Introduction
Ozone (O3) is a strong oxidizing agent
and has been widely used in the treatment of protrusion of lumbar
intervertebral disc (PLID), failed back surgery syndrome (FBSS),
soft tissue lesions and arthralgia (1–3).
However, Ginanneschi et al have reported ventral and dorsal
root injury after intervertebral disc infiltration of
O2O3 on L4-L5 disc herniation. Thus,
physicians must distinctly comprehend the risk of potential
complications (4). Therefore,
whether O3 has neurotoxicity in spinal cord neurons
(SCNs) has led to increasing attention to the clinical applications
of O3. Previous study results suggest that exposure to
O3 may cause SCNs death and that the neurotoxicity
caused by O3 is related to the increasing calcium
(Ca2+) release from endoplasmic reticulum (ER), thus
enhancing the activity of the Ca2+/calmodulin-dependent
protein kinase II (CaMKII)/mitogen-activated protein kinase (MAPK)
pathway (5).
The ER is a multifunctional organelle that manages
and participates in a wide range of cellular processes, which
include protein synthesis and protein folding, handling of
misfolded proteins and posttranslational modification, and delivery
of proteins to their eventual destination (6). The ER forms an interconnected network
of flattened, membrane-enclosed sacs in cytoplasm. It is an
organelle that can provide a place to synthesize proteins and
lipids. During ER stress, the intracellular folding protein and
(or) unfolded protein accumulates in the ER. The cell reacts to
obliterate these defective proteins, which is a process called
unfolded protein response (UPR) (7). Oxidative stress damage and
Ca2+ depletion, which are induced by a series of toxic
insults, bring about the accumulation of unfolded or misfolded
proteins in the ER and lead to the dysfunction of the UPR,
resulting in the onset of ER stress and cell death (8). In mammalian cells, several
ER-residing transmembrane proteins have been identified as
transducers of the ER stress signaling pathway, namely:
Inositol-requiring ER-to-nucleus signal kinase 1 (IRE1), PKR-like
ER kinase (PERK) and activating transcription factor 6 (ATF6)
(9). Following activation of UPR,
these transducers of ER stress are dimerized or cleaved to reduce
the UPR response. In particular, dimerization and phosphorylation
of IRE1 with RNase activity promotes the splicing of X box binding
protein-1 (XBP1s) mRNA in mammalian cells. ER stress activates the
RNase activity of IRE1α, leading to removal of 26 nucleotides from
XBP1s transcripts by unorthodox RNA splicing, which transforms the
inactive form of XBP1mRNA (XBP1u) into an active form (XBP1s)
(10). In addition, it has been
reported that processed XBP1 mRNA is rendered into the 54 kDa
processed XBP1s (11). XBP1s is a
basic leucine zipper (bZIP)-containing transcription factor, which
was initially recognized as a protein attaching to the cis-acting X
box region in the promoter region of the human major
histocompatibility complex class II genes. It is indispensable for
immunoglobulin secretion and the development of plasma cells in
mammals. The expression of GRP78 is induced by XBP1s through a
promoter which contains the ER response element (ERSE).
Accordingly, ER stress results in dimerization of PERK, resulting
in the activation of PERK, thereby inducing phosphorylation of
eIF2α, the general attenuation of protein translation, and
increased translation of the transcriptional factor ATF4 protein
(12). The increasing translation
of ATF4 can regulate the expression of C/EBP homologous protein
(CHOP), which is the downstream gene of ATF4. Both GRP78 and CHOP,
which are sentinel markers for ER stress under pathologic
conditions, bind to newly synthesized proteins transiently and are
then translocated into the ER along with permanent unfolded or
misfolded proteins (13).
It has been reported that XBP1s are effective
transcription factors with diverse targets specific for the
expression of ER chaperones and ER-associated degradation (ERAD)
components, both of which are crucial to discharge the ER burden
(14). However, ER stress may play
an essential role in neuronal apoptosis, and the role of XBP1s in
O3-induced ER stress remains unknown (15). The current study investigated
whether exposure to low concentrations of O3 induced
increased expression of GRP78 and XBP1s, and whether activation of
XBP1s is neuroprotective against O3-induced ER
stress.
Materials and methods
Animals and reagents
All programs involving the use of animals were
conducted in adherence to the guidelines of the National Institutes
of Health (NIH; Bethesda, MD, USA) and were approved by the Animal
Care and Use Committee of the School of Medicine of Shandong
University (Shandong, China). Neurobasal medium and B27 supplement
were purchased from Gibco; Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). Trypsin, phenylmethylsulfonyl fluoride (PMSF) and bovine
serum albumin (BSA) were obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). The following primary antibodies were
purchased from Cell Signaling Technology (Danvers, MA, USA): XBP1s,
CHOP and GRP78. Anti-β-actin antibody was purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA).
Construction of recombinant
adenovirus
The complementary DNAs encoding the full-length Rat
XBP1s proteins were amplified from isolated ovary total RNA by
reverse transcriptase polymerase chain reaction (RT-PCR). To clone
the full-length XBP1s cDNA (GenBank acc. no. NM_005080), the
following primers were used: Forward,
5′-TGAAGCTTTGCGTAGTCTGGAGCTATGG-3′ and reverse,
5′-TGCTCGAGATTGGATCATTCCTTAGACA-3′. The coding reading frame of the
cloned XBP1s gene was identified by sequencing and subcloned into
shuttle plasmid pShuttle-CMV (Agilent, US). The constructed
recombinant shuttle plasmid pShuttle-CMV-XBP1s was digested with
I-CeuI and I-SceI, and the obtained expression frame of the XBP1s
gene was linked to the vector carrying the pAdEasy adenovirus
backbone. The recombinant adenovirus backbone was linearized by
digestion with PacI and infected into HEK 293 cells for packing to
obtain recombinant adenovirus Adv-XBP1s. The adenovirus was
purified from the HEK 293 cell extracts by cesium chloride (CsCl;
Nacalai Tesque, Kyoto, Japan) density ultracentrifugation according
to the company manual (Takara Bio, Inc., Otsu, Japan). Viral stocks
were stored in 10% glycerol at −80°C and diluted with phosphate
buffered saline (PBS) to the appropriate concentration prior to
use. Ad-GFP, which encodes enhanced green fluorescent protein
(Clontech, Palo Alto, CA), was constructed similarly by overlap
recombination. The Adv-LacZ was made by the same procedure.
Primary spinal cord neuronal
cultures
Primary SCNs cultures were prepared from 30 Wistar
rats (1–2-days-old), as previously described (16) with minor modifications. The rats
were kept by breastfeeding during a 12-h dark/light diurnal cycle.
Briefly, the spinal cord from newborn rats within 48 h was digested
for 30 min with trypsin (0.125%; Sigma-Aldrich; Merck KGaA). 10%
fetal calf serum (FCS) was added to stop digestion and passed
through a 100-eye sieve. The cell suspension was centrifuged at
1,000 rpm for 8 min. Then, cells were washed three times with a
basic culture medium with 2 mmol/l L-glutamine, penicillin (100,000
U/l), streptomycin (100 mg/l) and 10% FCS. Cells were resuspended
in fresh Neurobasal medium with 2 mmol/l L-glutamine, 2% (v/v) B27
supplement, penicillin (100,000 U/l) and streptomycin (100 mg/l).
Cells were seeded on poly-L-lysine (0.1 g/l)-coated dishes at a
density of 0.125×106 cells/cm2 for the cell
counting kit-8 (CCK-8) assay or 0.33×106
cells/cm2 for protein extraction for western blot or
0.25×106 cells/cm2 for total RNA extraction
for reverse transcription-quantitative polymerase chain reaction
(RT-qPCR). For fluorescence experiments, neurons were mounted on
poly-L-lysine-coated glass coverslips at a density of
0.1×106 cells/cm2 or on poly-L-lysine-coated
dishes at 0.4×106 cells/cm2. After 24 h,
cells were treated with 0.1 mg/ml of cytosine arabinoside
(Sigma-Aldrich; Merck KGaA) to suppress glia proliferation. Fresh
growth medium was applied 2 times a week. Under these conditions,
glia growth was <10%.
Microtubular associated protein 2 (MAP2) is
specifically expressed in the neuron. We identified neurons by a
positive expression of MAP2 by immunofluorescence. The cells were
identified after culturing for 7 days. Briefly, cells were washed
with PBS three times and fixed with 4% paraformaldehyde for 15 min.
Then, 0.2% TritonX-100 was used for cell permeabilization for 5
min, after which cells were blocked with goat serum at room
temperature for 1 h and incubated with anti-MAP2 monoclonal (cat.
no. ab32454; 1:2,000; Abcam, Cambridge, UK) overnight at 4°C. The
next day, cells were incubated with secondary antibody (cat. no.
ab150077; 1:400; Abcam) for 1 h in a dark room. Stained neurons
were visualized using immunofluorescence microscopy.
Adenovirus transduction
Before being exposed to adenoviral vectors, the
primary SCNs were maintained for two days. Culture medium was added
to dilute high titer adenovirus to accomplish a multiplicity of
infection (MOI) of 20–400 plaque forming units (pfu)/cell. 100 ml
of culture medium was removed and replaced with 50 ml of culture
medium with adenovirus preparation for transduction. 50 ml of
medium was added to the culture after 2 h. Incubation with
adenovirus continued for 16 h. Then, the adenovirus medium was
removed and cells were cultured in fresh culture medium. Primary
SCNs were treated with adenovirus one day after plating. Cells were
incubated with adenovirus at an MOI of 0–500 for 2 h. Then, the
adenovirus-containing medium was replaced with fresh medium after 2
h.
O3 exposure
The O3 concentration was obtained through
the pre-experiment. CCK-8 assay result showed that the medical
O3 of 40 µg/ml can reduce neuronal cell viability
significantly (5). Cells were
exposed to 40 µg/ml O3 or tunicamycin (2 µg/ml) for 60
min. SCNs exposure to O3 at specified levels were
carried out in an O3 exposure chamber in vitro,
which was controlled by a computer (17). The SCNs stored a thin layer of
medium apically and were exposed to O3. Cells were
rocked to expose one side of the apical surface at the time to
O3 exposure. O3 concentration in the chamber
was detected accurately by an O3 analyzer (Model
MD-050-12-f-4; Perma Pure Inc., Toms River, NJ, USA) and adjusted
by a computerized system.
Isolation of RNA and RT-qPCR
Total RNA was prepared by TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions and was reverse transcribed to cDNA
using the MML-V reverse transcriptase kit (Takara Bio, Inc.). The
expression of XBP1s mRNA was examined by qPCR on a 7500-real time
RT-PCR system and software (Prism 7500; Applied Biosystems Inc.,
Foster City, CA, USA) using a Realtime PCR Master Mix kit (Thermo
Fisher Scientific, Inc., Pittsburgh, PA, USA). qPCR was performed
under the following conditions: 95°C for 30 sec followed by 40
cycles of 95°C for 5 sec and 60°C for 34 sec. GAPDH was used as an
internal standard. The Prime sequences are shown as follows, GAPDH
forward, 5′-CCCCCAATGTATCCGTTGTG-3′ and reverse,
5′-TAGCCCAGGATGCCCTTTAGT-3′; XBP1s forward,
5′-TGAAGCTTTGCGTAGTCTGGAGCTATGG-3′ and reverse,
5′-TGCTCGAGATTGGATCATTCCTTAGACA-3′. Samples were determined in
triplicate.
Western blot analysis
Cells were collected after transfection for 24 h.
Total protein was prepared according to the manufacturer's
directions. Briefly, rat SCNs were lysed in RIPA buffer with
protease inhibitors (in mmol/l: Leupeptin, 0.1 and PMSF, 0.3), and
stored on ice for 30 min and vortexed every 5 min. The supernatant
was collected after centrifugation at 14,000 rpm for 30 min at 4°C.
The protein concentration was detected by a Bio-Rad DC protein
assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). A total
of 50 µg protein was loaded in each lane of standard 4–12%
SDS-polyacrylamide gels to ensure loading consistency, as
previously described. Following electrophoresis and transfer of
proteins to nitrocellulose membranes, the membranes were blocked in
PBS containing 0.1% Tween (TBST) and 5% non-fat milk for 1 h.
Membranes were incubated in primary anti-GRP78 (ab108613; 1:1,000),
anti-CHOP (CST; cat. no. 2895; 1:1,000) and anti-XBP1s (cat. no.
ab37152; 1:1,000; all Abcam) antibody overnight at 4°C. The
membranes were incubated with horseradish peroxidase-labeled goat
anti-rabbit IgG (Thermo Fisher Scientific, Inc.) as a secondary
antibody (1:2,000) at room temperature for 1 h and then developed
using ECL (GE Healthcare, Chicago, IL, USA) for detection. The
bands densities were quantified by Image J image software v2.1.4.7
(NIH).
Evaluation of neuronal injury
Before exposure to O3 with the presence
or absence of 24 h Adv-XBP1s pretreatment, cells in 96-well plates
(2×104 cells/well) were washed with fresh medium and
were cultured. Then, fresh medium was used to incubate the cells.
Finally, the CCK-8 solution (10 µl) (Dojindo Laboratories,
Kumamoto, Japan) was added to each well for 2 h at 37°C. Samples
were measured by optical absorbance at 450 nm using an absorbance
microplate reader (ELx800 Absorbance Microplate Reader; Bio-Tek,
USA).
Statistical analysis
Data are presented as the mean ± standard deviation
(SD). The results were analyzed using one-way analysis of variance
(ANOVA), followed by the Student-Newman-Keuls test for multiple
comparisons with SPSS v13.0 Software (SPSS Inc., Chicago, IL, USA).
A P-value of P≤0.05 was considered statistically significant.
Results
Adenovirus mediated gene transfer into
the primary SCNs
Adenovirus transduction efficiency was first
optimized in the cultures. The primary SCNs were transduced with
different adenovirus MOI (5, 10, 50, 100, 150, 200, or 500) and
with varying adenovirus incubation times (2, 8, 18, 24 or 36 h)
(Fig. 1). We found the best
transduction efficiency was when the primary cultures were
transfected with adenovirus two days after planting and incubation
for 24 h. Under the above conditions, 83% XBP1s-positive cells
could be seen in the primary SCNs three days after transduction
with an MOI of 100 pfu/cell (Fig. 1A
and B). Cytotoxic effects began to appear as MOI of 500 pfu/
cell.
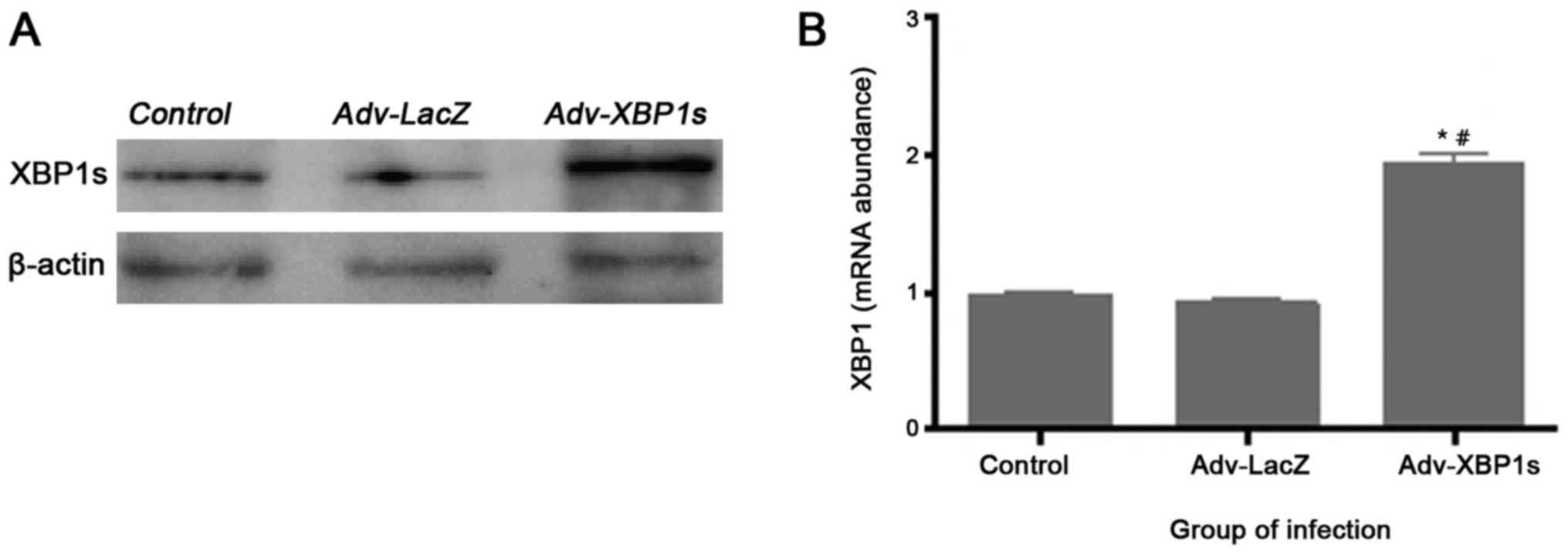
Adenovirus mediated overexpression of
XBP1s in primary SCNs
To examine in vitro XBP1s expression levels
following Ad vector infection, Western blotting and quantitative
Real-time-PCR analysis were performed. We observed an apparent
increase in the XBP1s expression after transduction with Adv-XBP1s
in primary SCNs (Fig. 2). The
control Ad vector Ad-LacZ did not increase XBP1s expression,
indicating that transduction with Ad vectors alone does not induce
any change in the cell systems.
O3 exposure induced the ER
stress response and XBP1splicing
ER stress, which is internally linked to cell
apoptosis, is one of the primary cell stress responses. Therefore,
neurons are hypersensitive to ER stress challenge, and the current
study investigated whether O3 (40 µg/ml O3
for 60 min) activated the ER stress response and expression of
GRP78 and CHOP, which are involved in ER stress in SCNs. As shown
in Fig. 3A and B, the ER stress
response of SCNs exposed to O3 was evidenced by an increase in
GRP78 and CHOP protein levels. These results show that
O3 (40 µg/ml O3 for 60 min) activates the ER
stress response.
Treatment of SCNs with O3 (40 µg/ml
O3 for 60 min) also increased the expression of XBP1s,
as detected by a specific antibody (Fig. 3C). Analysis of XBP1 mRNA revealed
O3 induced splicing of XBP1 mRNA. The splicing of XBP1
mRNA caused by O3 in SCNs was the same as that induced
by the classical ER stress inducer Tunicamycin (Fig. 3D). These results indicate that
O3 exposure increased XBP1s expression in SCNs.
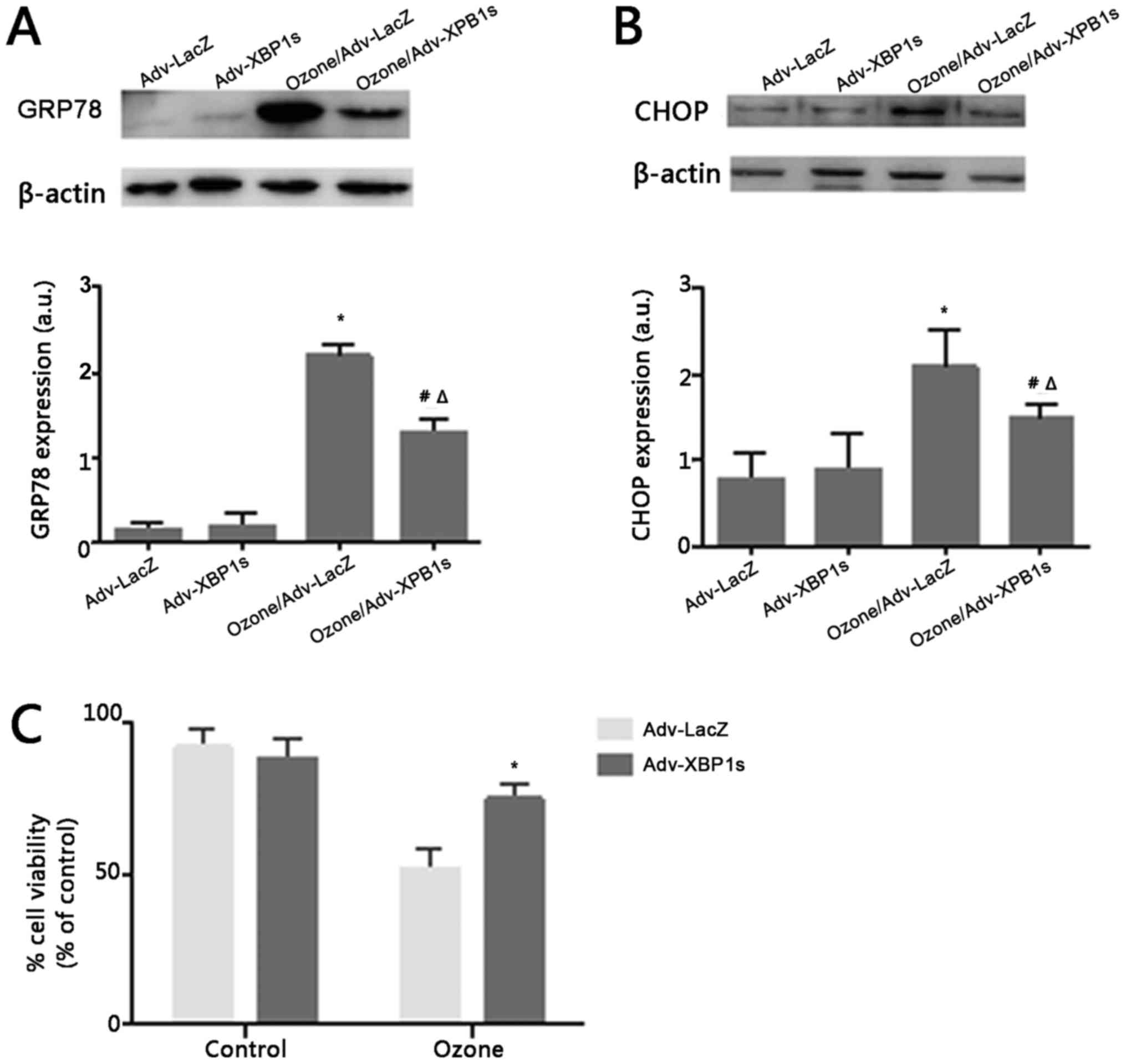
Adenovirus-mediated XBP1s
overexpression prevent ER stress and protects O3-induced
cell death in rat primary SCNs
During UPR, XBP1s regulate the expression of many ER
stress factors that include ER chaperones and ERAD components that
repair normal ER physiology. Thus, we conjecture whether XBP1s
overexpression could revert the ER stress induced by O3.
To detect ER stress independently of XBP1, we examined the activity
of GRP78 and CHOP. Before exposure to O3, both GRP78 and
CHOP expression was maintained at a lower level. However, after
exposure to O3, GRP78 and CHOP expression was
significantly higher (Fig. 4A and
B). This suggests that overexpression of XBP1s may play a
neuroprotective role in ER stress.
To investigate the effect of Adv-XBP1s on
O3-induced SCNs injury, SCNs were pretreated with 100MOI
Adv-XBP1s for 24 h, followed by 40 µg/ml O3 treatment
for 1 h. As shown in Fig. 4,
O3-induced loss of cell viability was significantly
reversed by the overexpression of XBP1s. These results indicate
that Adv-XBP1s have a protective effect against
O3-induced toxicity in SCNs.
Discussion
Previously, O3 treatment has been widely
applied in the treatment of PLID, FBSS, soft tissue lesions and
arthralgia (18). The effect of
O3 on the CNS, particularly biological mechanisms, has
been confirmed in a previous study conducted in the authors'
laboratory (5). The focus of the
present study was to investigate whether there is a novel and
effective method to prevent neurotoxicity induced by O3.
In the present study, SCNs were exposed to O3 to induce
GRP78 and XBP1 activation, resulting in increased cell death.
Interestingly, pretreatment of cells with Adv-XBP1s inhibited these
effects. The aim of the current study was to provide insight into
the protective role of XBP1s overexpression. To address this aim,
the role of XBP1s, a critical UPR effector, was investigated using
an in vitro rat primary SCNs model of O3
injury.
This study focuses on the protective role of XBP1s
in O3-induced spinal cord neuronal death. First, ER
stress was activated during O3-induced SCNs death. As a
result, an increase in GRP78 and CHOP levels as well as extensive
splicing of XBP1s appeared upon O3 treatment (Fig. 3A-C). Second, SCNs with XBP1s
overexpression prevented cell death, and partially reduced the
protein levels of GRP78 and CHOP (Fig.
4A and B). Strong activation of GRP78 was also observed in
previous studies during tunicamycin (an inhibitor of N-linked
glycosylation) and thapsigargin (an inhibitor of the ER
Ca2+-ATPase) treatment. In our study, we observed a
similar result where the transcription and protein expression
levels of XBP1s in SCNs were significantly increased after
tunicamycin treatment (Fig. 3C and
D). As a sentinel marker for ER stress under pathologic
conditions, GRP78 binds transiently to newly synthesized proteins
translocated into the ER and more permanently to unfolded or
misfolded proteins (19). GRP78
can trigger the UPR via dissociation from its interaction partners
PERK, ATF6 and IRE1a, which can subsequently lead to activation of
ER stress responses involving an induced expression of ER
chaperones to increase the folding capacity of the ER. When ER
stress occurs, sensor proteins from the ER membrane are dimerized
or cleaved to weaken this condition. IRE1, which is a sensor
protein with RNase activity, is dimerized and phosphorylated to
expedite splicing of mRNA for XBP1 in mammalian cells (14,20).
XBP1s protein from spliced mRNA induces GRP78 expression via a
promoter containing the ERSE.
In addition, CHOP is a transcription factor
belonging to the (c/EBP) family that induces cell cycle arrest and
apoptosis in ER stress and DNA damage response (21). Basal expression of CHOP is very low
under non-stressed conditions; however, its expression is induced
by a number of adverse physiological conditions. The expression of
CHOP is both cell- and stimulus-dependent, and thus has an
influence on the final result of ER stress. The expression of CHOP
is strongly induced via IRE1 and PERK signaling (22). Our research showed that SCNs
exposed to O3 had clearly raised the expression levels
of CHOP, which is an ER-related chaperone (Fig. 3A and B). This suggested that
O3 (40 µg/ml) might induce SCNs death through ER stress
and DNA damage.
In the UPR, the expressions of ER chaperones and
ERAD-related genes appear to be largely regulated by the
IRE1α-XBP1s pathway (20,23). The activated form of XBP1s
upregulates many ER chaperones and genes involved in ERAD, various
UPR ‘stress genes’, and enzymes involved in membrane biogenesis
(24). In addition, the
overexpression of XBP1s plays a protective role in a neuroblastoma
cell line against cell death, which is induced by proteasome
inhibition (25). XBP1s
overexpression protected against oxygen and glucose
deprivation/reoxygenation injury in rat primary hippocampal neurons
(26). This is meet with our
research. Besides, a recent study showed that overexpression of
XBP1s can promote proliferation in bone marrow-derived macrophages
(27), which is in accordance with
our results. Our research work found that overexpression of XBP1s
protected SCNs from O3-induced cell death, which may
play a protective role through the IRE1α-XBP1 pathway. Whether the
protective role of overexpression of XBP1s is involved in some
ER-related genes or enzymes will be investigated in future
research.
Taken together, it was hypothesized that the
activation of the XBP1s pathway and its downstream ER chaperones
could prevent cytotoxicity and play a protective effect. Therefore,
the impact on the activation of the IRE1α-XBP1s pathway in
opposition to cell death induced by these damages in addition to
representative ER stressors were investigated. As expected,
pretreatment with adenovirus-mediated overexpression of XBP1s
strongly reduced SCNs death induced by O3.
These results suggest that XBP1s activation could
potentially be applied to prevent ER stress-related neurotoxicity
and this cytoprotective effect was confirmed by XBP1s
overexpression. Furthermore, XBP1s and other components of the ER
stress response pathway could serve as potential drug targets in
preventative strategies for neurotoxicity associated with ER
stress. While the current study provides in vitro data
regarding the protective role of overexpression of XBP1 in SCNs,
in vivo therapeutic implications are still required.
Acknowledgements
The authors would like to thank the Central
Laboratory affiliated Shandong Provincial Hospital (Jinan,
China).
Funding
The present study was funded by grants from the
National Natural Science Foundation of China (grant no. 81271346)
and the Natural Science Foundation of Shandong Province, China
(grant no. ZR2010HM097).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZF, XL and YL conceived and designed the
experiments. YL and XZ performed the experiments. XJZ, JX and TS
conducted data analysis. YL and XZ produced the manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by Animal Care
and Use Committee of Shandong Provincial Hospital Affiliated to
Shandong University.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Elvis AM and Ekta JS: Ozone therapy: A
clinical review. J Nat Sci Biol Med. 2:66–70. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paoloni M, Di Sante L, Cacchio A, Apuzzo
D, Marotta S, Razzano M, Franzini M and Santilli V: Intramuscular
oxygen-ozone therapy in the treatment of acute back pain with
lumbar disc herniation: A multicenter, randomized, double-blind,
clinical trial of active and simulated lumbar paravertebral
injection. Spine (Phila Pa 1976). 34:1337–1344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Y, Ma Y, Jiang J, Ding T and Wang J:
Treatment of the lumbar disc herniation with intradiscal and
intraforaminal injection of oxygen-ozone. J Back Musculoskelet
Rehabil. 26:317–322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ginanneschi F, Cervelli C, Milani P and
Rossi A: Ventral and dorsal root injury after oxygen-ozone therapy
for lumbar disk herniation. Surg Neurol. 66:619–621. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Lin X, Zhao X, Xie J, JunNan W, Sun
T and Fu Z: Ozone (O3) elicits neurotoxicity in spinal cord neurons
(SCNs) by inducing ER Ca(2+) release and activating the CaMKII/MAPK
signaling pathway. Toxicol Appl Pharmacol. 280:493–501. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hetz C: The unfolded protein response:
controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Verkhratsky A and Petersen OH: The
endoplasmic reticulum as an integrating signalling organelle: From
neuronal signalling to neuronal death. Eur J Pharmacol.
447:141–154. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang CH, Chiu YC, Huang CF, Chen YW and
Chen PC: Arsenic induces cell apoptosis in cultured osteoblasts
through endoplasmic reticulum stress. Toxicol Appl Pharmacol.
241:173–181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang K and Kaufman RJ: Signaling the
unfolded protein response from the endoplasmic reticulum. J Biol
Chem. 279:25935–25938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin JH, Li H, Yasumura D, Cohen HR, Zhang
C, Panning B, Shokat KM, Lavail MM and Walter P: IRE1 signaling
affects cell fate during the unfolded protein response. Science.
318:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Paschen W, Aufenberg C, Hotop S and
Mengesdorf T: Transient cerebral ischemia activates processing of
xbp1 messenger RNA indicative of endoplasmic reticulum stress. J
Cereb Blood Flow Metab. 23:449–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu PD, Harding HP and Ron D: Translation
reinitiation at alternative open reading frames regulates gene
expression in an integrated stress response. J Cell Biol.
167:27–33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yoshida H, Yanagi H, Yura T and Mori K:
Identification of the cis-acting endoplasmic reticulum stress
response element responsible for transcriptional induction of
mammalian glucose-regulate proteins. Involvement of basic leucine
zipper transcription factors. J Biol Chem. 273:33741–33749. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshida H: Unconventional splicing of
XBP-1 mRNA in the unfolded protein response. Antioxid Redox Signal.
9:2323–33. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen J, Qin J, Liu X, Han Y, Yang Z, Chang
X and Ji X: Nitric oxide-mediated neuronal apoptosis in rats with
recurrent febrile seizures through endoplasmic reticulum stress
pathway. Neurosci Lett. 443:134–139. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marsala M, Kakinohana O, Hefferan MP,
Cizkova D, Kinjoh K and Marsala S: Synaptogenesis and amino acid
release from long term embryonic rat spinal cord neuronal culture
using tissue culture inserts. J Neurosci Methods. 141:21–27. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ahmad A, Ahmad S, Chang LY, Schaack J and
White CW: Endothelial Akt activation by hyperoxia: Role in cell
survival. Free Radic Biol Med. 40:1108–1118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Muto M, Ambrosanio G, Guarnieri G,
Capobianco E, Piccolo G, Annunziata G and Rotondo A: Low back pain
and sciatica: Treatment with intradiscal-intraforaminal O (2)-O (3)
injection. Our experience. Radiol Med. 113:695–706. 2008.(In
English, Italian). View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee AE, Iwakoshi NN and Glimcher LH: XBP-1
regulates a subset of endoplasmic reticulum resident chaperone
genes in the unfolded protein response. Mol Cell Biol.
23:7448–7459. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mihailidou C, Papazian I, Papavassiliou AG
and Kiaris H: CHOP-dependent regulation of p21/waf1 during ER
stress. Cell Physiol Biochem. 25:761–766. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamauchi T, Sakurai M, Abe K, Matsumiya G
and Sawa Y: Impact of the endoplasmic reticulum stress response in
spinal cord after transient ischemia. Brain Res. 1169:24–33. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sado M, Yamasaki Y, Iwanaga T, Onaka Y,
Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R,
Hashimoto T, et al: Protective effect against Parkinson's
disease-related insults through the activation of XBP1. Brain Res.
1257:16–24. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ibuki T, Yamasaki Y, Mizuguchi H and
Sokabe M: Protective effects of XBP1 against oxygen and glucose
deprivation/reoxygenation injury in rat primary hippocampal
neurons. Neurosci Lett. 518:45–48. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tian PG, Jiang ZX, Li JH, Zhou Z and Zhang
QH: Spliced XBP1 promotes macrophage survival and autophagy by
interacting with Beclin-1. Biochem Biophys Res Commun. 463:518–523.
2015. View Article : Google Scholar : PubMed/NCBI
|