoglio S, Gao DC, Camiolo M,
Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V,
et al: TGFb IL-6 axis mediates selective and adaptive mechanisms of
resistance to molecular targeted therapy in lung cancer. Proc Natl
Acad Sci USA. 107:15535–15340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tatnell PJ, Powell DJ, Hill J, Smith TS,
Tew DG and Kay J: Napsins: New human aspartic proteinases.
Distinction between two closely related genes. FEBS Lett.
441:43–48. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brasch F, Ochs M, Kahne T, Guttentag S,
Schauer-Vukasinovic V, Derrick M, Johnen G, Kapp N, Muller KM,
Richter J, et al: Involvement of napsin A in the C- and N-terminal
processing of surfactant protein B in type-II pneumocytes of the
human lung. J Biol Chem. 278:49006–49014. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ueno T, Linder S, Na CL, Rice WR,
Johansson J and Weaver TE: Processing of pulmonary surfactant
protein B by napsin and cathepsin H. J Biol Chem. 279:16178–16184.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suzuki A, Shijubo N, Yamada G, Ichimiya S,
Satoh M, Abe S and Sato N: Napsin A is useful to distinguish
primary lung adenocarcinoma from adenocarcinomas of other organs.
Pathol Res Pract. 201:579–586. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chuman Y, Bergman A, Ueno T, Saito S,
Sakaguchi K, Alaiya AA, Franzén B, Bergman T, Arnott D, Auer G, et
al: Napsin A, a member of the aspartic protease family, is
abundantly expressed in normal lung and kidney tissue and is
expressed in lung adenocarcinomas. FEBS Lett. 462:129–134. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schauer-Vukasinovic V, Bur D, Kling D,
Grüninger F and Giller T: Human napsin A: Expression,
immunochemical detection, and tissue localization. FEBS Lett.
462:135–139. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hirano T, Auer G, Maeda M, Hagiwara Y,
Okada S, Ohira T, Okuzawa K, Fujioka K, Franzén B, Hibi N, et al:
Human tissue distribution of TA02, which is homologous with a new
type of aspartic proteinase, napsin A. Jpn J Cancer Res.
91:1015–1021. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hirano T, Gong Y, Yoshida K, Kato Y,
Yashima K, Maeda M, Nakagawa A, Fujioka K, Ohira T, Ikeda N, et al:
Usefulness of TA02 (napsin A) to distinguish primary lung
adenocarcinoma from metastatic lung adenocarcinoma. Lung Cancer.
41:155–162. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ueno T, Linder S and Elmberger G: Aspartic
proteinase napsin is a useful marker for diagnosis of primary lung
adenocarcinoma. Br J Cancer. 88:1229–1233. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ueno T, Elmberger G, Weaver TE, Toi M and
Linder S: The aspartic protease napsin A suppresses tumor growth
independent of its catalytic activity. Lab Invest. 88:256–263.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Slack JL, Bi W, Livak KJ, Beaubier N, Yu
M, Clark M, Kim SH, Gallagher RE and Willman CL: Pre-clinical
validation of a novel, highly sensitive assay to detect
PML-RARalpha mRNA using real-time reverse-transcription polymerase
chain reaction. J Mol Diagn. 3:141–149. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thomson S, Buck E, Petti F, Griffin G,
Brown E, Ramnarine N, Iwata KK, Gibson N and Haley JD: Epithelial
to mesenchymal transition is a determinant of sensitivity of non
small cell lung carcinoma cell lines and xenografts to epidermal
growth factor receptor inhibition. Cancer Res. 65:9455–9462. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grunert S, Jechlinger M and Beug H:
Diverse cellular and molecular mechanisms contribute to epithelial
plasticity and metastasis. Nat Rev Mol Cell Biol. 4:657–665. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng JX, Guan SH, Xu Q, Liu JZ and Song
P: Inhibition of epithelial-mesenchymal transitionin A549 cell by
transfected Napsin A. Chin Med J (Engl). 125:2734–2740.
2012.PubMed/NCBI
|
|
28
|
Nakajima S, Doi R, Toyoda E, Tsuji S, Wada
M, Koizumi M, Tulachan SS, Ito D, Kami K, Mori T, et al: N-cadherin
expression and epithelial-mesenehymal transition in pancreatic
carcinoma. Clin Cancer Res. 10:4125–4133. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruoslahti E: RGD and other recognition
sequences for integrins. Annu Rev Cell Dev Biol. 12:697–715. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Juliano RL: Signal transduction by cell
adhesion receptors and the cytoskeleton: Functions of integrins,
cadherins, selectins, and immunoglobulin-superfamily members. Annu
Rev Pharmacol Toxicol. 42:283–323. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hynes RO: Integrins: Bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Yang J, Dai C, Wu C and Liu Y: Role
for integrin-linked kinase in mediating tubular epithelial to
mesenchymal transition and renal interstitial fibrogenesis. J Clin
Invest. 112:503–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bhowmick NA, Zent R, Ghiassi M, McDonnell
M and Moses HL: Integrin beta 1 signaling is necessary for
transforming growth factor-beta activation of p38MAPK and
epithelial plasticity. J Biol Chem. 276:46707–46713. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hauck CR, Sieg DJ, Hsia DA, Loftus JC,
Gaarde WA, Monia BP and Schlaepfer DD: Inhibition of focal adhesion
kinase expression or activity disrupts epidermal growth
factor-stimulated signaling promoting the migration of invasive
human carcinoma cells. Cancer Res. 61:7079–7090. 2001.PubMed/NCBI
|
|
35
|
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK,
Schaefer E, Damsky CH and Schlaepfer DD: FAK integrates
growth-factor and integrin signals to promote cell migration. Nat
Cell Biol. 2:249–256. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sieg DJ, Hauck CR and Schlaepfer DD:
Required role of focal adhesion kinase (FAK) for
integrin-stimulated cell migration. J Cell Sci. 112:2677–2691.
1999.PubMed/NCBI
|
|
37
|
Hauck CR, Hsia DA and Schlaepfer DD: The
focal adhesion kinase a regulator of cell migration and invasion.
IUBMB Life. 53:115–119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xie B, Zhao J, Kitagawa M, Durbin J, Madri
JA, Guan JL and Fu XY: Focal adhesion kinase activates Stat1 in
integrin-mediated cell migration and adhesion. J Biol Chem.
276:19512–19523. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nam SJ, Kim S, Kim JE, Lim MS,
Elenitoba-Johnson KS, Kim CW and Jeon YK: Aberrant expression
ofnapsinA in a subset of malignant lymphomas. Histol Histopathol.
31:213–221. 2016.PubMed/NCBI
|