Introduction
Endothelial progenitor cells (EPCs) enhance new
vessel formation and maintain homeostasis of the endothelium due to
their protective role in the cardiovascular system (1). Previous studies have shown that
homocysteine (Hcy) reduces the number of EPCs and impairs their
functional activities, including proliferation, migration, adhesion
and in vitro vasculogenesis capacity (2,3).
Furthermore, Hcy increases the levels of EPC apoptosis by
stimulating the production of reactive oxygen species (ROS) within
EPCs through the activation of NADPH oxidase (4). Analyze detection platforms such as
luminescence probe could be used in studying EPC function (5,6).
Pioglitazone (PIO), which has been used for the
treatment of type 2 diabetes for many years, has substantially more
potential beneficial effects than previously expected. PIO has been
shown to exert favorable cardiovascular effects by slowing the
progression of atherosclerosis progression (7), and may reduce the risk of myocardial
infarction, stroke and premature death in high-risk patients with
diabetes. PIO has been shown to exert beneficial effects in
vitro on EPCs isolated from patients with diabetes mellitus and
coronary artery disease (8), and
prevent apoptosis of EPCs and promote in vivo
neoangiogenesis in mice (9). In
addition, PIO ameliorates Ang II-induced senescence of EPCs
(10). However, to the best of our
knowledge, no previous study has investigated the role of PIO in
the regulation of EPC dysfunctions and its related potential
mechanisms under high levels of Hcy.
PKC activation has been demonstrated to be a common
signaling pathway through which Hcy exerts its pathogenic functions
in the vasculature. High levels of Hcy impair endothelial function
primarily through PKC activation (11). In monocytes, Hcy stimulates
phosphorylation of the NADPH oxidase subunits p47phox and p67phox
via activation (12). PKC
activation and NADPH oxidase phosphorylation may possibly be the
signaling pathways involved in Hcy-induced EPC dysfunctions. In our
previous study, we found that PIO mitigated Hcy-induced
downregulation of vascular endothelial growth factor (VEGF) and
interleukin (IL)-8 expression and secretion in EPCs via PKC and
NADPH oxidase (13).
On the basis of these findings, we speculated that
Hcy may impair the function of EPCs by initiating the activation of
PKC and NADPH oxidase, and the underlying protective effects of PIO
on the migration and adhesion of EPCs may result from the reduction
of oxidative stress produced by NADPH oxidase, and the inactivation
of PKC in EPCs.
Materials and methods
Materials
Peripheral blood mononuclear cells (PBMNCs) were
obtained from healthy volunteers. Written informed consent was
obtained from all volunteers, and the present study was approved by
the Ethics Review Board of Sir Run Run Shaw Hospital, College of
Medicine, Zhejiang University (Zhejiang, China). Volunteers
participated in this study are up to 10, but because of
contamination, EPCs were mainly collected from the blood of 8
volunteers. During this study, the doctors obeyed the ethical
principles and respected the volunteers' rights. All protocols are
in accordance with the Helsinki Declaration. Endothelial
Lympholyte®-H Cell Separation Media was acquired from
Cedarlane (Burlington, VT, USA). Cell Growth Medium-2 (EGM-2) was
purchased from Lonza (Walkersville, MD, USA). Fetal bovine serum
(FBS) was obtained from Gibco (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Hcy and Diphenyleneiodonium chloride (DPI) were
procured from Sigma Chemical (St. Louis, MO, USA). PIO was
generously provided by Huadong Medicine Co. Ltd (Hangzhou, China).
The reagent 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein
diacetate, acetyl ester (CM-H2DCFDA) was obtained from
Invitrogen (Thermo Fisher Scientific, Inc.). Lucigenin and NADPH
tetrasodium salt were purchased from Enzo Life Sciences, Inc.
(Farmingdale, NY, USA). Bisindolylmaleimide I (GF 109203×) was
acquired from Calbiochem (Darmstadt, Germany). Anti-gp91 (NOX2) was
purchased from Merck Millipore (Darmstadt, Germany). Anti-p67phox
was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). Anti-phospho-PKC α/βII was obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). HRP-conjugated monoclonal
mouse anti-GAPDH antibody was purchased from Kangchen (Shanghai,
China). PIO, DPI and GF109203× were dissolved in 0.1% dimethyl
sulfoxide (DMSO) and the vehicle (0.1% DMSO) was added to the
control samples.
Isolation and cultivation of EPCs
EPCs were isolated, cultured and characterized
according to previously described techniques (3). PBMNCs were isolated from healthy
volunteers using Ficoll density gradient centrifugation, and then
the cells were cultured on human fibronectin-coated dishes in
EGM-2, which contained 20% FBS, VEGF, fibroblast growth factor-2
(FGF-2), epidermal growth factor (EGF), insulin-like growth factor
(IGF) and ascorbic acid. Non-adherent cells were removed by washing
with phosphate-buffered saline (PBS) after 3 days in culture, and
adherent cells were maintained in new medium for a further 4
days.
Intracellular fluorescence measurement
of ROS
Intracellular ROS levels were measured by flow
cytometry using the fluorescent probe H2DCFDA. EPCs were
cultured for 7 days with EGM-2. The cells were treated with various
drugs and then loaded with 5 µM H2DCFDA in serum-free
medium excluding interference of phenol red at 37°C for 30 min.
After washing twice with PBS, cells were immediately monitored
using a flow cytometer (FACSCalibur; BD Biosciences Inc., Brea, CA,
USA) at an excitation wavelength of 488 nm and an emission
wavelength of 525 nm. The levels of ROS were determined by
comparing the changes in the fluorescence intensity with that of
the control.
Determination of NADPH oxidase
activity and anti-oxidase activity
To determine the activity of NADPH oxidase and
anti-oxidase, the lucigenin-derived enhanced chemiluminescence
assay was used. Briefly, quiescent cells were treated as indicated
and harvested. Following low-spin centrifugation, the pellet was
resuspended in ice-cold buffer (pH 7.0), containing 1 mmol/l
ethylene glycol tetraacetic acid (EGTA), 150 mmol/l sucrose and
protease inhibitor cocktail (Merck Millipore). Subsequently, the
cells were homogenized. The total protein concentration was
determined using a Bradford assay and adjusted to 1 mg/ml. Protein
samples (200 µl), including 500 µmol/l lucigenin as the electron
acceptor and 100 µmol/l NADPH as the substrate, were measured over
6 min in quadruplicate with a luminometer counter (Centro LB 960
Microplate Luminometer; Berthold Technologies GmbH & Co. KG,
Bad Wildbad, Germany). Data were collected at 2 min intervals in
order to measure relative changes in the levels of NADPH oxidase
activity.
Western blot analysis
After treatment, cells were washed three times with
ice-cold PBS and lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 150
mM NaCl, 1% Triton X 100, 0.5% sodium deoxycholate, 1 mM EDTA and 1
mM EGTA) supplemented with protease inhibitor cocktail (Merck
Millipore), 1 mM PMSF, 1 mM Na3VO4 and 10 mM
NaF.
The concentration of protein was determined by the
Bradford method. After denaturing at 95°C for 5 min, a total of 30
µg protein was loaded in each lane and subjected to 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The
protein was then transferred to a polyvinylidene difluoride (PVDF)
membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
membranes were blocked in 5% nonfat milk and then incubated
overnight with primary antibody at the appropriate dilution, before
incubation for 1 h with a secondary antibody conjugated to
horseradish peroxidase (1:10,000). After the reaction with the
enhanced chemiluminescence reagent (Amersham, Haemek, Israel), the
images were captured using the Image Reader LAS-4000 system
(Fujifilm, Tokyo, Japan).
Transwell migration assay
Following the indicated treatments, the culture
medium was removed and replaced with EBM-2 without any supplement.
After 12 h, EPC migration was evaluated by using a Transwell
migration assay. Briefly, 3×104 cells were suspended in
100 µl of EBM-2 supplemented with 0.1% BSA and placed in the upper
chamber of 8.0-µm pore size Transwell inserts (Merck Millipore).
VEGF in serum-free EBM-2 was placed in the lower compartment of the
chamber. After incubation for 24 h at 37°C in 5% CO2,
the cells that had not migrated were removed from the upper surface
of the filters using cotton swabs and those that had migrated to
the lower surface of the filters were fixed in 4% paraformaldehyde
and stained with 4′,6-diamidino-2-phenylindole (Roche Applied
Science, Indianapolis, IN, USA). Cells that had migrated into the
lower chamber were counted manually in five random microscopic
fields at a magnification, ×100.
Cell adhesion assay
After 24-h incubation with Hcy, EPCs were washed
with PBS and gently detached using 0.25% trypsin. Following
centrifugation and resuspension in EGM-2, identical cell numbers
were re-plated onto fibronectin-coated culture dishes and incubated
for 30 min at 37°C. Adherent cells were counted by independent,
blinded investigators.
Small interference (si)RNA
transfection
p67phox sense siRNA sequence is
5′-CAGGGAACAUUGUCUUUGUdTT-3′ and the anti-sense siRNA sequence is
5′-ACAAAGACAAUGUUCCCUGdTT-3′. The Nox2 sense siRNA sequence is
5′-CUCUGCGAUUCACACCAUUdTT-3′ and the anti-sense siRNA sequence is
5′-AAUGGUGUGAAUCGCAGAGdTT-3′. The negative control (NC) sense siRNA
sequence was 5′-UUCUCCGAACGUGUCACGUdTT-3′ and the NC anti-sense
siRNA sequence was 5′-ACGUGACACGUUCGGAGAAdTT-3′. siRNA duplexes
were synthesized by GenePharma (Shanghai, China). siRNA
transfection into the EPCs was performed using Hiperfect
Transfection Reagent (Qiagen AB, Sollentuna, Sweden) according to
the manufacturer's instructions. Briefly, 150 pmol of siRNA against
p67phox, Nox2 or NC siRNA was diluted in the appropriate volume of
serum-free EBM-2 to give a final volume of 500 ml. For complex
formation, 15 ml Hiperfect Transfection Reagent was added to the
diluted siRNA and then incubated for 10 min at room temperature.
Cells were incubated with the transfection complexes for 5 h under
normal growth conditions. After incubation, 1 ml fresh culture
medium containing serum was added to each well for further culture.
Verification of siRNA efficacy was achieved by western
blotting.
Statistical analysis
Data from at least three independent experiments are
expressed as the mean ± standard error of the mean. Data were
analyzed by unpaired Student's t-test for comparisons between two
groups or one-way analysis of variance with the
Student-Newman-Keuls pot hoc test for multiple comparisons.
Statistical analysis was performed using SPSS 19.0 software (IBM
Corp., Armonk, NY, USA). P<0.05 was considered to indicated a
statistically significant difference.
Results
PIO inhibits Hcy-induced PKC
activation
PKC acts as a major signaling system in response to
extracellular signals. In our previous study, we investigated the
phosphorylation levels of PKC in EPCs treated with Hcy and PIO
(13). Cells were treated with Hcy
at a concentration of 200 µM for 7.5, 15, 30, 60 and 120 min. The
results of the western blot analysis showed that the Hcy treatments
significantly increased the phosphorylation levels of PKC.
Pre-treatment with PIO (10 µM) inhibited phosphorylation of PKC in
EPCs.
PIO ameliorates Hcy-induced oxidative
stress possibly by inactivation of PKC
Intracellular ROS levels were measured by flow
cytometry using the fluorescent probe H2DCFH-DA. The levels of
NADPH oxidase activity were measured with lucigenin-enhanced
chemiluminescence in parallel. We also observed the effects of the
PKC inhibitor GF109203X (GF, 5 µM) and an NADPH oxidase inhibitor
(DPI, 5 µM). Pretreatment with PIO and GF profoundly repressed
Hcy-induced ROS production and NADPH oxidase activation, which was
consistent with the outcomes of pretreatment with DPI. The data are
also shown in our previous article (13).
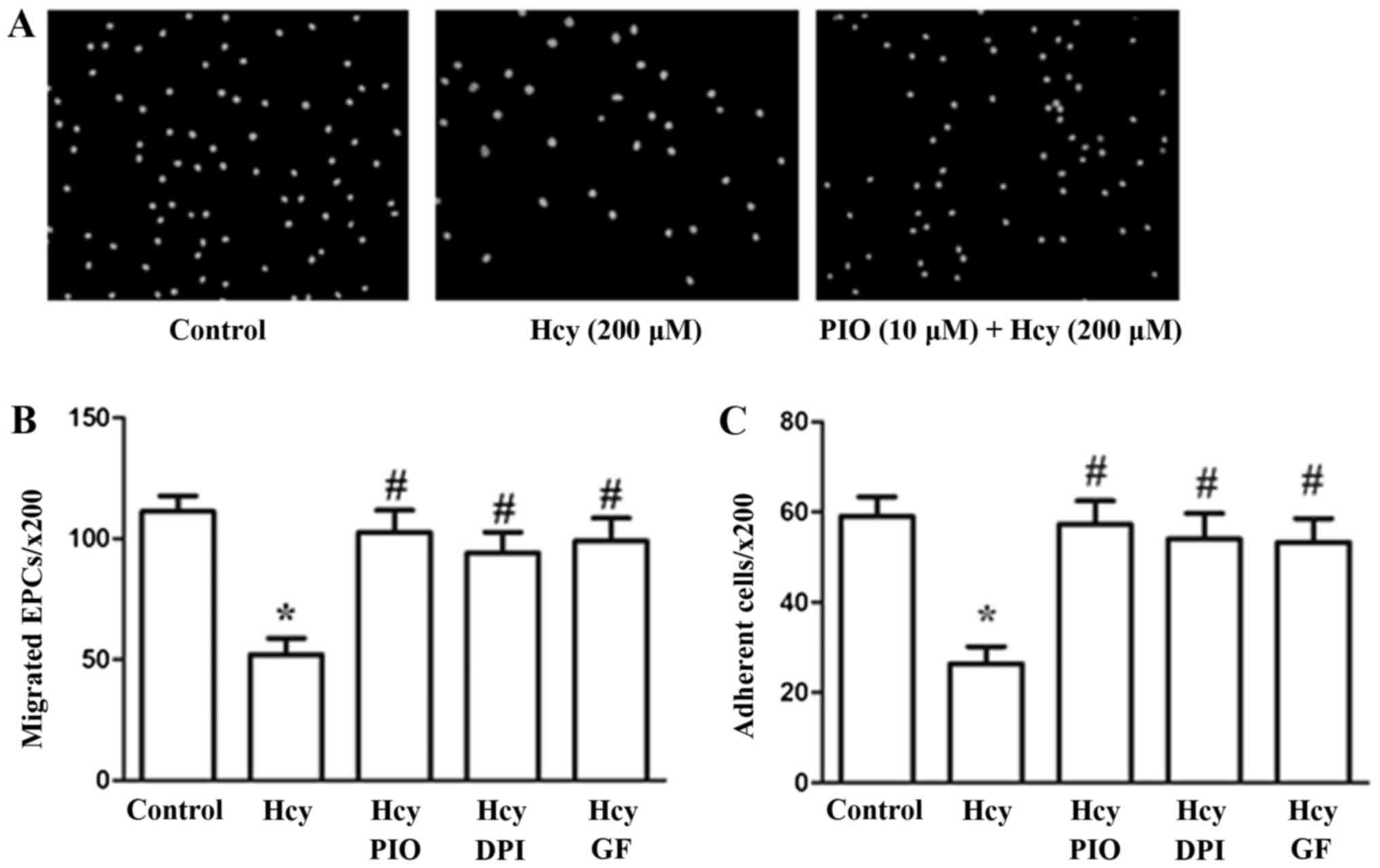
PIO may inhibit Hcy-induced reduction
of EPC migration and adhesiveness through inactivation of the PKC
pathway and reduction of oxidative stress
The effects of PIO, GF and DPI on EPC migration and
adhesion were assayed using 8.0-µm pore size Transwell membranes
and fibronectin-coated culture dishes, respectively. Hcy at a
concentration of 200 µM profoundly impaired cell migration and
adhesiveness in accordance with the results of our previous study.
PIO, GF and DPI suppressed the migration and adhesiveness
impairment induced by Hcy (Fig.
1).
The results revealed that PIO attenuates Hcy-induced
EPC dysfunctions such as migration and adhesiveness possibly by
inhibiting PKC activation and promoting antioxidant properties.
PIO inhibits Hcy upregulation of the
NADPH subunits Nox2 and p67phox
To verify whether the expression levels of NADPH
subunits were affected by Hcy, cells were treated with 0, 10, 50,
100 and 200 µM Hcy for 24 h. The western blot analysis showed that
the levels of Nox2 and p67phox were upregulated by Hcy, with a peak
in the levels after treatment with a concentration of 200 µM.
Pretreatment of EPCs with PIO (10 µM), GF (5 µM) and DPI (5 µM) for
30 min reduced Hcy-dependent Nox2 and p67phox expression (Fig. 2). The data suggests that PIO may
downregulate the levels of Nox2 and p67phox via the PKC
pathway.
Knockdown of Nox2 and p67phox inhibits
Hcy-induced dysfunction of EPCs
To further investigate the potential mechanisms by
which PIO restored Hcy-induced EPC dysfunction, Nox2-siRNA and
p67phox-siRNA transfections were applied to downregulate the
expression levels of these two NADPH subunits. Knockdown of the
expression levels was confirmed by Western blotting as compared
with the levels following treatment with the NC. The western
blotting data (protein level) to confirm the knock down of
Nox2-siRNA and p67phox-siRNA were shown in our previous article
(13). Our results showed that
EPCs transfected with Nox2 and p67phox siRNA exhibited
significantly higher levels of cell migration and adhesion compared
with cells transfected with control siRNA under stimulation with
Hcy (Fig. 3).
Discussion
The present study demonstrated that PKC and NADPH
oxidase play a major role in the protective effects of PIO against
EPC dysfunction induced by high concentrations of Hcy (HHcy).
Hyperhomocysteinemia has been demonstrated to be an
important pathological factor in vascular diseases, including
coronary artery, cerebrovascular and peripheral arterial occlusive
diseases. Hcy has been shown to inhibit the proliferation, adhesion
and migration of human CD34(+) endothelial colony-forming cells
(ECFCs) isolated from peripheral blood in a dose-dependent manner
(14). Hcy dose-dependently
impairs the proliferation, migration and in vitro
vasculogenesis capacity of EPCs (15). Moreover, PIO is convinced to its
use as an insulin sensitizer, PIO is believed to have ‘pleiotropic
effects’, including anti-apoptosis and anti-senescence (16). In our previous study, we confirmed
that Hcy induced downregulation of VEGF and IL-8 expression levels
and their secretion was normalized by PIO treatment. However,
whether PIO exerts the same protective effect on migration and
adhesion required further research.
Our previous data showed that treatment with Hcy
increased the phosphorylation levels of PKC in a time-dependent
manner. We found that PIO inhibited the PKC activation induced by
Hcy and suppressed Hcy-mediated ROS generation via the PKC/NADPH
oxidase signaling pathway. The results of the present study show
that PIO reverses the HHCy-induced inhibition of EPC migration and
adhesion. To explore the mechanism of this effect, the PKC
inhibitor GF and the NADPH oxidase inhibitor DPI were added prior
to treatment of the cells with Hcy. As a result, the Hcy-mediated
reduction in the levels of migration and adhesion was reversed,
which suggests that PIO attenuates Hcy-induced EPC dysfunction by
inhibition of PKC and NADPH oxidase. Additional signaling pathways
leading to Hcy-induced EPC dysfunction may be elucidated in further
studies.
Hcy seems to promote the formation of ROS primarily
through biochemical mechanism involving NADPH oxidase (Nox),
endothelial nitric oxide synthase (eNOS) and endothelial lipid
peroxidation (17). An increase of
ROS including hydrogen peroxide (H2O2) and
superoxide anion (O2-) was produced by activation of the
above in-vivo metabolism. At higher levels,
O2-will react with NO to form a cytotoxic peroxynitrite
(ONOO-) and to decrease NO that plays a critical role in
endothelial cell damage (18).
NADPH oxidases have emerged as major enzymes responsible for the
production of ROS in the blood vessel wall during cardiovascular
disease progression (19). Hcy
promotes the formation of ROS primarily via a biochemical mechanism
involving NADPH oxidase. The family of NAPDH oxidases comprises
seven members, each based on a distinct core catalytic subunit.
Nox2 (also known as gp91phox oxidase) was the first NADPH oxidase
to be identified, and p67phox is one of its cytosolic components
(20). The present study shows
that Hcy dose-dependently increased the expression levels of
p67phox and Nox2 and that PIO could inhibit overexpression of
p67phox and Nox2. We further demonstrated that p67phox siRNA and
Nox2 siRNA suppressed Hcy-impaired EPC function. This data suggests
that PIO attenuated Hcy-induced EPC dysfunction possibly by
inhibition of NADPH oxidase.
In the present study, we observed that PIO restored
Hcy-impaired EPC migratory and adhesive capacity via inhibition of
PKC and NADPH oxidase. Our previous study demonstrated that PIO
also normalized the production of VEGF and IL-8 in EPCs that had
been impaired by treatment with Hcy. These beneficial effects of
PIO make it a potential therapeutic strategy in EPC-based
cytotherapy for ischemic cardiovascular diseases such as hindlimb
ischemia in patients with diabetes and myocardial infarction.
EPC-driven neovascularization includes the migration of EPCs
through the bloodstream, the adhesion between EPCs and the
endothelium, and subsequent matrix degradation and migration of
EPCs towards sites of ischemia, where EPCs create an angiogenic
microenvironment through the secretion of cytokines and growth
factors and induce sprouting angiogenesis by the surrounding
endothelium (21). PIO may enhance
the angiogenesis inhibited by Hcy via restoration of the migratory,
adhesive and paracrine activity of EPCs (Fig. 4).
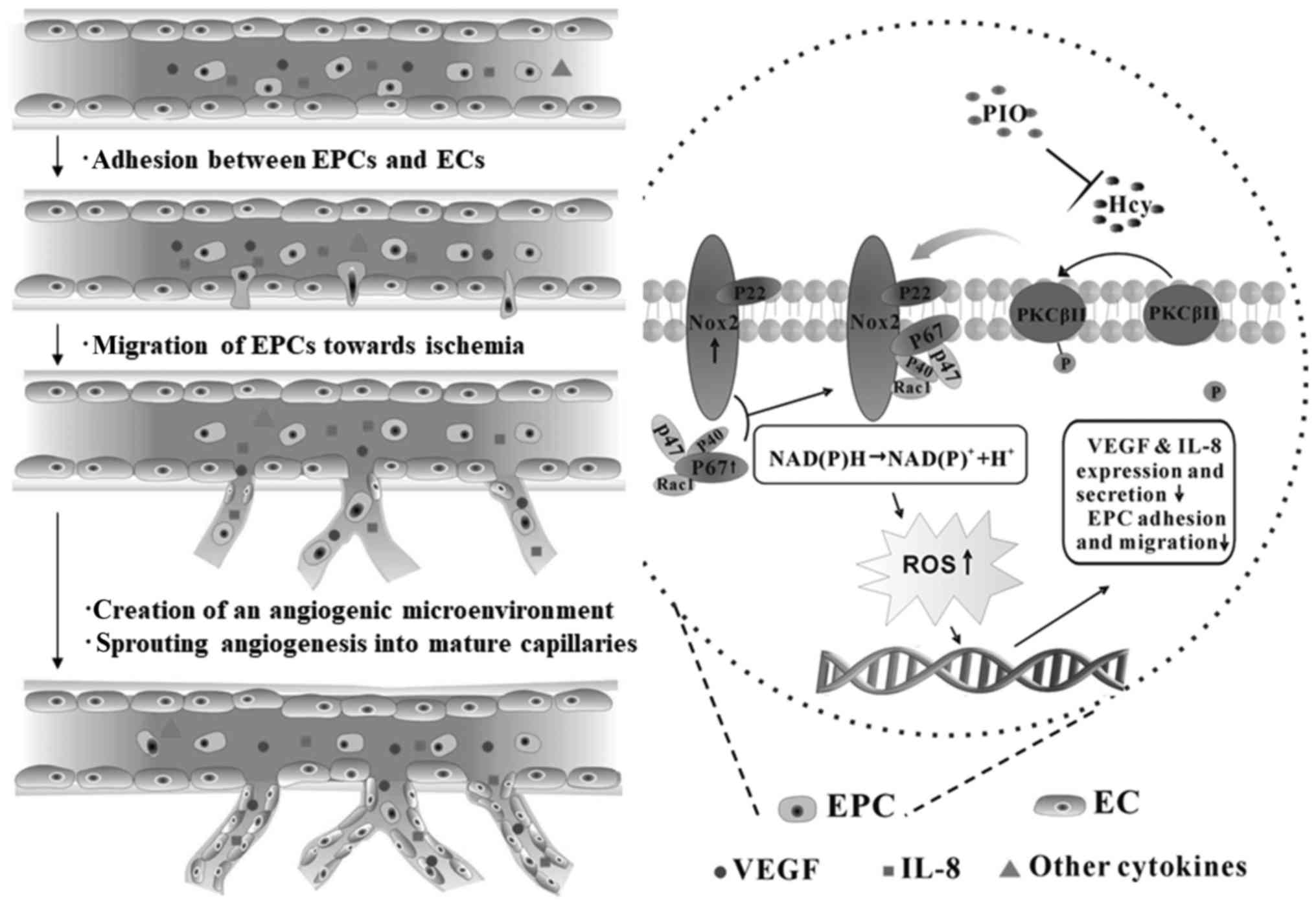 | Figure 4.Mechanisms underlying PIO attenuation
of Hcy-induced EPC dysfunction. Pretreatment of PIO restored EPC
migratory and adhesive activity. EPC-enhanced angiogenesis in sites
of ischemia via the following steps: i) EPCs adhere to the
endothelium; ii) transendothelial migration and degradation of the
endothelial basement membrane; iii) tunneling of EPCs create a
temporary capillary-like scaffold for the neovasculature; and iv)
EPCs continue to secrete multiple cytokines. In this process, VEGF
and IL-8 are key; however, other biomarkers are required for
further research. Hcy, homocysteine; EPC, endothelial progenitor
cell; PIO, pioglitazone; NADPH, nicotinamide adenine dinucleotide
phosphate; Nox2, NADPH oxidase 2; VEGF, vascular endothelial growth
factor; IL, interleukin; PKC, protein kinase C; ROS, reactive
oxygen species; EC, endothelial cells. |
In conclusion, the present study demonstrated that
PIO attenuates HHcy-induced EPC dysfunction, such as impaired
migratory and adhesive capacity. The mechanism of its protective
role on the migration and adhesion of EPCs was mediated by
inactivation of the PKC pathway and inhibition of the production of
intracellular oxidative products from NADPH oxidase. We further
confirmed the mechanism via knockdown of p67phox and Nox2. The
findings of the present study indicate the potential therapeutic
role of PIO in EPC dysfunction under HHcy stimulation and present a
possible EPC-based cytotherapy for patients with ischemic vascular
diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81500211) and
Natural Science Foundation of Zhejiang Province (grant no.
LY18H020001).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and SX conceived and designed the study. JZ, MW
and QL performed the experiments. JZ and YZ collected the data and
wrote the paper. LY and YZ analyzed the data, and LY interpreted
the data. SX reviewed and edited the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
participants. The present study was approved by the Ethics Review
Board of Sir Run Run Shaw Hospital, College of Medicine, Zhejiang
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abe Y, Ozaki Y, Kasuya J, Yamamoto K, Ando
J, Sudo R, Ikeda M and Tanishita K: Endothelial progenitor cells
promote directional three-dimensional endothelial network formation
by secreting vascular endothelial growth factor. PLoS One.
8:e820852013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu JH, Chen JZ, Wang XX, Xie XD, Sun J
and Zhang FR: Homocysteine accelerates senescence and reduces
proliferation of endothelial progenitor cells. J Mol Cell Cardiol.
40:648–652. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen JZ, Zhu JH, Wang XX, Zhu JH, Xie XD,
Sun J, Shang YP, Guo XG, Dai HM and Hu SJ: Effects of homocysteine
on number and activity of endothelial progenitor cells from
peripheral blood. J Mol Cell Cardiol. 36:233–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bao XM, Wu CF and Lu GP: Atorvastatin
inhibits homocysteine-induced oxidative stress and apoptosis in
endothelial progenitor cells involving Nox4 and p38MAPK.
Atherosclerosis. 210:114–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang W, Vellaisamy K, Li G, Wu C, Ko CN,
Leung CH and Ma DL: Development of a long-lived luminescence probe
for visualizing β-galactosidase in ovarian carcinoma cells. Anal
Chem. 89:11679–11684. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ko CN, Wu C, Li G, Leung CH, Liu JB and Ma
DL: A long-lived ferrocene-conjugated iridium(III) complex for
sensitive turn-on luminescence detection of traces of DMSO in water
and human serum. Anal Chim Acta. 984:193–201. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mazzone T, Meyer PM, Feinstein SB,
Davidson MH, Kondos GT, D'Agostino RB Sr, Perez A, Provost JC and
Haffner SM: Effect of pioglitazone compared with glimepiride on
carotid intima-media thickness in type 2 diabetes: A randomized
trial. JAMA. 296:2572–2581. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spigoni V, Picconi A, Cito M, Ridolfi V,
Bonomini S, Casali C, Zavaroni I, Gnudi L, Metra M and Dei Cas A:
Pioglitazone improves in vitro viability and function of
endothelial progenitor cells from individuals with impaired glucose
tolerance. PLoS One. 7:e482832012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gensch C, Clever YP, Werner C, Hanhoun M,
Böhm M and Laufs U: The PPAR-gamma agonist pioglitazone increases
neoangiogenesis and prevents apoptosis of endothelial progenitor
cells. Atherosclerosis. 192:67–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Imanishi T, Kobayashi K, Kuroi A, Ikejima
H and Akasaka T: Pioglitazone inhibits angiotensin II-induced
senescence of endothelial progenitor cell. Hypertens Res.
31:757–765. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang X, Yang F, Tan H, Liao D, Bryan RM
Jr, Randhawa JK, Rumbaut RE, Durante W, Schafer AI, Yang X and Wang
H: Hyperhomocystinemia impairs endothelial function and eNOS
activity via PKC activation. Arterioscler Thromb Vasc Biol.
25:2515–2521. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Siow YL, Au-Yeung KK and Woo CW OK:
Homocysteine stimulates phosphorylation of NADPH oxidase p47phox
and p67phox subunits in monocytes via protein kinase Cbeta
activation. Biochem J. 398:73–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu S, Zhao Y, Jin C, Yu L, Ding F, Fu G
and Zhu J: PKC/NADPH oxidase are involved in the protective effect
of pioglitazone in high homocysteine-induced paracrine dyfunction
in endothelial progenitor cells. Am J Transl Res. 9:1037–1048.
2017.PubMed/NCBI
|
|
14
|
Nelson J, Wu Y, Jiang X, Berretta R,
Houser S, Choi E, Wang J, Huang J, Yang X and Wang H:
Hyperhomocysteinemia suppresses bone marrow CD34+/VEGF receptor 2+
cells and inhibits progenitor cell mobilization and homing to
injured vasculature-a role of β1-integrin in progenitor cell
migration and adhesion. FASEB J. 29:3085–3099. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bao XM, Wu CF and Lu GP: Atorvastatin
inhibits homocysteine-induced dysfunction and apoptosis in
endothelial progenitor cells. Acta Pharmacol Sin. 31:476–484. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Werner C, Gensch C, Pöss J, Haendeler J,
Böhm M and Laufs U: Pioglitazone activates aortic telomerase and
prevents stress-induced endothelial apoptosis. Atherosclerosis.
216:23–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alvarez-Maqueda M, El Bekay R, Monteseirín
J, Alba G, Chacón P, Vega A, Santa María C, Tejedo JR, Martín-Nieto
J, Bedoya FJ, et al: Homocysteine enhances superoxide anion release
and NADPH oxidase assembly by human neutrophils. Effects on MAPK
activation and neutrophil migration. Atherosclerosis. 172:229–238.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ungvari Z, Csiszar A, Edwards JG, Kaminski
PM, Wolin MS, Kaley G and Koller A: Increased superoxide production
in coronary arteries in hyperhomocysteinemia: Role of tumor
necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide
synthase. Arterioscler Thromb Vasc Biol. 23:418–424. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sirker A, Zhang M and Shah AM: NADPH
oxidases in cardiovascular disease: Insights from in vivo models
and clinical studies. Basic Res Cardiol. 106:735–747. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: Physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krenning G, van Luyn MJ and Harmsen MC:
Endothelial progenitor cell-based neovascularization: Implications
for therapy. Trends Mol Med. 15:180–189. 2009. View Article : Google Scholar : PubMed/NCBI
|